

Introduction
Os anticorpos capazes de se dobrar e funcionar no ambiente intracelular são ferramentas promissoras para pesquisa e aplicações terapêuticas. Eles têm a capacidade de modular a actividade da proteína de ligação a uma proteína alvo dentro de células para prevenir interacções proteína-proteína, romper interacções proteína-ácido nucleico, ou impedir o acesso do substrato para enzimas 1-5.
Embora os anticorpos têm um grande potencial para aplicações intracelulares, engenharia-los para a dobragem correcta e a solubilidade no ambiente intracelular enquanto se mantém a capacidade de se ligar a um antigénio alvo é um desafio. O ambiente citoplasmático redução previne a formação das ligações dissulfureto normalmente necessários para a dobragem estável de anticorpos de comprimento completo e fragmentos de anticorpos, incluindo o anticorpo de cadeia única (scFv) Anticorpos 6,7. Um número de abordagens de evolução dirigida têm sido utilizados para conceber anticorpos com Hafinidades IGH para alvo antígenos 8-10. Estas abordagens geralmente usam exibição de fagos, display de superfície de levedura, ou a exposição da superfície bacteriana para rastrear grandes bibliotecas de anticorpos 11-13. Estes métodos são poderoso e eficaz para a identificação de anticorpos que se ligam a alvos, ainda que eles dependem da via de secreção para proteínas de transporte que vai ser exibido 14-16. A via secretora transloca proteínas desdobradas a partir do citoplasma para dentro do lúmen reduzindo retículo endoplasmático em levedura ou para o periplasma das bactérias. As proteínas, em seguida, dobrar sob condições de oxidação e são exibidas na superfície da célula ou empacotado em partículas fágicas para o rastreio de afinidade de ligação 17,18. Como resultado, os anticorpos isolados utilizando essas técnicas não irão necessariamente dobra bem no citoplasma, e a solubilidade intracelular deve muitas vezes ser manipuladas separadamente, se os anticorpos irão ser utilizados em aplicações intracelulares.
Melhorara eficiência de anticorpos de engenharia que são bem dobradas no citoplasma, que previamente relatado o sucesso de MAD-Trap (visualização ancorado à membrana para reconhecimento baseado-Tat de proteínas associando), um método para o rastreio de uma biblioteca de anticorpos scFv utilizando Escherichia coli inner- visor membrana 19. Bacterianas apresentam-membrana interna baseia-se na via de duplo-arginina translocação (TAT) para o transporte de anticorpos exibidos, em contraste com outros métodos de apresentação comum que utilizam a via de secreção. A via de Tat contém um mecanismo de controlo de qualidade que só permite, proteínas correctamente dobradas solúveis a serem transportados a partir do E. coli citoplasma, através da membrana interior, e para o periplasma 20,21. Substratos sobre-expressa Tat (ie., As proteínas direccionadas para a via de Tat com uma fusão N-terminal com o péptido de sinal Tat ssTorA) que são bem dobrada no citoplasma formar uma translocação de longa duração com o intermediário N-terminal in o citoplasma e o C-terminal no periplasma 19. Isto permite a exibição de substratos Tat correctamente dobradas, incluindo fragmentos de anticorpo, sobre a face periplasmática da E. coli membrana interna. Após a remoção da membrana externa por digestão enzimática para gerar esferoplastos, os anticorpos são expostos para o espaço extracelular (Figura 1). Isto permite que os substratos Tat indicadas na membrana interna para ser rastreados para a ligação a um alvo específico. Importante, aproveitando a via de Tat para a exposição da superfície da célula assegura que apenas os anticorpos da biblioteca que são bem dobradas no citoplasma será interrogado para a ligação, o que permite a engenharia simultânea de afinidade e de dobragem de ligação intracelular. Neste protocolo, descrevemos como exibir uma biblioteca de scFv sobre a E. membrana interna coli, deslocar a biblioteca contra um antigénio alvo, e executar uma tela secundária para identificar os componentes mais promissores da biblioteca. Enquanto nos concentramos o protocolo de scFvs, o método pode ser aplicado a engenharia de qualquer proteína cuja aplicação requer dobrar vinculativo e intracelular.
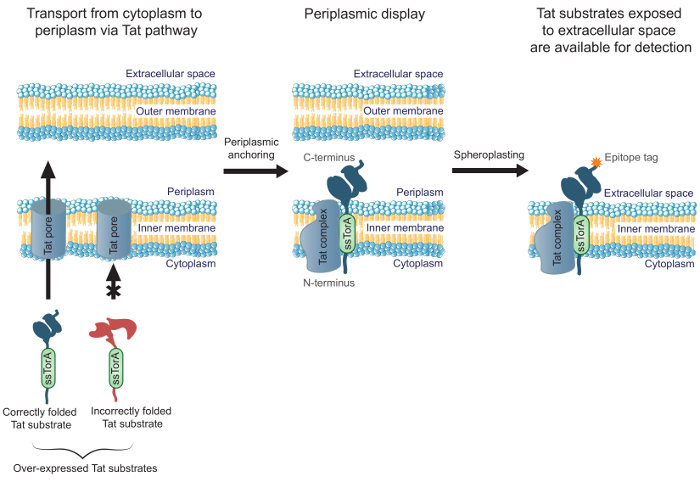
Figura 1. Tela interna da membrana Tat. Em E. coli, anticorpos scFv, expressos como uma fusão com a sequência sinal ssTorA e correctamente dobradas no citoplasma são transportados através da membrana interna. Uma translocação formas intermédias, onde os scFvs são ancoradas na membrana interna com o terminal N no citoplasma e o C-terminal no periplasma. A E. membrana externa de E. coli é digerido enzimaticamente para formar esferoplastos, expondo assim os anticorpos ancorados para o espaço extracelular e torná-los disponíveis para a detecção utilizando um anticorpo que se liga ao marcador de epítopo C-terminal fundido no anticorpo exibido.carga / 54583 / 54583fig1large.jpg "target =" _ blank "> Clique aqui para ver uma versão maior desta figura.
Protocol
1. Prepare a biblioteca scFv como uma fusão com a ssTorA Signal Sequence
- Obter uma biblioteca de ácido desoxirribonucleico (ADN) contendo variantes de um gene de scFv.
NOTA: A biblioteca também podem ser construídos utilizando qualquer modo adequado para gerar a diversidade ao longo de todo o gene de scFv ou domínios visados 22 (por exemplo, a terceira regiões determinantes da complementaridade, as CDR3.). - Inserir a biblioteca de ADN no plasmídeo pIMD (Figura 2), utilizando métodos de clonagem molecular padrão 23.
NOTA: Este plasmídeo expressa scFv como uma fusão genética para a sequência de sinal ssTorA (N-terminal para scFv) e o epitopo marcador FLAG (do terminal C para o scFv). O design do plasmídeo para a exposição da membrana interna foi descrito anteriormente 19. O plasmídeo pIMD está disponível a partir dos autores.

Figura 2. visor plasmídeo da membrana interna (pIMD) mapa (Passos 1.2 a 1.3). Este plasmídeo contém um promotor lac, a origem ColE1 de replicação, e um gene de resistência ao cloranfenicol. O gene scFv inserido está fundido com a sequência de sinal para direccionar o ssTorA scFv para a via de Tat e a uma etiqueta de epitopo FLAG, com todos os três na mesma grelha de leitura. sítios de enzimas de restrição estão indicados. Para uma biblioteca inserida entre os locais de enzimas de restrição XbaI e NotI, o tamanho do plasmídeo é 2219 pb, mais o tamanho do scFv. Por favor clique aqui para ver uma versão maior desta figura.
- Transformar o plasmídeo de ADN contendo a biblioteca em E. MC4100 23 células de E. coli. Recuperar e crescer esta forma bacteriana da biblioteca. Centrifugar a 4000 × g durante 15 min à temperatura ambiente para recolher as células. Remover o sobrenadante e ressuspender as células recolhidasem 25% de glicerol em Luria-Bertani (LB) de mídia. alíquotas armazenar a -80 ° C até ser necessário, ou prosseguir para a Etapa 2.
NOTA: O protocolo foi verificada com células MC4100, embora outras E. Também se espera que as estirpes de E. coli para ser compatível com o protocolo. A electroporação é o método preferido para a transformação, devido à sua elevada eficiência de transformação. A biblioteca devem normalmente consistir em pelo menos 10 9 scFv variantes, nesta fase, e cada alíquota deverá conter suficientes células de tal modo que a biblioteca está coberta de 100 vezes.
2. Expressar a Biblioteca e preparar esferoplastos
- Descongelar uma alíquota da biblioteca bacteriana (a partir do Passo 1,3) à temperatura ambiente, e adicionar a alíquota para um frasco contendo 100 ml de meio LB com 20 ug / ml de cloranfenicol (Cm). Crescer durante 3 h a 37 ° C e 225 rpm num agitador incubadas.
- Após 3 h, remover o recipiente de recolha do agitador incubadas a 37 ° C. Permitir a expressão da biblioteca de scFvproceder O / N durante 15 a 22 h a 20 ° C e 225 rpm num agitador incubadas.
NOTA: Não indutor é necessária para utilizar o plasmídeo pIMD, como o promotor é furado. Note-se que as células MC4100 não sobre-expressam o repressor lac (Lacl e não é encontrada no plasmídeo).

Figura 3. células de E. coli e esferoplastos. (A) E. células de E. coli são de forma cilíndrica. (B) Após spheroplasting utilizando EDTA e lisozima, a membrana exterior da E. células de E. coli é rompido, e os esferoplastos resultantes são de forma esférica. Diferencial de contraste de interferência (DIC) imagens de microscopia foram obtidos usando uma objectiva 100X de um microscópio invertido. Por favor Clamber aqui para ver uma versão maior desta figura.
- Prepare os esferoplastos biblioteca.
NOTA: Os esferoplastos são formadas pela ruptura da membrana exterior da E. coli e são de forma esférica (Figura 3).- Prepare os tampões necessários.
NOTA: Todos os buffers deve ser estéril.- Prepare 1 × solução salina tamponada com fosfato (PBS; pH 7,4) por dissolução de 8 g de NaCl, 0,2 g de KCl, 1,44 g de Na 2 HPO 4, e 0,24 g de KH 2 PO 4 em H2O destilada a um volume final de 1000 ml. Manter em gelo.
- Prepare PBS com 0,1% (w / v) de soro de albumina de bovino (BSA) por dissolução de 0,2 g de BSA em 200 mL de 1 x PBS. Manter em gelo.
- Preparar o tampão de fraccionamento (FB) por mistura de 7,5 ml de H esterilizada por filtração a 1 M de sacarose, 1 ml de tampão Tris 1 M (pH 8,0), e 1,5 ml de destilado 2 O. Manter em gelo.
- Prepare 1 mM de ácido etilenodiaminotetra-acético (EDTA) por adição de 30 ul de OF 0,5 M de EDTA para 14,97 ml de H2O destilada
- Prepare 0,5 M MgCl2 por dissolução de 4,76 g de MgCl 2 em 100 ml de H2O destilada Manter em gelo.
- Retirar o frasco do agitador, e medir a densidade óptica (DO) a 600 nm utilizando um espectros otómetro para determinar a densidade celular. Calcular o volume da cultura induzida necessária de modo a que cada amostra para spheroplasting tem 1 x 10 10 células.
NOTA: A aproximação de uma DO 600 de 1, indicando uma concentração de 10 9 células / ml para E. coli pode ser utilizado 24. - Centrifugar o volume calculado de cultura induzida num tubo de microcentrífuga de 1,5 ml a 12.000 x g à temperatura ambiente durante 5 min. Preparar pelo menos duas amostras, no caso de surgir um problema na preparação da amostra.
- Remover o sobrenadante das culturas centrifugadas e ressuspender cada pelete de células em 100 ul de FB gelada. Centrifugar a 12.000 ×g à temperatura ambiente durante 1 min, e, em seguida, remover o sobrenadante por pipetagem. Ressuspender cada sedimento em 350 ul de FB arrefecido em gelo suplementado com 3,5 uL de 10 mg / ml de lisozima.
- Lentamente vortex cada tubo enquanto se adiciona, gota a gota, 700 mL de EDTA a 1 mM, e depois incubação dos tubos à temperatura ambiente durante 20 min enquanto gira lentamente num rotor tubo para misturar as amostras. Retire os tubos da rotador, adicionar 50 ul de gelado 0,5M MgCl2 a cada tubo, e incubar em gelo durante 10 min. Centrifugar os tubos a 11.000 x g a 4 ° C durante 10 min.
- Isolar o sedimento de esferoplastos.
- Use uma micropipeta com uma ponta de 1 ml para puxar lentamente parte de pellet. Mantendo o tubo em ângulo com a abertura directamente acima de um novo tubo de 1,5 mL, lentamente levantar a ponta da pipeta para fora do sobrenadante e o sedimento deslizar no novo tubo.
- Se um volume significativo de sobrenadante é transferido para o novo tubo, removê-lo por pipetagem. Se a pastilha não é firme ENOurg para transferir, re-centrifugar a 11000 × g durante 2 min e tentativa de isolamento de novo pelete.
- Ressuspender o sedimento em esferoplastos a cada tubo em 1 ml de gelado 1 × PBS. Alternam entre pipetagem e lentamente vórtex num suporte vortex até que o sedimento é ressuspenso completamente. Não manter as amostras fora do gelo durante mais de 2 minutos de cada vez, e retornar para o gelo durante pelo menos 5 minutos antes da remoção do gelo novamente. Mantenha os esferoplastos a 4 ° C (até 2 dias) até ser usado para filtração no Passo 4.
- Prepare os tampões necessários.
3. imobilizar o antigénio alvo em esferas magnéticas
- Biotinilar o antigénio alvo in vivo durante a produção recombinante em E. células de E. coli. Como alternativa, use a conjugação química 25 ou compra antigénio alvo que já foi biotinilado, e continue na Etapa 3.2.
- Adicione 816 g bicina a 50 ml de água para fazer 10 × bicina buffer. Dilui-se a buffer para 1 × em H2O destilada e aquecer a 50 ° C. Adicionar 14,7 mg de biotina e 12 ml de tampão aquecida a 1 × bicina para fazer uma solução de biotina que é biotina 5 mM em 10 mM de tampão bicina. Armazenar a -20 ° C até ser necessário.
- Express e biotinilar a proteína alvo utilizando o plasmídeo pAK400cb BCCP-26, que permite a produção do antigénio alvo como uma fusão com a proteína transportadora de biotina carboxilo (BCCP).
NOTA: E. células de E. coli nativa biotinilar BCCP, eliminando a necessidade de purificar e quimicamente biotinilar a proteína alvo antes da imobilização em esferas revestidas com estreptavidina. O E. nativa coli biotina ligase BirA é suficiente para biotinilação da proteína de fusão.- Crescer E. coli contendo o plasmídeo de biotinilação (com o antigénio alvo inserido como uma fusão com o terminal N de BCCP) O / N durante 15 a 18 horas em 5 ml de meio LB suplementado com 20 ug / ml de Cm, a 37 ° C sob agitação à 225 rpm.
- Medir a OD a 600 nm utilizando um espectrofotómetro e calcular o volume de cultura necessário (v Adicionar) para subcultura a uma OD inicial de 0,05 em 25 ml de meio LB fresco com 20 ug / ml cm, usando a equação: V add = (0,05 × 25 mL) / (OD 600-0,05), em que OD 600 é a densidade óptica da cultura o / N e V de adição é o volume de o / N cultura para adicionar ao LB fresco Subcultura e crescer até uma densidade óptica de 0,5 a 0,8 em um agitador incubadas a 37 ° C e 225 rpm.
- Adicionar β-D-1-tiogalactopiranósido para uma concentração final de 100 uM e biotina até uma concentração final de 5 uM de isopropilo. Induzir a expressão num agitador incubadas durante 15 a 22 h a 20 ° C e 225 rpm.
- Colheita bactérias por centrifugação a 4000 x g a 4 ° C durante 10 min. Remover o sobrenadante. Armazenar o sedimento a -20 ° C até que esteja pronto para uso.
- Adicionar1 ml de um detergente de lise de células por 0,2 g de sedimento de células. Ressuspender por pipetagem e rodar cuidadosamente durante 20 min para lisar as células. Após a lise, centrifugar a 16000 x g e 4 ° C durante 20 min. Pipetar o lisado solúvel (sobrenadante) para um novo tubo de 1,5 mL.
- Usar uma coluna peso molecular de corte de 3 kDa, para remover a biotina não ligada. Pipetar o lisado na coluna, e centrifuga-se a 20 ° C de acordo com as instruções do fabricante. Lava-se com 1 x PBS até a biotina no lisado foi diluída 100 vezes e o volume do lisado lavado é igual ao volume original de lisado. Transferir o lisado para um novo tubo.
- Imobilizar o antigénio alvo biotinilado em esferas magnéticas revestidas com estreptavidina.
- Prepare 1 × PBS e 1 x PBS com BSA a 0,1% (v / w) tal como descrito no passo 2.3.1.
- Prepare os grânulos magnéticos.
NOTA: Este requer a utilização de uma cremalheira de separação magnética.- Ressuspender streptavIDIN-pérolas magnéticas revestidas no seu recipiente original. De qualquer vórtice durante pelo menos 30 seg ou girar durante 5 min.
- Transferir 7-10 × 10 9 esferas para um tubo de 1,5 ml.
NOTA: O volume necessário será dependente da concentração de talão fornecido pelo fabricante. - Colocar o tubo contendo os grânulos sobre o bastidor íman durante 2 minutos para recolher as esferas, no lado do tubo. Com o tubo ainda no ímã, remova cuidadosamente o sobrenadante por pipetagem sem perturbar as contas.
- Para lavar, remover o tubo do ímã, e ressuspender as contas em 1 ml de 1 × PBS por pipetagem sem gerar bolhas. Devolver o tubo para o ímã por 2 min para recolher as contas, e remova cuidadosamente o sobrenadante por pipetagem. Repetir o processo mais duas vezes para um total de três lavagens. Certifique-se de que nenhum líquido é deixado no tubo após a lavagem final.
- Adicionar o ligado contendo o antigénio biotinilado para o BEA magnéticads.
- Remover o tubo do íman e voltar a suspender as pérolas em 1 ml de lisado (a partir do passo 3.1.5). Incubar à temperatura ambiente durante 30 min enquanto rotação suave.
- Colocar o tubo sobre o íman durante 3 min para recolher as esferas revestidas com antigénio. Lavam-se as esferas revestidas com cinco vezes com 1 x PBS com BSA a 0,1% da mesma maneira como descrito nos Passos 3.2.2.3 a 3.2.2.4. Após a lavagem final, voltar a suspender as pérolas em 1 × PBS com 0,1% de BSA até o mesmo volume utilizado no passo 3.2.2.2.
- Se o antigénio alvo imobilizado é estável a 4 ° C, armazenar as pérolas revestidas a 4 ° C até ser necessário para a filtração. Caso contrário, vá para a Etapa 4.
4. Tela Biblioteca scFv por Efeito panning contra o antigénio alvo (Figura 4)

Figura 4. Efeito panning (Passo 4). Antigen-pérolas magnéticas revestidas com are incubadas com esferoplastos que expressam variantes biblioteca de anticorpos. O DNA de plasmídeo a partir de esferoplastos ligado a pérola é recuperada e utilizada para gerar uma sub-biblioteca, que é rastreada utilizando o ecrã secundário baseado em ELISA. Correspondentes etapas do protocolo são anotados. Por favor clique aqui para ver uma versão maior desta figura.
- Incubar as contas revestidas com esferoplastos.
- Usar um grânulo de esferoplastos a proporção de aproximadamente 5: 1. Adicionar 4 x 10 esferoplastos 9 e 8 × 10 8 esferas para um tubo estéril de 15 ml.
NOTA: Assume-se que não há células foram perdidas durante o processo de spheroplasting, de modo que a concentração é ainda 1 × 10 10 esferoplastos / ml. - Adicionar 1 × PBS com BSA a 0,1% para levar o volume total a 4 ml. Aliquota em quatro tubos de 1,5 ml com 1 ml de cada vez. Incubam-se as reacções a 4 ° C durante 5 h, enquanto se suavemente rotativa.
- Usar um grânulo de esferoplastos a proporção de aproximadamente 5: 1. Adicionar 4 x 10 esferoplastos 9 e 8 × 10 8 esferas para um tubo estéril de 15 ml.
- Prepare os esferoplastos-bound grânulo por reacção em cadeia da polimerase (PCR).
- Colocar os tubos de reacção panning no íman durante 3 min. Remover o sobrenadante por pipetagem, e lava-se os esferoplastos-bound grânulo quatro vezes com gelo frio 1 × PBS com BSA a 0,1% da mesma maneira como descrito nos Passos 3.2.2.3 a 3.2.2.4. Ressuspender as esferoplastos-bound talão em cada um dos tubos em 25 ul de H2O destilada Armazenar os grânulos à temperatura de -20 ° C ou prosseguir para a Etapa 4.3.
- Executar todo o plasmídeo de PCR em esferoplastos ligado com pérolas para amplificar os plasmídeos contendo os genes de scFv ligado a grânulos.
- Obter iniciadores com as seguintes sequências: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(iniciador directo) e 5'TAGCTCTTGATCCGGCAAACAAA3' (iniciador inverso).
NOTA: Estes irão ligar-se de ponta a ponta em cadeias opostas do plasmídeo pIMD (Figura 2) e são concebidos para emparelhar com uma característica comum de pIMD, de modo a amplificação irá ocorrer independentemente doscFv sequência variante. - Fosforilar os iniciadores.
NOTA: Sem fosforilação, re-ligação não irá ocorrer. Iniciadores também podem ser encomendados com 5'-fosforilação, em vez de usar este método fosforilação neste protocolo.- Em um tubo de 0,5 ml, configurar uma reacção de fosforilação para o iniciador de PCR para a frente, como descrito na Tabela 1. Repetir este processo para o iniciador inverso.
- Incubam-se as reacções a 37 ° C durante 1 h. Em seguida, incubam-los a 65 ° C durante 20 minutos para desactivar a quinase de polinucleótidos de T4 (PNK). Armazenar os iniciadores fosforilados a -20 ° C.
- Executar PCR.
- Num tubo de PCR, preparar a reacção de PCR tal como descrito na Tabela 2.
NOTA: Vários reações podem ser preparados para maior rendimento. Os esferoplastos ligado com pérolas não utilizados podem ser armazenados a -20 ° C. - Aquecer as reacções de PCR a 98 ° C durante 15 minutos num termociclador para assegurar a lise completa dos esferoplastos. Retire os tubos do termociclador, e adicionar 0,5 mL de uma polimerase de alta fidelidade de cada. Retorno tubos para o termociclador e executado utilizando o programa pormenorizado na Tabela 3.
- Reúnem-se os produtos de PCR conforme apropriado. Armazenar a -20 ° C ou prosseguir para a etapa 4.4.
- Num tubo de PCR, preparar a reacção de PCR tal como descrito na Tabela 2.
- Obter iniciadores com as seguintes sequências: 5'CCAACTCTTTTTCCGAAGGTAACTG3 '(iniciador directo) e 5'TAGCTCTTGATCCGGCAAACAAA3' (iniciador inverso).
Tabela 1. PNK reacção de fosforilação (Passo 4.3.2.1).
| Reagente | Volume (uL) |
| H2O destilada | 15 |
| 10x T4 ADN tampão de reacção de ligase | 2 |
| iniciador 100 uM | 2 |
| Polinucleotídeo cinase de T4 (PNK) | 1 |
| Reagente | Volume (uL) |
| H2O destilada | 28,5 |
| 5x de alta fidelidade tampão polimerase | 10 |
| iniciador de sentido directo 10 uM fosforilado | 2.5 |
| iniciador inverso 10 uM fosforilado | 2.5 |
| 40 mM mistura de dNTP (10 mM de cada dNTP) | 1 |
| esferoplastos ligado-Bead | 5 |
Tabela 3. programa de PCR (passo 4.3.3.2).
| Passo | Temperatura (° C) | Time (min: seg) | Número de ciclos |
| desnaturar inicial | 98 | 00:30 | 1 |
| Desnaturar | 98 | 00:10 | 35 |
| Recozimento | 69 | 00:30 | |
| Extensão | 72 | 00:30 por kb | |
| extensão final | 72 | 06:00 | 1 |
| Aguarde | 12 | Infinito | 1 |
- Re-circularizar os produtos de PCR a todo o plasmídeo, e usar o produto ligado para transformar MC4100 de E. células de E. coli.
- Purifica-se o produto de PCR, executando a reacção de PCR num gel de agarose a 23, a coloração do ADN no gel 23, e utilizando umkit de limpeza de gel para purificar o plasmídeo linearizado, seguindo as instruções fornecidas pelo fabricante. Medir a concentração, utilizando um espectrofotómetro a 260 nm. Armazenar o fragmento purificado a -20 ° C até ser necessário, ou continuar para o passo 4.4.2.
- Re-circularizar plasmídeo a partir do produto de PCR.
- Para evitar a ligação intermolecular do produto de PCR, levar a cabo a reacção de ligação com uma baixa concentração 27 de 1 ng / ul do produto de PCR. Calcular o volume necessário para a preparação de uma reacção de ligação de 800 ul a esta concentração.
- Preparar a reacção de ligação em gelo. Num tubo, adicionar o volume calculado no passo 4.4.2.1 do produto de PCR, 80 pi de tampão ligase 10 x de DNA, e H2O destilada até 800 ul. Adicionar 4 ul de DNA-ligase de T4, e colocar imediatamente os tubos a 16 ° C num banho de água ou termociclador. Incubar a 16 ° CO / N durante 14 a 18 h. Armazenar as reacções de ligação completaram a -20 ° Caté que seja necessário, ou prosseguir para a etapa 4.4.3.
- Coloque a reacção de ligação num bloco de calor a 65 ° C durante 15 minutos para aquecer-inactivar a ligase de ADN. Em seguida, use um kit de membrana de microdiálise ou limpeza DNA de-sal o DNA ligado. Armazenar a 20 ° C ou prosseguir para a etapa 4.4.4.
- Use todo o, produto de ligação com sal-de inactivado pelo calor para transformar MC4100 de E. 23 células de E. coli. Preparar stocks de glicerol, conforme descrito no Passo 1.3, das células que contêm a sub-biblioteca panned resultante, e alíquotas armazenar a -80 ° C.
- Repita Passo 4 na sua totalidade usando uma alíquota do Passo 4.4.4 para fazer um segundo deslizamento sobre a sub-biblioteca.
NOTA: A segunda panning ajuda a enriquecer, para componentes de biblioteca que se ligam bem ao antígeno alvo 19.
5. Execute uma tela secundária Usando um Immunosorbent Método de ensaio ligado a enzima para identificar clones promissores para posterior caracterização (Figura 5) </ P>

Rastreio baseado em ELISA Figura 5. secundário (Passo 5). Variantes (A) a partir da sub-biblioteca da biblioteca enriquecida durante pan são inoculadas em poços individuais de uma placa de cultura para o crescimento e expressão. (B) Uma placa de ELISA revestida com o antigénio alvo. (C) As variantes de bibliotecas são rastreadas utilizando o ecrã secundário à base de ELISA descrito no protocolo. Após a análise de dados obtidos a partir do ecrã secundário, variantes de interesse são seleccionados e caracterizado pelo facto adicional. Correspondentes etapas do protocolo são anotados. Por favor clique aqui para ver uma versão maior desta figura.
- Descongelar um tubo da sub-biblioteca criticado (a partir do Passo 4.4.4) e placa em placas de agar LB. Placa várias diluiçõesem concentrações suficientemente baixas para garantir colónias individuais (por exemplo, 10 2 - 10 6 vezes de diluições). Incubar as placas durante 15 a 18 horas a 37 ° C. Armazenar as placas a 4 ° C ou prosseguir para a Etapa 5.2.
- Cultura e induzem colônias da sub-biblioteca criticado. Executar todas as etapas sob condições estéreis. Usar uma pipeta de canais múltiplos para as etapas que envolvem as placas de 96 poços.
- Adicionar 200 mL de LB com 20 ug / ml de Cm para cada poço de uma placa de cultura de 96 poços de fundo redondo.
- Escolher uma colónia individual da placa de agar com uma ponta de pipeta, coloque a ponta no primeiro poço da placa de 96 poços, e agitar suavemente para inocular. Utilize uma nova ponta para cada poço. Inocular uma colónia em cada poço. Como controlo, incluem, pelo menos, um controlo de esterilidade bem sem colónias inoculadas.
- Repita os passos 5.2.1 e 5.2.2 para inocular várias placas de 96 poços.
- Colocar as placas de 96 poços num agitador de microplacas a 310 rpm. Incubar a 376; C durante 20 a 24 horas para expressar o scFv.
- Para cada placa de cultura preparada no Passo 5.2, um revestimento de 96 cavidades de placas de ELISA com o antigénio alvo.
- Diluir antigénio alvo purificada até uma concentração apropriada (por exemplo, 1 ng / ml a 4 mg / ml) em 1 × PBS para fazer a solução de revestimento. Adicione 5 ml de solução de revestimento para cada placa de 96 poços.
NOTA: A concentração apropriada depende do antigénio específico a ser usado e podem necessitar de ser ajustadas. - Adicionar 50 ul da solução de revestimento, para cada poço de uma placa de ELISA de poliestireno de 96 poços clara de alta ligação. Bater levemente a placa sobre a superfície da bancada para garantir que toda a superfície de cada poço é revestido. Repita o procedimento para cada placa. Incubar as placas a 4 ° CO / N.
- Diluir antigénio alvo purificada até uma concentração apropriada (por exemplo, 1 ng / ml a 4 mg / ml) em 1 × PBS para fazer a solução de revestimento. Adicione 5 ml de solução de revestimento para cada placa de 96 poços.
- Replicar as colónias das placas de cultura de 96 cavidades em placas de agar.
- Coloque um replicador de poliestireno estéril para os poços de uma placa de cultura para recolher uma pequena quantidade de Liquidentidade. Cuidadosamente elevar o replicador e transferir para um LB placa de agar 15 cm de tal modo que todas as dicas estão tocando a placa. Uma vez que o líquido tenha transferido, levante o replicador para cima. Repita para cada placa de cultura.
- Rotular a placa de agar com a orientação correcta de modo que os resultados a partir do ecrã secundário na placa de 96 poços pode ser combinado com a colónia replicado correcta sobre a placa, se caracterização adicional é desejado. Crescer a 37 ° C durante 15 a 18 horas, e, em seguida, armazenar a 4 ° C até ser necessário.
- Executar a tela secundária ELISA.
- Preparar a solução de bloqueio, fazendo 2% (w / v) de leite em pó em PBS 1 ×. Esvaziar a solução de revestimento a partir das placas de ELISA. Adicionar 100 ul da solução de bloqueio a cada poço. Incubar à temperatura ambiente durante pelo menos 2 h, ou bloco de O / N a 4 ° C.
- Preparar o tampão de lavagem através da adição de polissorbato 20 a uma concentração final de 0,05% em 1 x PBS. Adicione 250 ml por placa de ELISA.
- Adicionar 20 ul de umaconcentrado de detergente de lise celular a cada poço da placa de cultura de fundo redondo, e incuba-se a placa de cultura num agitador de microplacas à temperatura ambiente durante 15 a 20 min. Comece a lise, ao mesmo tempo que o bloqueamento das placas de ELISA é completa, de modo que a lise e a lavagem Passo 5.5.4 podem ser executados simultaneamente.
- Esvaziar a solução de bloqueamento das placas de ELISA. Lavam-se as placas de ELISA bloqueadas quatro vezes com 200 ul de tampão de lavagem por poço por lavagem. Esvaziar o tampão de lavagem dos poços.
- Transferir 50 uL de cada poço da placa de lise de células para o correspondente poço da placa de ELISA, utilizando-se uma nova ponta de cada poço. Incubar a placa para ELISA a TA, durante 1 a 2 horas.
- Prepare a solução de anticorpos para detectar scFvs ligados.
- Utilize uma peroxidase de rábano (HRP) -conjugated anticorpo primário que se liga ao marcador de epítopo FLAG fundido com os scFvs da biblioteca.
- Dilui-se o anticorpo para a diluição apropriada para usar num ensaio ELISA (ver fornecedor & #39; s recomendações) em 2% (w / v) de leite seco em 0,05% de polissorbato 20 em 1 × PBS. Prepare 5 ml para cada placa.
- Lavam-se as placas de ELISA quatro vezes com tampão de lavagem, tal como descrito no Passo 5.5.4.
- Adicionar 50 ul de solução de anticorpo a cada poço da placa de ELISA. Incubar durante 1 a 2 horas à temperatura ambiente.
- Prepara-se o substrato de HRP por dissolução de comprimidos de dicloridrato de o-fenilenodiamina (OPD) em H2O destilada acordo com o protocolo do fabricante, evitando a luz. Prepare 20 ml por placa ELISA.
- Preparar 3 H 2 SO 4 por diluição de H 2 SO 4 concentrado com H2O destilada como necessário. Prepare 5 ml por placa ELISA.
Cuidado: H 2 SO 4 é um ácido forte. Certifique-se de usar equipamento de protecção individual adequado. - Lavam-se as placas de ELISA quatro vezes com tampão de lavagem, tal como descrito no passo 5.5.4.
- Incubar as placas de ELISA com o substra HRPte.
- Adicionar 200 ul de substrato de HRP a cada poço. Para minimizar a exposição à luz, adicionar o substrato a uma placa de ELISA de cada vez, e embrulhar com uma folha de alumínio, antes de prosseguir para o próximo placa. Incubar as placas durante 30 a 60 min à temperatura ambiente no escuro.
- Após os primeiros 30 minutos, as placas para verificar escurecimento do substrato, e incuba-se mais longo, se necessário para visualizar o desenvolvimento de cor.
- Adicionar 50 ul de 3 H 2 SO 4 a cada poço para parar a reacção. Usando uma dica diferente para cada bem, misture a solução nas cavidades cuidado pipetando para cima e para baixo sem formação de espuma. Por razões de consistência e para evitar a saturação, adicionar H 2 SO 4 e rapidamente com cuidado para todas as placas de ELISA antes de se misturar a solução para cada placa.
- Medir a absorvância da solução nos poços de cada placa a 492 nm utilizando um leitor de placas.
- Analisar os dados de absorvância para identificar scFv variantes que exhib-lo prometendo sinais de ligação e caracterizar estes scFvs promissores. Escolha scFvs que apresentam sinais de absorvância maior do que o sinal de fundo e mais elevadas do que a média do sinal em cada placa.
Observação: O nível de absorvância será dependente das propriedades do antigénio e o anticorpo anti-FLAG utilizado, juntamente com a força das variantes de scFv que foram isolados no rastreio.
Representative Results
O mecanismo de controlo da qualidade da proteína intracelular dobragem da via Tat em E. coli limita o transporte através da membrana celular interna de proteínas que são bem dobradas no ambiente citoplasmático redutor. Por sobre-expressando uma fusão de um scFv com a sequência de sinal ssTorA (a sequência de sinal da proteína Tora, que é transportado naturalmente Tat pela via 20), a translocação está parado, o que resulta na exibição de scFv sobre a membrana interna 19. Após interrupção enzimática da membrana externa, os anticorpos exibidos são disponibilizados para o rastreio para a actividade de ligação ao antigénio. A capacidade para tirar vantagem da via de Tat para exibição de scFv foi demonstrada por Karlsson et al. 19 (Figura 6). Os anticorpos scFv scFv13 scFv13.R4 e foram fundidas com quer a sequência nativa ou uma ssTorA ssTorA modificado que carece do par de resíduos arginina-arginina reconhecido pelaa via de Tat. scFv13.R4 foi manipulado por Martineau et al., a partir de scFv13 através de quatro ciclos de evolução dirigida e é conhecido para dobrar bem no citoplasma 9. Este scFv foi apresentado na membrana interna, mas apenas quando expresso como uma fusão com a sequência sinal nativa ssTorA (Figura 6). Contrariamente, scFv13 não é bem dobrada citoplasmaticamente 9, de modo que não é apresentada também na membrana interna, independentemente de a sequência de sinal a qual se encontra fundido. Além disso, se os scFvs foram expressos em células que não tinham a proteína TATC, um componente vital da máquina Tat 20,28, display não foi observado, mostrando a importante ligação entre a tela do interior da membrana e da via Tat. Estes resultados demonstram que apenas as proteínas que contêm o péptido de sinal de Tat e que são correctamente dobrado no citoplasma são exibidas na membrana interna, permitindo o transporte através da via Tat para funcionar como uma tela para fol intracelularding.

Figura 6. Detecção de scFv exibidos sobre a membrana interna. A citometria de fluxo foi realizada a análise para detectar a exposição de mal dobrado scFv13 e bem dobrada scFv13.R4 na membrana interna. Os scFv foram fundidos com ssTorA nativa ou ssTorA (KK), em que o par Arg-Arg na sequência ssTorA foi modificado para Lis-Lis. As etiquetas de epitopo FLAG C-terminal as scFv foram detectados com um isotiocianato de fluoresceína (FITC) -conjugated anticorpo anti-FLAG. As células sem a proteína TATC (ΔtatC) e ssTorA-scFv13 sem o marcador FLAG foram testados como controlos. H indica o valor de fluorescência média. Reproduzido de referência 19 com permissão. Por favor clique aqui para ver uma versão maior desta figura.
19. Para demonstrar isto, uma biblioteca de PCR propensa a erro com base na scFv13, que tem um baixo grau de afinidade de ligação para β-galactosidase, foi deslocada contra o antigénio alvo β-galactosidase usando o visor e panning método descrito no protocolo. scFv 1-4 foi isolado depois de um ciclo de mutagénese e deslizamento, e exibiu maior afinidade de ligação para a p-galactosidase do que scFv13 (Figura 7A) e um nível mais elevado de solubilidade citoplasmático (Figura 7B).
Uma nova biblioteca, com base em 4/1 de scFv, foi feito usando propensa a erros de PCR, e panning desta biblioteca de segunda geração contraβ-galactosidase foi feito utilizando uma modificação do protocolo descrito. O panning contra β-galactosidase para a segunda fase da evolução foi feito na presença de purificada, solúvel scFv 14 como um concorrente para melhorar a probabilidade de isolar clones com maior afinidade do que scFv 1-4. Após esta segunda rodada de mutagênese e panning, scFv 2-1 e 2-3 scFv foram isolados usando a triagem secundária baseada em ELISA. Estes scFvs não só exibiu maior afinidade de ligação para β-galactosidase que scFv13, mas também exibiu uma melhor ligação do que o clone primeira rodada scFv 1-4. scFv 2-1 exibiram β-galactosidase de ligação comparável à do scFv13.R4 (Figura 7A). scFv 2-3 também mostra um aumento adicional na solubilidade em comparação com o scFv citoplasmática 14, com destaque para a engenharia simultânea de solubilidade e de ligação ao antigénio. Uma vez que a afinidade e a expressão solúvel dos scFvs são rastreados para simultaneamente, é possível que um scFv seleccionados foi modsolubilidade rar, mas de alta versa ligação ou vice. Por exemplo, o scFv tem 2-1 expressão solúvel scFv menor do que 3/2, mas apresenta maior afinidade de ligação para p-galactosidase.

Figura 7. alvo de ligação e expressão citoplasmática de scFv variantes isolado usando o visor interior da membrana. ScFv (A) foram expressas no citoplasma da E. coli (por exemplo., sem a sequência de sinal ssTorA) com uma hexa-histidina (6 × His) tag e purificado usando ácido spin-colunas de níquel-nitrilotriacético. A ligação dos scFvs purificados a p-galactosidase foi medida com um ensaio ELISA. scFv purificadas foram carregadas em placas de ELISA revestidas com β-galactosidase, e os scFvs ligados foram detectados com um anticorpo anti-His 6 ×. Os dados são uma média de seis replicados, e a barra de erro mostra o erro padrão da média.(B) As fracções solúveis e insolúveis dos lisados celulares a partir de células que expressam scFvs citoplasmaticamente foram analisados por Western blot sondada com um anticorpo anti-His 6 ×. A concentração total de proteína foi utilizada para normalizar o carregamento das amostras. Reproduzido (A) e adaptado (B) a partir da referência 19 com permissão. Por favor clique aqui para ver uma versão maior desta figura.
Discussion
Engenharia anticorpos para a actividade citoplasmática é uma tarefa difícil, devido ao ambiente redutor do citoplasma, o que impede a formação de ligações dissulfureto de estabilização 6,7. Isto faz com que a maioria dos anticorpos a ser inactiva no citoplasma, a menos que são concebidos para a estabilidade e a solubilidade no citoplasma, para além de ser manipulado para a afinidade de ligação. Os métodos existentes de exibição de fagos, exibição de superfície bacteriana, e métodos de exposição de superfície de levedura todos utilizam a via secretora 14-16 para a exibição de anticorpos manipulados, mas estes métodos não têm meios para dobragem engenheiro intracelular. Os anticorpos manipulados utilizando visor interior da membrana citoplasmática melhoraram a estabilidade e solubilidade porque o controlo da via da Tat qualidade dobragem previne a translocação de anticorpos que são mal dobrados e instáveis no citoplasma. Este método simplifica o processo iterativo de engenharia de anticorpos intracelulares para uma afinidadeND solubilidade, como as duas propriedades são concebidos em um passo. Embora este método foi concebido para engenharia de anticorpos com a solubilidade no ambiente intracelular de redução, também poderia ser aplicada para anticorpos de engenharia para funcionar em condições não redutoras, uma vez que as proteínas por engenharia utilizando este método de manter a sua dobragem no ambiente oxidante do periplasma.
Embora esta técnica simplifica o processo de engenharia de anticorpos com elevada afinidade e alta solubilidade citoplasmática, várias limitações são importantes a considerar quando se usa este protocolo. Quando se analisam os sinais de ELISA ecrã secundário para identificar variantes de scFv promissor, o limiar para discernir entre as variantes potencialmente interessantes e aqueles que não podem apresentar adequada de ligação do antigénio não é provável que seja aparente até depois de vários clones foram ainda caracterizados. É importante olhar para a ligação melhorada ao longo do anticorpo parental; Contudo,um sinal anormalmente elevada pode ser um indicativo de avidez de 29 ou efeitos de agregação 30, um desafio que não é exclusivo para a abordagem de triagem visor interno da membrana. Uma limitação chave para recordar quando se usa este protocolo é a incapacidade de recuperar esferoplastos depois de percorrer, como eles são não viáveis (dados não publicados). Isto necessita os passos de amplificação de ADN e transformação para recuperar os plasmídeos que codificam anticorpos.
Vários passos críticos do protocolo permitir a engenharia simultânea de dobragem e de ligação de anticorpos. Para o rastreio para ser bem sucedido, a biblioteca de scFv serem projectados devem ser expressos como uma fusão com o péptido sinal ssTorA. Sem esta sequência, os anticorpos não irão ser dirigidos para a via de Tat e, portanto, não vai ser translocada para o periplasma 19. Além disso, é imperativo que uma etiqueta de epitopo no terminal C está fundido com os anticorpos para permitir a detecção dos anticorpos exibidos no escaninhoensaios de ding. Claramente, a E. estirpe de E. coli utilizada para expressar os scFvs também deve ter a maquinaria necessária via de Tat, mas isto é verdade para a E. utilizada estirpes de E. coli.
Modificações a este protocolo é possível melhorar o seu potencial para isolar anticorpos com as características desejadas. Um passo panning subtrativo pode ser concluída antes do panning contra o antigénio alvo para esgotar a biblioteca scFv de constituintes não-desejada. Os esferoplastos de bibliotecas podem ser incubados com esferas magnéticas revestidas com BCCP sozinho ou revestido com uma proteína não desejada, e os esferoplastos que se ligam a essas esferas pode ser descartada antes de rastreio dos restantes esferoplastos não ligadas para a ligação ao alvo desejada. Como mencionado nos resultados representativos, um método para melhorar a afinidade de um scFv isolado é incluir um competidor solúvel na reacção de deslizamento para competir com os scFv exibidos sobre os esferoplastos. Porque a amostra solúveletitor é uma proteína purificada, sem ADN é amplificado a partir dele, de modo que apenas sequências dos scFv exibidos sobre os esferoplastos são recuperados na reacção de PCR. Além disso, este método pode ser alargado a outros tipos de engenharia de anticorpos ou a proteínas de ligação que não anticorpos.
E. visor interior da membrana coli é uma plataforma poderosa para anticorpos de engenharia com afinidade elevada e elevados níveis de solubilidade intracelular. Este método é particularmente adequado para a engenharia eficiente de anticorpos concebidos para funcionar no ambiente intracelular. Estes anticorpos intracelulares já estão a ser explorados como potenciais terapêuticos em vários campos, incluindo doenças neurodegenerativas, câncer e infecções virais 31. Esta técnica permitirá uma utilização mais generalizada de anticorpos intracelulares como ferramentas de pesquisa e medicina nestes domínios e qualquer outro campo onde estudar é desejado um alvo proteínas in situ.
Materials
| Name | Company | Catalog Number | Comments |
| scFv library | Varies | A suitable scFv library should be obtained from a commercial or academic source. | |
| MC4100 E. coli cells | Coli Genetic Stock Center | 6152 | Cells need to be chemically competent or electrocompetent, depending on the selected transformation method. |
| Glycerol | Fisher Scientific | BP229-4 | |
| Difco dehydrated culture media LB Broth, Miller (Luria-Bertani) | BD | 244610 | |
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | |
| Sodium chloride (NaCl) | Fisher Scientific | BP358-1 | |
| Potassium chloride (KCl) | Fisher Scientific | BP366-500 | |
| Sodium phosphate, dibasic (Na2HPO4) | Fisher Scientific | BP332-500 | |
| Potassium phosphate, monobasic (KH2PO4) | Fisher Scientific | BP362-500 | |
| Bovine serum albumin (BSA) | Fisher Scientific | BP9706-100 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| Tris base | Fisher Scientific | BP1521 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5 M | Fisher Scientific | BP2482-500 | |
| Magnesium chloride (MgCl2) | Fisher Scientific | BP214-500 | |
| Lysozyme | Sigma Aldrich | L3790-10X1ML | |
| Vortex mixer | VWR | 97043-564 | |
| Bicine | Fisher Scientific | BP2646100 | |
| D-Biotin | Fisher Scientific | BP232-1 | |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | BP1755-1 | |
| BugBuster Master Mix (cell lysis detergent) | EMD Millipore | 71456 | |
| Vivaspin 2 MWCO, 3,000 daltons | GE Healthcare Sciences | 28932240 | |
| Target antigen | Varies | N/A | Purified target antigen may be purchased or produced/purified. |
| Dynabeads MyOne Streptavidin T1 | Invitrogen | 65601 | |
| Dynamag-2 magnet | Invitrogen | 12321D | |
| Tube rotator | VWR | 13916-822 | |
| PCR primers | IDT | N/A | Primer sequences are as described in the protocol. |
| 10x T4 DNA ligase reaction buffer | New England BioLabs | B0202S | |
| T4 Polynucelotide kinase (PNK) | New England BioLabs | M0201S | Make sure the T4 ligase buffer used in the primer phosphorylation reaction contains 1 mM ATP. |
| 5x Phusion HF buffer pack | New England BioLabs | B0518S | |
| Deoxynucleotide (dNTP) solution mix, 10 mM each dNTP | New England BioLabs | N0447L | |
| Phusion DNA polymerase | New England BioLabs | M0530S | Other high-fidelity polymerases may be used as an alternative, but the annealing temperature in Table 3 must be adjusted. |
| C1000 Touch thermal cycler with dual 48/48 fast reaction module | Bio-Rad | 185-1148 | |
| Agarose | Promega | V3121 | |
| SYBR Safe DNA gel stain | Invitrogen | S33102 | |
| Wizard SV gel and PCR clean-up system | Promega | A9281 | |
| T4 DNA ligase | New England BioLabs | M0202S | |
| Microdialysis membrane filter | EMD Millipore | VSWP04700 | |
| Agar | BD | 214030 | |
| 96-well polystyrene round-bottom cell culture plates | VWR | 10062-902 | |
| Costar general polystyrene assay plate lids | Corning | 3931 | |
| Microtitre plate shaker | VWR | 12620-926 | |
| Costar 96 well EIA/RIA Easy Wash clear flat bottom polystyrene high bind microplate | Corning | 3369 | |
| Bel-blotter polycarbonate 96-well replicating tool | Bel-Art Products | 378760002 | |
| Instant nonfat dry milk | Quality Biological | A614-1000 | |
| Tween 20 (polysorbate 20) | Fisher Scientific | BP337-500 | |
| PopCulture reagent (concentrated cell lysis detergent) | EMD Millipore | 71092-3 | |
| Monoclonal ANTI-FLAG M2-Peroxidase(HRP) antibody produced in mouse | Sigma Aldrich | A8592 | |
| SigmaFast OPD | Sigma Aldrich | P9187-50SET | |
| Sulfuric acid (H2SO4), 10 N solution | Fisher Scientific | SA200-1 | |
| Reynolds Wrap aluminum foil | VWR | 89079-075 | |
| BioTek Epoch microplate spectrophotometer | Fisher Scientific | 11120570 |
References
- Biocca, S., Pierandrei-Amaldi, P., Campioni, N., Cattaneo, A. Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (NY). 12 (4), 396-399 (1994).
- Chen, S. Y., Bagley, J., Marasco, W. A. Intracellular antibodies as a new class of therapeutic molecules for gene-therapy. Hum. Gene Ther. 5 (5), 595-601 (2008).
- Gargano, N., Biocca, S., Bradbury, A., Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J Virol. 70 (11), 7706-7712 (1996).
- Mhashilkar, A. M., et al. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. Embo J. 14 (7), 1542-1551 (1995).
- Strube, R. W., Chen, S. Y. Characterization of anti-cyclin E single-chain Fv antibodies and intrabodies in breast cancer cells: enhanced intracellular stability of novel sFv-F-c intrabodies. J. Immunol. Meth. 263 (1-2), 149-197 (2002).
- Mössner, E., Koch, H., Plückthun, A. Fast selection of antibodies without antigen purification: adaptation of the protein fragment complementation assay to select antigen-antibody pairs. J. Mol. Biol. 308 (2), 115-122 (2001).
- Wörn, A., et al. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 275 (4), 2795-2803 (2000).
- Knappik, A., Plückthun, A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 8 (1), 81-89 (1995).
- Martineau, P., Jones, P., Winter, G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 280 (1), 117-127 (1998).
- Steipe, B., Schiller, B., Plückthun, A., Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240 (3), 188-192 (1994).
- Daugherty, P. S. Protein engineering with bacterial display. Curr Opin Struct Biol. 17 (4), 474-480 (2007).
- Lener, M., et al. Diverting a protein from its cellular location by intracellular antibodies. Eur. J. Biochem. 267 (4), 1196-1205 (2000).
- Lynch, S. M., Zhou, C., Messer, A. An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 377 (1), 136-147 (2008).
- Gai, S. A., Wittrup, K. D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struc. Biol. 17 (4), 467-473 (2007).
- Kieke, M. C., et al. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. USA. 96 (10), 5651-5656 (1999).
- Steiner, D., Forrer, P., Stumpp, M. T., Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 24, 823-831 (2006).
- Pugsley, A. P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57 (1), 50-108 (1993).
- Rapoza, M. P., Webster, R. E. The filamentous bacteriophage assembly proteins require the bacterial SecA protein for correct localization to the membrane. J. Bacteriol. 175 (6), 1856-1859 (1993).
- Karlsson, A. J., et al. Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Molec. Biol. 416 (1), 94-107 (2012).
- DeLisa, M. P., Tullman, D., Georgiou, G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 100 (10), 6115-6120 (2003).
- Fisher, A. C., Kim, W., DeLisa, M. P. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 15 (3), 449-458 (2006).
- Maynard, J., Georgiou, G. Antibody engineering. Annu Rev Biomed Eng. 2, 339-376 (2000).
- Green, M. R., Sambrook, J. Molecular Cloning: A Laboratory Manual. 1, Fourth, Cold Spring Harbor Laboratory Press. (2012).
- Milo, R., Jorgensen, P., Moran, U., Weber, G., Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750-D753 (2010).
- Hermanson, G. T. Bioconjugate Techniques. , Third, Elsevier/Academic Press. (2013).
- Tayapiwatana, C., Chotpadiwetkul, R., Kasinrerk, W. A novel approach using streptavidin magnetic bead-sorted in vivo biotinylated survivin for monoclonal antibody production. J Immunol Methods. 317 (1-2), 1-11 (2006).
- Zhu, G., Song, L., Lippard, S. J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73 (14), 4451-4460 (2013).
- Bogsch, E. G., et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 273, 18003-18006 (1998).
- Julian, M. C., et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28 (10), 339-350 (2015).
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug. Discov. 13 (11), 799-801 (2014).
- Marschall, A. L., Dübel, S., Böldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs. 7 (6), 1010-1035 (2015).
