

Summary
Представлено сравнение и оптимизация двух методов растительного органелларного ДНК: традиционное дифференциальное центрифугирование и фракционирование общей гДНК на основе статуса метилирования. Мы оцениваем полученное количество и качество ДНК, демонстрируем эффективность в короткометражном секвенировании следующего поколения и обсуждаем потенциал для использования в долгочитаемом одномолекулярном секвенировании.
Abstract
Органные органные геномы растений содержат большие повторяющиеся элементы, которые могут подвергаться спариванию или рекомбинации с образованием сложных структур и / или подгеномных фрагментов. Органелларные геномы также существуют в смесях в пределах определенного клеточного или тканевого типа (гетероплазмы), а обилие подтипов может изменяться во время развития или при стрессе (субстехиометрическое смещение). Для более глубокого понимания структуры и функции органелларного генома необходимы технологии секвенирования следующего поколения (NGS). Традиционные исследования секвенирования используют несколько методов для получения органеллярной ДНК: (1) Если используется большое количество исходной ткани, оно гомогенизируется и подвергается дифференциальной центрифугированию и / или градиентной очистке. (2) Если используется меньшее количество ткани ( т. Е. Если семена, материал или пространство ограничены), тот же процесс выполняется, как и в (1), за которым следует амплификация всего генома для получения достаточной ДНК. (3) Анализ биоинформатики может быть использован дляСуммарную геномную ДНК и разобрать органолептические прочтения. Все эти методы имеют присущие проблемы и компромиссы. В (1) может быть трудно получить такое большое количество исходной ткани; В (2) усиление цельного генома может привести к смещению последовательности; И в (3) гомология между ядерными и органелларными геномами может мешать сбору и анализу. В растениях с крупными ядерными геномами выгодно обогащать органеральную ДНК, чтобы снизить затраты на секвенирование и сложность последовательности для анализа биоинформатики. Здесь мы сравниваем традиционный метод дифференциального центрифугирования с четвертым методом, адаптированным подходом CpG-methyl pulldown, для разделения общей геномной ДНК на ядерные и органеарные фракции. Оба метода дают достаточную ДНК для NGS, ДНК, которая сильно обогащена для органеллярных последовательностей, хотя и при различных соотношениях в митохондриях и хлоропластах. Мы представляем оптимизацию этих методов для ткани листьев пшеницы и обсуждаем основные преимущества и dЯвляется преимуществом каждого подхода в контексте ввода образца, упрощения протокола и последующего приложения.
Introduction
Секвенирование геномов является мощным инструментом для анализа лежащей в основе генетической основы важных признаков растений. В большинстве исследований секвенирования генома основное внимание уделяется содержанию ядерного генома, так как большинство генов расположено в ядре. Тем не менее, органелл геномов, в том числе митохондрии (через эукариот) и пластид (в растениях; специализированной форме, хлоропластов, работает в фотосинтезе) способствуют значительной генетической информации , необходимой для организменном развития, реакции на стресс и общей пригодности 1. Органелларные геномы обычно включаются в общую экстракцию ДНК, предназначенную для секвенирования ядерного генома, хотя также используются методы уменьшения количества органелл до экстракции ДНК 2 . Во многих исследованиях были использованы результаты секвенирования от полных выделений гДНК для сборки органеллярных геномов 3 , 4 , 5 ,Xref "> 6 , 7. Однако, когда целью исследования является фокусирование на органелларных геномах, использование общей гДНК увеличивает затраты на секвенирование, потому что многие чтения« теряются »для последовательностей ядерных ДНК, особенно у растений с большими ядерными геномами Кроме того, из-за дублирования и переноса органелларных последовательностей в ядерный геном и между органеллами правильное отображение положения секвенирования, считываемое в соответствующий геном, является биоинформационно сложным 2 , 8. Очистка органелларных геномов от ядерного генома является одной Стратегия по уменьшению этих проблем. Дальнейшие стратегии биоинформатики могут использоваться для разделения чтений, которые сопоставляются с областями гомологии между митохондриями и хлоропластами.
В то время как органелларные геномы многих видов растений были секвенированы, мало что известно о широте разнообразия органелларного геномаДоступных в диких популяциях или в культивируемых племенных пулах. Известно, что органелларные геномы являются динамическими молекулами, которые подвергаются значительной структурной перестройке из-за рекомбинации между повторяющимися последовательностями 9 . Кроме того, в каждой органелле содержится несколько копий органомерного генома, и в каждой клетке содержится множество органелл. Не все копии этих геномов идентичны, что называется гетероплазмой. В отличие от канонической картины «мастер-кругов», в настоящее время растут доказательства более сложной картины органелларных структур генома, включая субгеномные круги, линейные хромосомы, линейные конкатэмеры и разветвленные структуры 10 . Сборка растительных органелларных геномов дополнительно осложняется их относительно большими размерами и существенными инвертированными и прямыми повторами.
Традиционные протоколы для органолептической изоляции, очистки ДНК и последующего генома E sequencing часто громоздки и требуют больших объемов ввода ткани, с несколькими граммами до более сотни граммов молодой листовой ткани, необходимой в качестве отправной точки 11 , 12 , 13 , 14 , 15 , 16 , 17 . Это делает секрецию органелларного генома недоступным, когда ткань ограничена. В некоторых ситуациях количество семян ограничено, например, когда необходимо последовательность на основе поколений или в мужских стерильных линиях, которые необходимо поддерживать путем скрещивания. В этих ситуациях органелларная ДНК может быть очищена, а затем подвергнута амплификации цельного генома. Однако амплификация цельного генома может привести к существенному смещению последовательности, что является особой проблемой при оценке структурных изменений, подгеномных структур и уровней гетероплазмы> 18. Недавние успехи в подготовке библиотек для технологий короткого чтения с чередованием преодолели барьеры с низким входным потенциалом, чтобы избежать усиления цельного генома. Например, набор для подготовки библиотеки Illumina Nextera XT позволяет использовать всего лишь 1 нг ДНК для ввода 19 . Тем не менее, стандартная подготовка библиотек для приложений с длительным чтением, таких как технологии секвенирования PacBio или Oxford Nanopore, по-прежнему требует относительно большого количества входной ДНК, что может создать проблему для секвенирования органелларного генома. Недавно были разработаны новые пользовательские протоколы последовательного кодирования с длительным чтением для уменьшения количества вводимых количеств и для облегчения секвенирования генома в образцах, где получение микрограммовых количеств ДНК затруднено 20 , 21 . Однако получение высокомолекулярных чистых органелларных фракций для подачи в эти библиотечные препараты остается проблемой.
Мы искали tO сравнить и оптимизировать органические методы обогащения и выделения ДНК, подходящие для NGS, без необходимости амплификации цельного генома. В частности, наша цель заключалась в определении оптимальных методов обогащения высокомолекулярной органелларной ДНК из ограниченных исходных материалов, таких как подвыборка листа. В этой работе представлен сравнительный анализ методов обогащения органелларной ДНК: (1) модифицированный традиционный протокол дифференциального центрифугирования по сравнению с (2) протоколом фракционирования ДНК на основе использования коммерчески доступного подхода к распаду ДНК CpG-метилсвязывающего домена 22 применяется к растительной ткани 23 . Мы рекомендуем лучшие методы для выделения органелларной ДНК из ткани листьев пшеницы, которая может быть легко распространена на другие растения и типы тканей.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Генерация растительных материалов для выщелачивания и выделения ДНК
- Стандартный рост посевов пшеницы
- Семена растений в вермикулите в небольших квадратных горшках с 4-6 семенами на угол. Перенос в теплицу или камеру роста с 16-часовым светом, 23 ºC днем / 18 ºC ночь.
- Вода растения каждый день. Оплодотворить растения с ¼ чайной ложкой зернистого удобрения 20-20-20 NPK при прорастании и через 7 дней после прорастания.
- Альтернативная этиоляция проростков пшеницы
- Следуйте шагу 1.1, но поместите горшки в темную камеру роста, 23 ° C в течение 16 ч / 18 ° С в течение 8 часов. Альтернативно, закройте растения в теплице ( например, с контейнером для хранения, однако необходимо поддерживать надлежащую вентиляцию).
- Рост и сбор тканей
- Растите растения на 12-14 дней. Для большинства генотипов пшеницыS, 75 - 100 саженцев дают около 10-12 г ткани, что достаточно для двух органелл. Экстракций с использованием метода дифференциального центрифугирования (раздел 2); Только одно растение необходимо, если использовать подход, основанный на распаде ДНК на основе CpG-метилирования, для фрактального органеллара из ядерной ДНК (раздел 3).
- При использовании метода дифференциального центрифугирования собирайте свежую ткань и немедленно приступайте к обработке образцов, как описано в разделе 2.
- Если вы используете подход CpG-methyl pulldown, урожай 20 мг секций молодой листовой ткани в микроцентрифужные пробирки (используйте стандартную или этиолитированную ткань, см. Представительские результаты ). Заморозить на жидком азоте и замораживать при -80 ºC до использования. Перейдите к раскрытию фракционирования ДНК, как описано в разделе 3.
2. Способ № 1: экстракция ДНК с использованием дифференциального центрифугирования (DC)
ПРИМЕЧАНИЕ.Протокол стандартного центрифугирования был модифицирован из двух публикаций, в которых оптимизированы условия для выделения обоих органелл, но обогащаются митохондриями 17 , 24 . Полученный протокол менее трудоемкий и использует меньше токсичных химических веществ, чем предыдущие методы. В частности, мы внесли изменения в буферы и стадии промывки, включая добавление поливинилпирролидона (ПВП) в буфер для экстракции STE и удаление заключительной стадии промывки в буфере NETF, который содержит фторид натрия (NaF).
Внимание: Подготовка и использование буфера STE следует выполнять под химическим вытяжным шкафом с надлежащим оборудованием для индивидуальной защиты, так как этот буфер содержит 2-меркаптоэтанол (BME).
- Что нужно сделать перед запуском
- Убедитесь, что все оборудование очень чистое, и автоклавируйте любое оборудование, которое может быть автоклавировано ( например, шлифовальные цилиндры, высокоскоростные центриФуги и т . Д. ).
ПРИМЕЧАНИЕ. Рекомендации по фильтрации рекомендуется для всех этапов, требующих пипетирования, чтобы избежать перекрестного загрязнения. - См. Список необходимого оборудования и реагентов и подготовьте необходимые буферы и рабочие запасы для метода № 1 ( таблица 1 ). Охладите криогенные шлифовальные блоки до -20 ºC, а роторы и буферы - до 4 ºC, установите микроцентрифугу на 4ºC и включите водяную баню с температурой 37ºC.
- Убедитесь, что все оборудование очень чистое, и автоклавируйте любое оборудование, которое может быть автоклавировано ( например, шлифовальные цилиндры, высокоскоростные центриФуги и т . Д. ).
- Выделение органелл
- Уберите 5 г свежей ткани и промойте ее холодной, стерильной водой в охлажденном стакане на льду.
ПРИМЕЧАНИЕ. Всегда держите образцы на льду во время всех операций и перевозок в центрифуги, вытяжные шкафы и т. Д. Кроме того, работайте в холодной комнате, если есть доступ к достаточному пространству и оборудованию для выполнения протокола. - Используя ножницы, срезайте ткань листьев на части размером ~ 1 см непосредственно в трубку объемом 50 мл, содержащую два керамических шлифованияцилиндры.
ПРИМЕЧАНИЕ. Очистите или замените ножницы между образцами, чтобы избежать перекрестного загрязнения. - Если нет тканевого гомогенизатора, используйте ступку и пестик и следуйте инструкциям для замены шагов 2.2.4 - 2.2.9.
- Обрежьте ткань листа в предварительно охлажденный раствор на льду. Измельчите образцы в течение 2 - 3 минут в 15 мл STE (в вытяжном шкафу).
- Вылейте буфер (оставить ткань в ступке) через воронку, содержащую один слой предварительно мокрой стерильной фильтровальной ткани (размер пор от 22 до 25 мкм, см. Основной протокол для деталей) в другую трубку объемом 50 мл , Добавьте еще 10 мл STE в ступку и пестик и снова гомогенизируйте.
- Налейте гомогенизированную ткань и буфер в ту же воронку. Промойте раствор и пестик 10 мл STE и вылейте его в воронку. Сожмите и выжмите фильтровальную ткань в воронку, чтобы восстановить как можно больше жидкости.
ПРИМЕЧАНИЕ. Измените перчатки между образцами, чтобы избежать перекрестного загрязнения. ПродолжайтеTocol на этапе 2.2.10.
- Добавьте 20 мл STE (в вытяжной шкаф) к каждой трубке объемом 50 мл.
- Поместите образцы в предварительно охлажденные криогенные мелющие блоки в тканевый шлифовальный аппарат и измельчите образцы в течение 2 х 30 с при 1750 об / мин. Поверните позиции образца и поместите образцы на лед в течение ~ 1 мин между шлифовальными машинами.
ПРИМЕЧАНИЕ. На этой стадии можно использовать ступку и пестик, блендер или другое устройство для измельчения / гомогенизации ткани. Тем не менее, каждый метод будет влиять на качество ДНК в разной степени, и поэтому длину и качество ДНК следует оценивать, прежде чем продолжить применение в нисходящем направлении. - Вставьте воронку в чистую пробирку объемом 50 мл, помещенную во льду. Поместите один слой фильтровальной ткани в воронку и предварительно промокните ее 5 мл STE. Не отбрасывайте поток.
- Налейте гомогенизированную ткань в воронку. Промойте шлифовальную трубку 15 мл STE, повторите и поверните трубку, чтобы ополоснуть стенки и крышку, и влейте в нееэл.
- Осторожно удалите керамические камни, а затем вдавите и выжмите фильтровальную ткань в воронку.
ПРИМЕЧАНИЕ. Измените перчатки между образцами, чтобы избежать перекрестного загрязнения. - Оберните колпачки с парафильмом, чтобы избежать утечки. Центрифуга при 2000 мкг в течение 10 мин при 4 ° С.
- Осторожно аспирируйте супернатант, используя серологическую пипетку (не мешайте таблетке) и поместите ее в высокоскоростную центрифужную пробирку объемом 50 мл (если трубки не имеют плотных уплотняющих прокладок, заверните пробку с парафильмом, чтобы избежать утечки). Отбросьте гранулы.
- Балансируйте трубки до 0,1 г с помощью STE и центрифугируйте полученный супернатант в течение 20 минут при 18000 xg и 4ºC. Чтобы сбалансировать трубки, поместите на балансе небольшой стакан льда, помассируйте весы и взвешивайте образцы на льду, чтобы они были холодными. В качестве альтернативы, используйте в холодильнике баланс и вытяжной шкаф.
- Отбросьте супернатант. Добавьте 1 мл ST в таблетку и осторожно положитеС мягкой кистью. Добавьте 24 мл ST (конечный объем 25 мл) и смешайте / закрутите ( т. Е. Нажмите кисть на стороне трубки, чтобы удалить всю жидкость).
- Балансируйте трубки до 0,1 г, используя ST. Центрифуга в течение 20 мин при 18000 xg и 4 ºC. Между тем, подготовьте решение DNaseI (см. Таблицу 1 для рецептов на складе и рабочих решений). Для каждого образца сделайте одну 200 мкл аликвоту в 1,5 мл пробирке.
- Удалите супернатант, промокните пробирку и повторно суспендируйте гранулу (все еще в высокоскоростной центрифужной пробирке) в 300 мкл ST, используя мягкую кисть. Поместите кисть в предварительно приготовленную пробирку объемом 1,5 мл, содержащую 200 мкл раствора DNaseI, и закрутите кисть, чтобы удалить остатки гранул, застрявших в щетке. Внесите раствор DNaseI обратно в высокоскоростную центрифужную пробирку и осторожно смешайте.
- Инкубируйте при 37 ° C в течение 30 минут на водяной бане (оберните парафильм вокруг верхней части трубки, чтобы предотвратить утечку конденсатаГ в колпачок). Аккуратно перемешайте, закручивая 2 раза во время инкубации.
- Осторожно пипеткой таблетированной смеси из трубки с помощью наконечника пипетки с широким отверстием и поместите его в 1,5 мл трубки с низким связыванием. Добавьте 500 мкл 400 мМ ЭДТА, pH 8,0, в высокоскоростную центрифужную пробирку и аккуратно пипеткой, чтобы вытащить из остатка остаточный осадок. Перенесите ЭДТА в ту же 1,5-мл трубку с низким связыванием в виде смеси гранул и осторожно перемешайте путем инверсии.
- Центрифуга при 18000 мкг в течение 20 мин при 4 ° С. Отбросьте супернатант, промокните пробирку и сразу используйте для изоляции ДНК. При необходимости замораживать гранулы при -20 ºC, но это может привести к снижению урожайности, так как остаточная ДНКаза может ухудшить ДНК образца, если она не будет немедленно обработана.
- Уберите 5 г свежей ткани и промойте ее холодной, стерильной водой в охлажденном стакане на льду.
- Выделение ДНК из изолированных органелл с использованием коммерческого подхода на основе столбцов
ПРИМЕЧАНИЕ. См. Руководство по набору для полного протокола 25 и см. Ниже для внесения изменений. PrПредпочтительнее переходить непосредственно из органелларной изоляции в экстракцию ДНК. Повторное замораживание и оттаивание уменьшают размеры фрагментов ДНК и приводят к деградации ДНК остаточной ДНКазой. Ограничьте вихревую или энергичную пипетирование, так как это может сдвинуть ДНК. Рекомендуется использовать микроцентрифужные трубки с низким связыванием, чтобы максимизировать восстановление ДНК.- Процедура экстракции ДНК
ПРИМЕЧАНИЕ. Прочитайте подробный коммерческий протокол 25, прежде чем начинать следить за тем, чтобы буферы были надлежащим образом изготовлены / сохранены и что процедуры колонкового столба понятны.- Добавьте 180 мкл буфера ATL непосредственно в пробирку с гранулой (оттаивают, если предварительно замораживают и уравновешивают до комнатной температуры на столешнице).
- Следуйте шагу 3 в протоколе «Очистка ДНК от тканей» в справочнике по набору со следующими изменениями: 30-минутный лизис на стадии 3 включает необязательное расщепление РНКазой А и элюирование в 3 × 200 мкл AE ( Каждый вИ затем объединить элюции).
- Сохраните аликвоту (не менее 20 мкл) для qPCR (см. Шаг 4.1). Для количественной оценки до концентрирования, сохраните дополнительные 1 мкл для высокочувствительной количественной оценки.
- При желании, протекайте с концентрацией проб.
- Процедура экстракции ДНК
- Концентрация проб с использованием коммерческих фильтров
ПРИМЕЧАНИЕ. Дополнительную информацию см. В коммерческом протоколе 26 . В зависимости от использования в нисходящем потоке может не потребоваться концентрация проб ( например, для конечных точек PCR и приложений qPCR). Однако для построения библиотеки NGS, вероятно, потребуется сконцентрировать разбавленную органелларную ДНК, полученную после экстракции ДНК.- Обработка колонны
- Осторожно предварительно взвешивайте (см. Таблицу 2 ) пустой блок фильтра (без трубки) на чистом куске взвешивающей бумаги на цифровом аналитическом балансе. Запишите вес.
- ПиПлетте комбинированные элюирования в блок фильтра и тщательно взвешивайте снова.
ПРИМЕЧАНИЕ. В коммерческом руководстве 26 говорится, что максимальный объем блока фильтра составляет 500 мкл, но до 575 мкл можно одновременно добавлять в блок без переполнения. - Осторожно поместите заполненный фильтрующий блок в трубку (снабженную колонками). Центрифугируйте при 500 xg в течение желаемого времени для достижения требуемого объема концентрата. Для объема образца, равного ~ 575 мкл, 20-минутный спин обычно приводит к концентрации концентрата 15-30 мкл.
- Выньте фильтр из трубки и снова взвешивайте. Используйте таблицу, чтобы определить, достигнут ли желаемый объем концентрата. Если нет, снова центрифугируйте при 500 xg в течение более короткого промежутка времени и снова взвешивайте; Повторите до тех пор, пока не будет достигнут желаемый объем концентрата.
- Поместите новую трубку (снабженную колоннами) поверх блока фильтра и инвертируйте. Центрифуга в течение 3 мин при 1000 мкг для передачи coНейтралитет к трубке.
- Определите объем восстановленного. Обычно это будет на 3 - 5 мкл меньше расчетного объема из-за сохранения фильтра. Если чрезмерно концентрированный, разбавленный стерильной водой или TE для достижения желаемого объема.
- Количественное определение ДНК с использованием высокочувствительной количественной оценки (согласно инструкциям производителя).
- Обработка колонны
3. Способ №2: Метил-фракционирование (MF) Подход к обогащению для органеллярной ДНК из общей геномной ДНК
ПРИМЕЧАНИЕ. Этот протокол был модифицирован из разработанного пользователем протокола извлечения ДНК Genomic Tip Kit для растений и грибов 27 и коммерческого протокола 28 для микробиологического обогащения ДНК. Теоретически, любой протокол выделения ДНК, который дает высокомолекулярную ДНК, может быть использован для раскрытия. Для секвенирования с коротким считыванием любая экстракция, дающая преимущественно фрагменты размером более 15 т.п.н., подходит для использования в раскрывающемся списке. Для loNg-read, могут потребоваться более крупные фрагменты. Поэтому мы оптимизировали этот протокол для получения высокомолекулярной ДНК.
- Выделение полной ДНК
ПРИМЕЧАНИЕ. См. Список необходимого оборудования и реагентов и подготовьте необходимые буферы и рабочие запасы для Метода № 2 ( Таблица 1 ). Добавьте лизирующие ферменты в лизисный буферный буфер, чтобы сделать рабочий раствор лизирующего буфера. Включите термомиксер и установите его на 37 ° C. Включите водяную баню до 50 ° C и положите буфер QF в ванну. Поместите 70% EtOH в морозильник и установите микроцентрифуг на 4 ° C.- Полная экстракция ДНК с использованием коммерческих колонок для экстракции ДНК
ПРИМЕЧАНИЕ. Перед началом ознакомьтесь с коммерческим справочником 29 для получения подробной информации об использовании колонок анионного обмена гравитационного потока. Столбы могут быть установлены с использованием специализированной стойки или размещены над трубами с использованием прилагаемых пластиковых колец. Все этапы, включая gЭномические наконечники, должны пропускаться под действием силы тяжести, а остаточная жидкость НЕ должна быть пропущена.- Измельчить 20 мг замороженной ткани в жидком азоте в 2-мл трубке с низким связыванием с использованием ручных мелющих пестиков, предназначенных для 2-мл пробирки.
- Добавьте 2 мл рабочего раствора лизирующего буфера (трубки будут очень полными).
- Инкубировать в термомиксере при 37 ° С в течение 1 ч при осторожном перемешивании со скоростью 300 об / мин. Если термомиксер недоступен, подходящей альтернативой является инкубация на тепловом блоке и перемешивание путем мягкого щелчка каждые 15 минут.
- Добавьте 4 мкл РНКазы А (100 мг / мл, конечная концентрация 200 мкг / мл). Инвертировать для смешивания и инкубации в термомиксере в течение 30 мин при 37 ° С с осторожным перемешиванием со скоростью 300 об / мин.
- Добавить 80 мкл протеиназы К (20 мг / мл, конечная концентрация 0,8 мг / мл), инвертировать для смешивания и инкубировать в термомиксере в течение 2 ч при 50 ° С с осторожным перемешиванием со скоростью 300 об / мин.
- Центрифуга в течение 20 мин при 4 ° С и 15000 xg для гранулирования нерастворимых обломков.
- В то время как образцы центрифугируют, уравновешивают колонки с 1 мл буфера QBT и позволяют колонне пустить гравитационным потоком.
- Используйте наконечник пипетки с широким отверстием, чтобы быстро применить образец (избегать гранулы) до уравновешенной колонки и дать ему полностью протекать через колонку. Если образец снова станет облачным, фильтром или центрифугой перед нанесением на столбец (см. Коммерческое руководство для деталей 29 ).
- После того, как образец полностью вступил в смолу, промойте колонку 4 х 1 мл буфера QC.
- Приостановить колонку на чистой, 2 мл, с низкой связующей микроцентрифужной пробиркой. Элюируют геномную ДНК с 0,8 мл буферного QF, предварительно нагретого до 50 ° C.
- Осаждайте ДНК путем добавления 0,56 мл (0,7 объема буфера для элюирования) изопропанола с комнатной температурой к элюированной ДНК.
- Перемешивают путем инверсии (10X) и центрифугируют в течение 20 мин при 15000 × и 4 ° С. ЗаботаПолностью удалите супернатант, не нарушая стекловидного, слабо прикрепленного гранулы.
- Промыть центрифугированную ДНК-таблетку 1 мл холодного 70% этанола. Центрифуга в течение 10 мин при 15000 xg и 4 ° C.
- Осторожно удалите супернатант (будьте осторожны с этим шагом), не нарушая осадок. Сушили на воздухе в течение 5-10 мин и ресуспендировали ДНК в 0,1 мл буфера для элюирования (ЕВ). Растворяют ДНК в течение ночи при комнатной температуре. Избегайте пипетирования, который может срезать ДНК.
- Количественное определение образцов с использованием высокочувствительного анализа количественной оценки ДНК (согласно инструкциям производителя).
- Полная экстракция ДНК с использованием коммерческих колонок для экстракции ДНК
- Фракционирование на основе бисера метилированной и неметилированной ДНК
ПРИМЕЧАНИЕ. В недавней публикации было продемонстрировано использование коммерчески доступного набора 28 , в котором используется раскрытый подход, в котором используется CpG-специфический белок с метилсвязывающим доменом, слитый с фрагментом Fc человеческого IgG (белок MBD2-Fc) до фракции(Неметилированный) из ядерного генома (сильно метилированного) содержания 23 . Эффективность фракционирования в образцах пшеницы ранее не тестировалась с использованием этого коммерческого набора MF 28 .- Что нужно сделать перед запуском
- Свежеприготовить 80% этанола (по меньшей мере 800 мкл на реакцию). Устанавливают 5-кратный буфер связывания / промывки для оттаивания на льду и готовят 5 мл 1-кратного буфера на образец (разбавленный буфер 5X стерильной, без нуклеазной воды и оставляют на льду во время протокола).
- Подготовьте MBD2-Fc белковые магнитные бусины
- Подготовьте необходимое количество наборов шариков. Масштабируйте реакции, чтобы использовать от 1 до 2 мкг общей входной ДНК, требуя 160 - 320 мкл шариков. Обратите внимание, что перечисленные ниже реакции рассчитаны на 1 мкг общей входной ДНК, поэтому они требуют 160 мкл шариков. Масштабируйте реакции в соответствии с потребностями.
- Используя наконечники с широким отверстием, аккуратно пипетируйте белок A Magnetic BЧтобы создать однородную суспензию. В качестве альтернативы осторожно вращайте трубку шариков в течение 15 минут при 4 ° C.
ПРИМЕЧАНИЕ. Не встряхивайте шарики. - Приступайте к указаниям изготовителя 28 .
- Захват метилированной ядерной ДНК
- Для каждого отдельного образца добавьте 1 мкг входной ДНК в пробирку, содержащую 160 мкл связанных с MBD2-Fc магнитных гранул.
- Добавьте 5x связующий / промывочный буфер, если необходимо, с учетом объема входного образца ДНК для конечной концентрации 1x (объем 5x связующего / промывочного буфера для добавления (мкл) = объем входной ДНК (мкл) / 4). Пипетируйте образец вверх и вниз несколько раз, чтобы перемешать, используя наконечник пипетки с широким отверстием.
- Поверните трубки при комнатной температуре в течение 15 мин. Осторожно пипетируйте образцы с помощью наконечника пипетки с широким отверстием и прокрутите образцы 2 - 3 раза на протяжении всей инкубации, чтобы предотвратить скопление шариков.
ПРИМЕЧАНИЕ. Пипетка и фликкиNg имеет решающее значение для обеспечения эффективного раскрытия метилированной ДНК.
- Собирать обогащенную, неметилированную органелларную ДНК
- Кратко спин трубки, содержащей ДНК и MBD2-Fc-связанной магнитной шариковой смеси. Поместите трубку на магнитную стойку не менее 5 минут, чтобы собрать бусины сбоку от трубки. Решение должно выглядеть ясным.
- Используя наконечники с широким отверстием, осторожно удалите очищенный супернатант, не нарушая бусины. Перенесите супернатант (содержащий неметилированную, обогащенную органеллами ДНК) в чистую, с низкой связью, 2-мл микроцентрифужную пробирку. Храните этот образец при -20 или -80 ° C или переходите непосредственно к шагу 3.2.6 для очистки.
- Элюат захватил ядерную ДНК из магнитных гранул MBD2-Fc
- Если ядерная фракция также желательна, следуйте инструкциям изготовителя 28, чтобы элюировать ядерную ДНК из связанных с MBD2-Fc магнитных гранул; Очистить, как описано на этапе 3.2.7.
- Очистка нуклеиновой кислоты на основе бисера
- Убедитесь, что гранулы очистки находятся при комнатной температуре и тщательно перемешаны. Выполните протокол в соответствии с инструкциями в руководстве MF kit 28 .
ПРИМЕЧАНИЕ. Образец теперь можно использовать для построения библиотеки NGS или другого последующего анализа.
- Убедитесь, что гранулы очистки находятся при комнатной температуре и тщательно перемешаны. Выполните протокол в соответствии с инструкциями в руководстве MF kit 28 .
4. Количественная оценка и контроль качества
- Анализ qPCR для оценки органического обогащения
ПРИМЕЧАНИЕ. Параметры реакции и анализа qPCR, перечисленные здесь, были предназначены для использования на Roche LightCycler 480 и, возможно, их необходимо настроить для различного оборудования и реагентов. Если qPCR недоступен, конечная точка PCR и визуализация на агарозном геле могут быть использованы в качестве качественного показателя чистоты образца, используя те же самые праймеры и условия, описанные здесь. Размеры Amplicon будут ~ 150 bp для всех наборов праймеров. См. Таблицу 3 для секвенирования праймераCes и спаривания.- Настройка реакции qPCR
- Чтобы настроить отдельную реакцию 20 мкл qPCR, осторожно пипеткой в одну лунку 96-луночного планшета qPCR: 10 мкл 2x SYBR Green I Master; 2 мкл 10 мкМ смеси прямого и обратного праймеров (для конечной концентрации 0,5 мкМ); 2 мкл шаблона (в пределах стандартной кривой); И 6 мкл стерильного, не содержащего нуклеазы H 2 O. Чтобы уменьшить ошибки пипетирования, предпочтительно сделать мастер-смесь со всеми компонентами реакции, кроме шаблона. Добавьте мастер-микс к пластине qPCR, а затем добавьте шаблон, представляющий интерес для каждой лунки. Для минимизации последствий ошибки пипетирования необходимо выполнить три технических повторения для каждого образца.
ПРИМЕЧАНИЕ. В конечном счете, отношение ядерных циклов квантования атомов к органеллам сравнивается между образцами, поэтому допустимы незначительные различия в концентрации. Однако концентрации ДНК должны быть примерно в пределах eaCh другой. - Уплотните пластину высококачественной уплотнительной пленкой qPCR. Аккуратно встряхните образцы, стараясь избегать создания пузырьков. Кратко открутите планшет в течение 2 минут при 4 ° C для сбора образца и устранения любых мелких пузырьков.
- Загрузите пластину в машину. Запустите программу qPCR согласно приведенным ниже рекомендациям.
- Чтобы настроить отдельную реакцию 20 мкл qPCR, осторожно пипеткой в одну лунку 96-луночного планшета qPCR: 10 мкл 2x SYBR Green I Master; 2 мкл 10 мкМ смеси прямого и обратного праймеров (для конечной концентрации 0,5 мкМ); 2 мкл шаблона (в пределах стандартной кривой); И 6 мкл стерильного, не содержащего нуклеазы H 2 O. Чтобы уменьшить ошибки пипетирования, предпочтительно сделать мастер-смесь со всеми компонентами реакции, кроме шаблона. Добавьте мастер-микс к пластине qPCR, а затем добавьте шаблон, представляющий интерес для каждой лунки. Для минимизации последствий ошибки пипетирования необходимо выполнить три технических повторения для каждого образца.
- Параметры реакции qPCR
ПРИМЕЧАНИЕ. Это параметры по умолчанию, за исключением цикла отжига на стадии усиления. Отрегулируйте эту настройку, чтобы разместить конкретные праймеры, если те, которые используются, отличаются от праймеров, представленных в этом протоколе.- Предварительно инкубировать при 95 ° С в течение 5 мин с частотой рампы 4,4 ° С / с.
- Выполните 45 циклов амплификации (1) 95 ° C в течение 10 с с частотой рампы 4,4 ° C / с; (2) 60 ° С в течение 20 с, с разбросом 2,2 ° С / с; И (3) 72 ° C в течение 10 с, с разбросом 4,4 ° C / с (данные, полученные во время (3)).
- Используйте опциюЦикл кривой расплава в расплаве 95 ° С в течение 5 с при скорости рампы 4,4 ° С / с; 65 ° С в течение 1 мин, с разбросом 2,2 ° С / с; И 97 ° C, с непрерывным режимом сбора.
- Используйте цикл охлаждения 40 ° C в течение 30 с при скорости рампы 1,5 ° C / с.
- Параметры анализа
- Выберите шаблон SYBR. Проверьте параметры программы на кнопке «Эксперимент». После загрузки пластины анализ можно запустить, и настройки могут быть отрегулированы во время анализа.
- Назначьте образцы с помощью редактора образцов. Выберите Abs Quant в качестве рабочего процесса и укажите образцы как неизвестные, стандарты или отрицательные элементы управления. Назначьте репликации и заполните имена образцов первого из каждого репликации. Добавьте концентрации и единицы в стандарты.
- Настроить подмножества для анализа; Они назначаются в редакторе подмножества.
- Для анализа выберите Abs Quant / 2nd Derivative Max из списка «Создать новый анализ».Импортируйте внешнюю сохраненную стандартную кривую (если применимо), а затем нажмите вычисление; Отчет будет содержать выбранную информацию.
- Для выполнения точной абсолютной количественной оценки для определения количества копий или концентрации используйте стандартную кривую, которая является репрезентативной для тестируемого образца ( например, органелларная ДНК, выделенная из вышеуказанных методов). Поскольку количество митохондриальной ДНК, необходимое для подготовки стандартной кривой, слишком велико для достижения достаточного количества ткани, не используйте вычисления количества копий, предоставляемые программным обеспечением, а вместо этого проверяйте значения точки пересечения (Cp) для определения относительного обогащения Органеллара по сравнению с ядерной ДНК в образцах. Сравните эти относительные количества с суммарной геномной ДНК (см. Представительские результаты ). Эффективность тестового праймера при пяти разведениях 1:10 общей геномной ДНК из полностью светлых, двухнедельных семян пшеницы (репрезентативная эффективность, указанная вE легенда на рисунке 2 ).
- Настройка реакции qPCR
- Электрофорез в импульсном поле (PFGE)
ПРИМЕЧАНИЕ. Этот протокол основан на рекомендациях производителя для выполнения PFGE для разрешения высокомолекулярной ДНК. См. Таблицу материалов.- Подготовка геля и образцов
- Следуйте рекомендациям по подготовке геля и образцов и адаптируйте их к доступной системе.
- Запуск параметров
- Следуйте рекомендациям по настройке системы электрофореза и используйте следующие параметры: начальное время переключения 2 с, конечное время переключения 13 с, время работы 15 ч и 16 мин, В / см 6 и включенный угол 120 ° ,
- Пятно и изображение геля
- Пятно гель с красителем выбора ( например, бромид этидия или подходящая альтернатива) и изображение с подходящей системой документации на гель.
- Подготовка геля и образцов
- Используйте 1 нг ДНК в качестве входного сигнала для набора ДНК-библиотеки Prep Kit в соответствии с инструкциями производителя.
- Штрих-код и объединить образцы для секвенирования за один проход. Выполните последовательность в соответствии с рекомендациями производителя.
ПРИМЕЧАНИЕ. Параметры пула и последовательности могут быть изменены в зависимости от интересующих видов, желаемого уровня покрытия и платформы, используемой для последовательности библиотек. Например, полоса HiSeq имеет значительно больший выход, чем дорожка MiSeq, поэтому многие другие образцы могут быть мультиплексированы. Последовательность меньшего подмножества выборок, чтобы определить, достаточны ли уровни охвата органеллогенных геномов для анализа ниже по течению.- Изучите качество чтения с помощью FastQC 31, чтобы определить степень обрезки и фильтрации, требуемую для данных.
- Обрезать и фильтровать необработанные чтения с помощью Trimmomatic 32 или другой сопоставимой программы. Используйте следующие настройки: ILLUMINACLIP 2:30:10 (для удаления адаптеров), ВЕДУЩИЙ 3, ТРЕНИРОВКА 3, СКОЛЬЖЕНИЕ ВИДЕО 4:10 и МИНЛЕН 100.
- Сопоставьте обработанную по качеству фильтрованную и адаптированную по частям парную конец (PE), чтобы прочитать митохондрию китайской пружины (NCBI Reference Sequence NC_007579.1 33 ), хлоропласт (NCBI Reference Sequence NC_002762.1 34 ) и ядерные 35 эталонных геномов с использованием Bowtie2 36 , Со следующими настройками: -I 0 -X 800 - чувствительный.
- Преобразуйте файлы выравнивания sam в формат bam (samtools) и отсортируйте файлы bam. Используйте файлы bam, чтобы рассчитать охват генома и покрытие каждой базой с помощью постельных принадлежностей. Визуализируйте результаты с помощью функции R-plot.
- Что нужно сделать перед запуском
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Протоколы, представленные в этой рукописи, описывают два разных метода обогащения органелларной ДНК из растительной ткани. Представленные здесь условия отражают оптимизацию для ткани пшеницы. Сравнение основных этапов протоколов, требуемого ввода ткани и выхода ДНК описано на рисунке 1 . Этапы протестированного нами протокола DC следуют аналогичным условиям, описанным ранее ( рис. 1А ). Собранные ткани должны обрабатываться свеже и подвергаться дифференциальному центрифугированию и / или градиентам, чтобы изолировать интактные органеллы. Ядерная ДНК удаляется до того, как органеллы будут лизированы, и, наконец, ДНК извлекается и используется для последующих приложений. Напротив, в протоколе MF растительная ткань может собираться и храниться перед использованием, а интактные органеллы не требуются. Вместо этого ядерная и органелларная ДНК фракционируют из общей гДНК на основе Статус метилирования ДНК. Оба протокола дают примерно одинаковое количество органелларной ДНК ( рис. 1C ). Что касается общего выхода органелларной ДНК по отношению к вводу ткани, протокол MF является предпочтительным, когда ткань ограничена, так как может быть использован небольшой образец из одного растения, и растение может быть разрешено расти для дальнейшего анализа. Как правило, в протоколах постоянного тока требуются все воздушные ткани многих сеянцев, и эти растения отбрасываются. Тем не менее, метод DC может быть оптимизирован специально для обогащения одного типа органелл над другим, что невозможно с помощью подхода MF. Стоит отметить, что общее время для каждого протокола примерно эквивалентно, хотя в подходе MF меньше времени на использование.
Оба метода обогащают органическую ДНК, хотя и с разными пропорциями митохондрий и пластовых последовательностей:
«> Очень низкие количества очищенной органелларной ДНК получают из любого метода (порядка ~ 50-100 нг, рис. 1С ). Для оценки уровней обогащения органелларного генома и заражения ядерным геномом в ДНК, выделенной из DC и MF Методах был использован анализ qPCR. В этом анализе относительные количества трех ампликонов ( т.е. ядерно-специфических, ACTIN , митохондриальных, NAD3 и Хлоропласт-специфичный, PSBB ) оценивали в полной геномной ДНК, и фракция органелларной ДНК была получена из обоих методов ( рисунок 2 ). Циклы количественного определения (C q ) были исследованы для каждого образца ( рис. 2A ), а поскольку C q определяется как цикл ПЦР, при котором флуоресценция от амплификации мишени возрастает выше уровня фоновой флуоресценции, C q и численность мишени имеют Обратная зависимость. ВОбразец DC, C q NAD3 и PSBB , соответственно, ~ 17 и ~ 15 циклов раньше ACTIN (который имеет C q ~ 36) (см. Рисунок 2B для значений C q и уровней обогащения). Это соответствует теоретическим 167,181- и 47,790-кратным обогащениям для NAD3 и PSBB , соответственно, Относительно ACTIN в образце DC ( рисунок 2B , см. Легенду на рисунке 2 для расчета). В образце общей геномной ДНК фоновые обогащения для NAD3 и PSBB относительно ACTIN составляют только 158 и 10 701 соответственно. Нет ничего удивительного в том, чтобы найти более высокое содержание органорных ампликонов относительно ядерного ампликона в полной геномной ДНК, учитывая, что органелларные геномы существуют в большем количестве копий на клетку, чем ядерный геном 37, и что количество органелл peR может различаться в зависимости от типа ткани или стадии 38 , 39 развития . В целом, данные показывают, что метод DC предпочтительно обогащает митохондрии, что следует ожидать, поскольку скорости центрифугирования оптимизированы для селективной изоляции митохондрий и уменьшения загрязнения ядер и хлоропластов.Неметилированная фракция общей гДНК MF также демонстрирует значительное обогащение обоих органеллярных ампликонов и, как ожидается, сохранит нативные относительные количества этих мишеней. Фракционные обогащения для NAD3 и PSBB относительно ACTIN в неметилированной фракции составляют 20 551 и 1 703 253 соответственно ( фиг. 2A и 2B ). В метилированной фракции фоновые обогащения для NAD3 и PSBB относительно ACTIN составляют 31 и 823 соответственно, indiЧто белок MBD2-Fc является высокоэффективным в раскрытии метилированной ядерной ДНК. Поскольку у кристалла хлоропласта более высокое содержание, чем митохондриальный ампликон, в общей геномной ДНК (ранее ~ 6 C q ), ранее метилированная фракция (~ 5 C q ранее) и неметилированная фракция (~ 6 C q ранее), это говорит о том, что Натуральное обилие этих ампликонов существенно не изменяется с помощью раскрытия MDB2. Мы фокусируемся здесь на неметилированной (органелларной) фракции из-за интереса к секвенированию этих геномов. Однако, если ядерный геном является основным интересом, MF и последующее секвенирование метилированной фракции дадут гораздо более высокий охват ядерным геномом, чем полное секвенирование геномной ДНК, из-за уменьшения «загрязнения» органелларной ДНК.
Стоит отметить, что если qPCR недоступен, конечная точка PCR (с использованием тех же праймеров, что и для qPCR) обеспечивает качествоОценки органической чистоты. В этом случае образцы чистой органелларной ДНК будут демонстрировать амплификацию для митохондриальных и пластидных ампликонов, но не обнаруживается амплификация ядерного ампликона на агарозном геле, тогда как общая геномная ДНК показывает амплификацию для всех трех наборов праймеров, как показано в предыдущих исследованиях 11 , 12 .
Органальная ДНК, выделенная из обоих методов, подходит для NGS:
Обрезанные и очищенные чтения секвенирования PE (см. Шаг 4.3) были сопоставлены с ранее опубликованными органоэлларными ссылочными геномами пшеницы, а количество чтений, используемых для сопоставления каждого образца, варьировалось от ~ 800 000 до 1100 000 ( рисунок 3I ). Результаты картирования de novo Последовательность Illumina, которая читается в доступных хлоропластах пшеницы и геномах митохондрий, согласуется с qpCR resС использованием метода DC, дающего ДНК, которая более обогащена митохондриальной ДНК ( рис. 3A и 3B , ~ 80% и ~ 10% от читаемой карты относятся к митохондриальным (mt) и хлоропластовым (cp) геномам соответственно) и МФ-методу Которая, по-видимому, отражает местное обилие двух органелларных геномов ( рис. 3А и 3В , ~ 20% и ~ 80% карт считывания к геномам мт и ср соответственно). В обоих методах теоретический охват (см. Легенду рис. 3 для расчета) обоих органелларных геномов пшеницы превышает 100X-охват (и охватывает до 2000 облучений для генома хлоропластов в неметилированной фракции из МФ-метода), даже Когда 12 библиотек мультиплексированы ( рис. 3C и 3D ; 6 библиотек, включенных в этот анализ, были объединены с дополнительными 6 библиотеками для отдельного анализа, в общей сложности 12 библиотекОбъединенный в одну последовательность последовательности). Более детальный взгляд на охват был достигнут путем изучения доли генома, покрытого на определенной глубине, а также уровней охвата каждой базовой зоны ( рис. 3Е- 3I ). Для метода МФ среднее значение для базовой зоны составляло ~ 300 - 450X для генома mt и 4000-5000X для гена cp. Для метода постоянного тока среднее значение для каждой базовой зоны составляло ~ 900 - 1300 и ~ 500 - 700X для геномов mt и cp соответственно. Однако была небольшая доля как геномов mt, так и cp, которые имели чрезвычайно низкий или высокий уровень охвата, и это было обнаружено в органелярной ДНК, полученной из обоих методов ( фиг. 3I ). Регионы более высокого, чем средний охват, вероятно, соответствуют регионам гомологии между органелларными геномами, а области с низким охватом могут указывать SNP или другие небольшие варианты между сортами, которые мы секвенировали, и опубликованными ссылками. В поддержку этого понятия эти шипыВысокого охвата были наиболее выражены для ДНК mt, полученной из метода MF ( фиг. 3E и 3I ), вероятно, из-за высокого охвата гена cp в этом методе. Необъяснимо, что охват гена cp более неравномерен в методе MF, чем метод DC ( рис. 3G и 3H ), что может быть связано с небольшими смещениями в MBD2-Fc-распаде вдоль ДНК cp. Дальнейшие эксперименты потребуются, чтобы определить, почему это так. Несмотря на это, геномы mt и cp имели относительно равномерный охват обоими методами и отсутствовали большие области отсутствия покрытия, что может быть продемонстрировано путем изучения доли геномов, упорядоченных на заданной глубине ( рис. 3E -3H ). Кроме того, уровни охвата для обоих геномов считаются достаточными для последующего анализа, такого как вариантный анализ. Если это будет необходимо для анализа редких вариантов, уменьшение числаR объединенных образцов достигнет большего охвата. В качестве альтернативы, гораздо большее количество выборок может быть объединено на дорожке HiSeq, достигая еще большей глубины последовательности, хотя и при жертвовании длиной последовательности, поскольку библиотеки HiSeq в настоящее время ограничены длиной PE150 в отличие от библиотек MiSeq PE300.
Для изучения уровней заражения ядерным геномом с использованием подхода, связанного с отображением, были изучены категории считывания ПЭ. Чтение PE может отображать опорный геном в различных конфигурациях. Когда вы читаете 1 и 2, выровняйтесь по ссылке в режиме «голова к голове» с определенным «ожидаемым» расстоянием между двумя помощниками (в зависимости от среднего размера вставки библиотеки и обычно указывается в качестве входного параметра в программном обеспечении сопоставления ), Эти PE-чтения, как говорят, сопоставляются «согласованно». Напротив, «несогласованное» сопоставление - это ситуация, когда сопоставление карт с меньшим или большим, чем ожидалось,(См. Рис. 1). В зависимости от того, где находится геном или карта в альтернативных конфигурациях (от головы до хвоста или от хвоста до хвоста). Если только один помощник выравнивается с эталонным геномом, тогда считается, что чтение PE не соответствует или несогласованно с эталонным геномом. Во всех трех категориях чтения-отображения, PE-чтения могут выравниваться с эталонным геномом один или несколько раз.
Как для DC-и MF-выделенной органелларной ДНК, считывание картирования в митохондриальный геном было преимущественно в согласованной согласованной одноразовой категории ( фиг. 4A ), тогда как считывания отображались на геном хлоропластов в относительно равных пропорциях согласованно один раз и соответствовало более чем Одно время ( рис. 4B ), вероятно, из-за больших инвертированных повторов, присутствующих в геноме хлоропластов, а также с чрезвычайно высокими уровнями охвата. Тем не менее, меньшее количество ПЭ читается, сопоставленное с ядерным геномом и в значительной степени сопоставляемое более одного раза вНи согласованной, ни дискордантной моды ( т. Е. Только один матер способен сопоставлять). Скорее всего, это сопоставление «вне цели» с последовательностями в ядерном геноме, которые гомологичны органелларным геномам или неправильно разобранным регионам. Только незначительное количество считываний (<5%) сопоставляется с ядерным геномом в согласии, что указывает на низкий уровень загрязнения ядерного генома в органной ДНК, выделенный из метода DC или MF ( рисунок 4C ), что также отражается в результатах qPCR ( рисунок 2А ). Ядерную фракцию после выведения MBD2-Fc из китайских неэтилированных тканей весны также секвенировали, чтобы определить, насколько эффективна пулдаун при удалении неметилированной ДНК. Менее 1% читается в библиотеке, полученной из ядерной фракции, отображаемой в органолептические эталонные геномы, тогда как ~ 45% всех читаемых изображений сопоставлены с ядерным геномом ( рисунок 4 ). Тем не менее, большинство читаемых изображений несогласованно, wВероятно, отражает высокие уровни неправильной сборки и фрагментации в ядерном контрольном геноме пшеницы. Независимо от того, что результаты показывают, что раскрытие MBD2-Fc является высокоэффективным при удалении неметилированной органелларной ДНК из метилированной ядерной ДНК. Следует отметить, что, поскольку обогащенная органелларными ДНК, полученная в результате этих методов, содержит смесь митохондрий и хлоропластов, а также потому, что сходства последовательностей, возникающие в результате переноса древних генов между этими органеллами, остаются в их геномах, правильное назначение чтений для конкретных Геномы должны решаться биоинформативно.
Этиоляция листовой ткани не вызывает значительного изменения содержания органических веществ:
Традиционно этиолированные ткани являются предпочтительными для выделения митохондриальной ДНК растений, чтобы уменьшить уровни фенольных и крахмалов, что может мешать экстрактуN или ниже по потоку 13 . Чтобы определить, можно ли изменить или улучшить уровни обогащения органелларного генома в условиях роста, как этиолированные, так и неэтилированные ткани были подвергнуты протоколу MF и секвенированию. Интересно отметить, что этиоляция значительно не изменила процент прочтений, которые были отображены на органоэлементные эталонные геномы ( фиг. 3A и 3B ) или на покрытие по каждой базовой линии ( фиг. 3I ) по сравнению с неэтилированными условиями. Мы также выделили органелларную ДНК с использованием дифференциального центрифугирования как с этиолированными, так и с неэтилированными тканями, и была обнаружена небольшая разница в обогащении между различными тканями с использованием qPCR (данные не показаны). Это говорит о том, что для исследований органеллярного секвенирования можно использовать более физиологически релевантные неэтилированные ткани без существенного изменения обогащения.
Контроль качества предполагает, чтоДНК МФ наиболее подходит для долговременного секвенирования:
Поскольку длительное чтение последовательности становится более доступным для исследователей, изоляция высокомолекулярной ДНК становится все более важной. Для оценки органеллярной ДНК, изолированной любым методом для неповрежденности и качества, применяли PFGE. Общая геномная ДНК обычно мигрирует как диффузный мазок в PFGE, а молекулярная масса определяется протоколом и как ДНК хранится и обрабатывается после экстракции. Общая геномная ДНК, выделенная геномными кончиками, должна превышать 50 кб, что было проверено с использованием PFGE ( рисунок 5 , дорожка 2). Общая геномная ДНК из геномных наконечников используется в качестве входных данных в набор для обогащения микробиома для фракционирования ядра от органной ДНК. Ядерная фракция, полученная после фракционирования, уменьшается по размеру, но остается в центре около 50 кб ( рис. 5 , дорожка 4). Это не суПри условии, что относительно более грубая обработка ядерной фракции в качестве элюирования из MBD2-Fc-связанных гранул требует теплового и протеиназного расщепления K. Из-за ограниченной массы органелларная фракция не запускалась на PFGE, но последующий анализ с TapeStation показал ДНК> 50 кб (данные не показаны). Органная ДНК, полученная с дифференциальным центрифугированием, имеет среднюю массу ~ 20 кб, что, вероятно, вызвано протоколом расширенной органолептической изоляции и последующей экстракцией и концентрацией ДНК на основе колонок. Методы органеллральной изоляции и альтернативные методы экстракции ДНК на основе градиента могут поддерживать больший размер фрагментов ДНК. Независимо от того, что ДНК размером, полученным в этом протоколе, может быть использована для генерации секвенирования 10 или 15 кб, если в процессе подготовки библиотеки проводится осторожность.
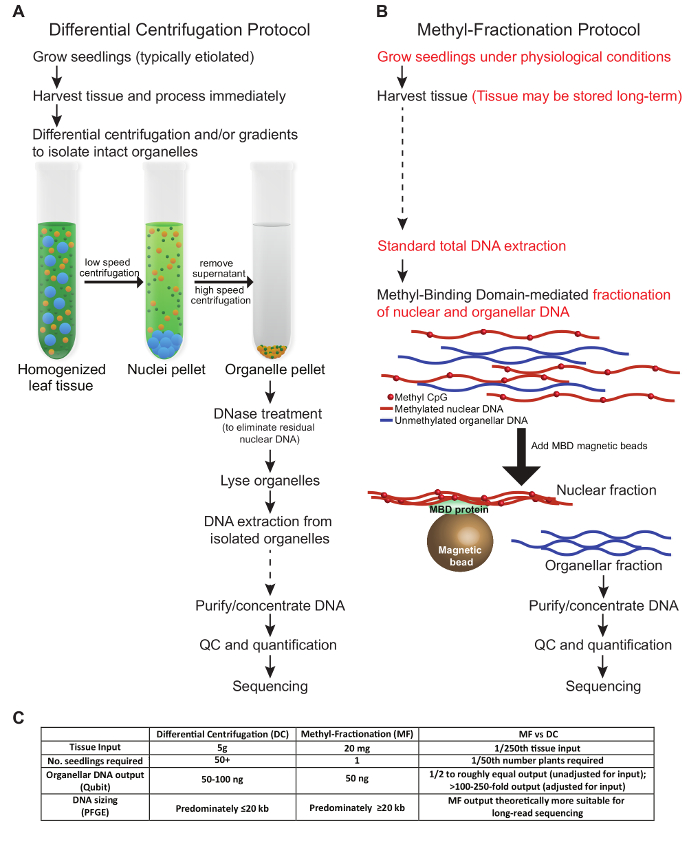
Рисунок 1: Сравнительный взгляд на два методаDs для обогащения органической ДНК растений. Традиционный DC-протокол ( A ) противопоставляется протоколу MF ( B ). Рекомендуется избегать замораживания и оттаивания образцов; Однако этапы, на которых образцы могут храниться долговременно, обозначены пунктирными стрелками ( А и В ). Основные различия между протоколами выделены красным цветом ( B ). ( C ) В таблице сравниваются методы с точки зрения ввода ткани, количества необходимых растений, выхода ДНК и результирующего размера ДНК. Нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 2: Оценка ядерного заражения ДНК в органической ДНК, выделенной с использованием двух методов. (
( B ) В таблице показаны значения C q , которые показаны на графике в ( A ), и обогащение складок органорных ампликонов относительно ACTIN . * Свертывание обогащения = 2 ( Cq ACTIN - Cq Target) . Формула предполагает идеальную эффективность 2 для каждого набора праймеров, поскольку малый девиатИон каждого набора праймеров из 2 является незначительным и мало повлияет на расчет и общий тренд ( ACTIN = 1,961 , NAD3 = 1,95 и PSBB = 1,989 ). Эффективность праймера оценивали путем создания стандартной кривой с серией из пяти разведений 1:10 общей геномной ДНК. Нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 3: Чтение карт и теоретическое освещение хромопластов и митохондриальных геномов. Процент считываний, сопоставленных с митохондриальными ( A ) или хлоропластовыми ( B ) китайскими весенними справочными геномами. Соответствующее теоретическое освещение митохондриального ( C ) или хлоропласта ( D ) китайского источникаMes, предполагая, что размеры генома 450 и 135 кб соответственно вычисляются с использованием общих чисел считывания и процента считывания картирования в разных геномах. Широкое распространение генома для органелларной ДНК по методу MF ( E и G ) или DC-метод ( F и H ). Данные в панелях E- H взяты из эталонного образца китайской весны, но все остальные образцы показали аналогичную тенденцию. ( I ) Средний, самый низкий и самый высокий уровень охвата каждой базой для всех образцов в панелях A- D . Типовые метки, включая «E», обозначают этиолированные образцы, а «NE» обозначает неэтилированные образцы. DC обозначает ДНК, выделенную с помощью метода дифференциального центрифугирования, и неметилированный указывает ДНК, которая находится в неметилированной фракции после раскрытия с MBD2-Fc (протокол MF). Образцы с надписью «Chris» обозначают пшеницу Triticum aestivum'Крис.' CS обозначает образцы пшеницы Triticum aestivum 'Chinese Spring. Примечание. Из-за гомологии последовательностей между хлоропластом, митохондриями и ядерными геномами, полученными в результате переноса древних генов между органелларными геномами, а также между органелларными и ядерными геномами, небольшой процент необработанных чтений может отображаться в нескольких геномах. Кроме того, считывания, которые не относятся к органеральному эталонному геному, не представлены на этом рисунке. Следовательно, показанные здесь проценты ( А и В ) не составляют 100%. Нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 4: PE Read Mapping к ядерному геноту пшеницы. Процент категорий PE Считывают типы отображения в митохондриальные (A) , хлоропласты (B) или ядерные (C) китайские весенние справочные геномы. - E обозначает этиолированные образцы и - NE обозначает неэтилированные образцы. DC обозначает ДНК, выделенную с помощью метода дифференциального центрифугирования. Неметилированный указывает ДНК, которая находится в неметилированной фракции после раскрытия с MBD2-Fc в протоколе MF, а метилированный обозначает ядерную фракцию после раскрытия MBD2-Fc. Образцы с надписью «Chris» обозначают пшеницу Triticum aestivum «Chris». CS обозначает образцы пшеницы Triticum aestivum 'Chinese Spring.' Неотображаемые чтения не отображаются. Нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
Oad / 55528 / 55528fig5.jpg "/>
Рисунок 5: Исследование качества ДНК с использованием PFGE. Общая геномная ДНК пшеницы (дорожка 2), органелларная ДНК пшеницы, полученная из дифференциального центрифугирования (дорожка 3), и ядерная фракция после MF с подходом MBD2-Fc (дорожка 4) подвергались PFGE на 1% агарозном геле с 1 kb удлиненная лестница, используемая в качестве маркера (дорожки 1 и 5). Нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
| Имя буфера | Рецепт | Заметки | метод |
| Буфер STE | 400 мМ сахарозы, 50 мМ Трис, рН 7,8, 20 мМ ЭДТА, рН 8,0, 0,6% (мас. / Об.) Поливинилпирролидона (ПВП), 0,2% (мас. / Об.) Бычьего сывороточного альбумина (БСА), 0.1% (об. / Об.) Β-меркаптоэтанола (BME) | Буферную смесь, содержащую только сахарозу, трис и ЭДТА, можно приготовить до месяца вперед и выдерживать при 4 ° С. PVP, BSA и BME следует добавлять к аликвоте необходимого количества буфера непосредственно перед использованием. | Способ №1 |
| Буфер ST | 400 мМ сахарозы, 50 мМ Трис, pH 7,8, 0,6% (мас. / Об.) Поливинилпирролидона (ПВП), 0,1% (мас. / Об.) Бычьего сывороточного альбумина (БСА) | Буферную смесь, содержащую только сахарозу и трис, можно приготовить до месяца вперед и хранить при температуре 4 ° C. Обратите внимание, что буфер ST не содержит EDTA или BME и содержит более низкую концентрацию BSA. | Способ №1 |
| Источник ДНКазы | 2 мг / мл ДНКазы в 0,15 М NaCl до концентрации 2 мг / мл | Хранить 200 мкл аликвоты при -20 ° C. Для приготовления рабочего раствора ДНКазы (200 мкл раствора ДНКазы на образец) см.Таблица 1 ниже. См. Полный протокол ниже для получения полной информации о переваривании ДНКазы. Рабочее решение DNase должно быть подготовлено свежим. Для прекращения реакции ДНКазы требуется 400 мМ раствора EDTA pH 8,0 (конечная концентрация, необходимая для прекращения реакции, составляет 0,2 М ЭДТА, см. Полный протокол для подробностей). | Способ №1 |
| Рабочее решение DNase | 0,25 мг / мл ДНКазы и 20 мМ MgCl 2 в буфере ST | Подготовьте свежий 200 мкл на образец. Концентрации показаны для конечного объема реакции, поэтому смесь: 62,5 мкл 2 мг / мл ДНКазы (на основе конечного 500 мкл объема реакции), 4 мкл 1 М MgCl 2 (в расчете на 200 мкл раствора раствора ДНКазы) и 133,5 мкл ST-буфера для Конечный объем 200 мкл. | Способ №1 |
| Буфер для лизиса | 20 мМ ЭДТА рН 8,0; 10 мМ Трис, pH 7,9; 500 мМ гуанидин-HCl; 200 мМ NaCl; 1% Triton X-100; 0,5 мг / мл лизирующих ферментов изTrichoderma harzianum | Смешайте все ингредиенты, за исключением лизирующих ферментов, и храните их при комнатной температуре. Лизирующие ферменты следует добавлять свежими в небольшую аликвоту для немедленного использования. | Способ №2 |
Таблица 1: Рецепты домашних буферов и рабочих запасов.
| Лист концентрации | |||||||
| ИМЯ ОБРАЗЦОВ | Вес пустого устройства (г) | Вес заполненного устройства (г) | Заполненный объем (ul, заполненный минус пустой вес) | Вес после 1-го вращения (20 мин *, г) | Объем после 1-го спина (ul, заполненныйМинус пустые веса) | Вес после 2-го вращения (X мин *, г) | Объем после 2-го спина (ul, заполненный минус пустых весов) |
| Обратите внимание, что фактический восстановленный объем будет на несколько ul меньше расчетного объема. | |||||||
Таблица 2: Рабочий лист концентрации.
| имя | Специфичность генома | Источник последовательности генов | Последовательность (5 '- 3') |
| Ta_ACTIN - F | ядерной | Gramene Scaffold IWGSC_CSS_1AS_scaff_3272162: 10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | ядерной | То же, что и выше | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | Митохондриальная | Присоединение NCBI EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | Митохондриальная | То же, что и выше | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | хлоропластов | Присоединение NCBI KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | хлоропластов | То же, что и выше | GCTGCCTGTTTCCTTGTAGTT |
Таблица 3: Список праймеров qPCR.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
На сегодняшний день большинство исследований органолептических секвенирования сосредоточены на традиционных методах DC, чтобы обогатить специфическую ДНК. Были описаны способы выделения органелл из различных растений, включая мох 40 ; Однодольные, такие как пшеница 15 и овес 11 ; И двудольные растения, такие как арабидопсис 11 , подсолнечник 17 и рапс 14 . Большинство протоколов фокусируются на листовой ткани 13 , 14 , 15 , 16 , 17 , причем некоторые из них были адаптированы для различных типов тканей, включая семена 11 . Было также продемонстрировано выделение органелл из протопластов 41 . Тем не менее, это не поддается всем системам, и это невозможно, когда ткань, представляющая интерес, ограничена. Многие из этих орговНелекарственные методы изоляции были разработаны для восстановления интактных органелл для конкретных экспериментов, таких как физиологические исследования. Эти протоколы громоздки и обычно требуют использования градиентов плотности, таких как градиенты сахарозы или Перколла, которые очень эффективны при выделении специфических органелларных фракций, но требуют большого ввода ткани ( т.е. более 5 г и выше килограммов, в зависимости от Тип ткани). Однако метод DC может быть оптимизирован для обогащения конкретных клеточных фракций, таких как митохондрии или хлоропласты, путем изменения скорости вращения и градиентов плотности. Напротив, подход MF требует гораздо меньшего количества исходного материала (20 мг), но митохондриальная и пластидная ДНК будут присутствовать на их относительное количество в ткани, используемой для извлечения ДНК. Тем не менее протокол MF предлагает альтернативный подход для выделения смешанной органелларной ДНК и особенно полезен для начала с небольшим количеством ткани.
T O оценить чистоту образца после выделения органелл, в большинстве исследований на сегодняшний день используется только конечная ПЦР и гель-электрофорез 11 , 12 . Это дает справедливую качественную меру чистоты образца. Однако низкие уровни амплификации могут не отображаться на агарозном геле. Несколько отчетов включают более количественные меры контроля качества, такие как qPCR 14 . Для количественной оценки чистоты образца ДНК, выделенной из обоих методов, мы использовали qPCR и секвенирование для определения количества ядерной ДНК в образце, а также относительных пропорций митохондриальной и хлоропластной ДНК. Оба метода, оцененные здесь, эффективны при удалении ядерной ДНК. Оба метода дают смесь митохондриальной и хлоропластной ДНК, хотя и в разных пропорциях.
Сообщается, что выращивание растений в темноте (этиоляции) помогает облегчить органеральную изоляцию из-за уменьшения фенольныхRef "> 13. Однако в этом сравнении мы не нашли заметного преимущества для работы с этиолитированной тканью над светлыми образцами. Хотя доля специализированных хлоропластов, вероятно, будет выше при светлом выражении, общее количество пластиды, как Отраженный в пропорции считывания картирования к геному хлоропластов, не изменяется при различных условиях освещенности. Поэтому для последующих функциональных анализов, таких как оценка гетероплазмы в разных тканях или при разных стрессорах или для анализа экспрессии, мы рекомендуем выполнять геномное секвенирование на Растения, выращенные в физиологически значимых условиях.
Для применения с технологиями секвенирования с коротким считыванием обе методики, сравниваемые здесь, дают достаточное количество и качество ДНК. Однако для достижения длительных чтений> 20 кб для приложений с одномолекулярным секвенированием требуется большее количество ДНК более высокого качества. Например, в идеале,> 1 мкг чистой оргиДНК неллярной пшеницы с молекулярной массой> 20 кб необходима для внутренних протоколов с низким входным сигналом для 20-kb-библиотек вставки библиотеки 42 . Новые разработанные пользователем протоколы с низким входным сигналом могут снизить требования к ДНК ( то есть до 50 нг или даже меньше 20 ), но проблема заключается в том, что в библиотечные препараты входит высококачественная высокомолекулярная ДНК. Существенно, что большая часть ДНК составляет> 20 кб, так как меньшие фрагменты будут предпочтительно вставлены в SMRTbell и сбросить распределение по размеру библиотеки 43 . Мы пробовали ряд протоколов извлечения домашней ДНК и ряд коммерческих протоколов для извлечения ДНК (не показаны). Для ткани листьев пшеницы наилучший баланс между количеством и качеством ДНК, особенно длиной, был получен с использованием коммерческого набора 27 , 29 . В зависимости от видов растений и интересующей ткани, alternatiПротоколы добычи могут быть одинаково пригодными или более плодотворными. Тем не менее, мы заключаем, что полная экстракция высокомолекулярной геномной ДНК размером> 50 кб с последующим фракционированием с подходом 28 MBD2-Fc вниз может поддаваться длительному считыванию из ограниченного исходного материала. Будущая работа должна проверять пределы исходного материала, необходимого после фракционирования для подготовки библиотеки с длинной вставкой и последующего длительного чтения. Критически этот подход может обеспечить надежный метод выделения ДНК из подвыборки одного листа, который подходит для длительного чтения, без амплификации цельного генома. Мы ожидаем, что этот подход будет легко адаптирован к дополнительным типам тканей и широко применим к другим видам растений. Это будет особенно полезно в ситуациях, когда количество тканей ограничено, например, секвенирование у отдельных поколений в схеме пересечения или в более редких типах тканей.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Авторы заявляют, что у них нет конкурирующих интересов.
Упоминание торговых наименований или коммерческих продуктов в этой публикации исключительно для целей предоставления конкретной информации и не подразумевает рекомендации или одобрения Департамента сельского хозяйства США. USDA является поставщиком равных возможностей и работодателем.
Acknowledgments
Мы хотели бы отметить финансирование со стороны Министерства сельского хозяйства США по сельскохозяйственным исследованиям и Национального научного фонда (IOS 1025881 и IOS 1361554). Мы благодарим Р. Каспера за техобслуживание и уход за теплицами. Мы также благодарим Университет Миннесотского геномического центра, где были подготовлены и упорядочены библиотеки Illumina. Мы также благодарны за комментарии редакторов журнала и четырех анонимных рецензентов, которые еще больше укрепили нашу рукопись. Мы также благодарим ОЭСР за стипендию SK по интеграции этих протоколов для совместных проектов с коллегами из Японии.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
