

Summary
A comparação e otimização de dois métodos de enriquecimento de DNA orgânico de plantas são apresentadas: centrifugação diferencial tradicional e fracionamento do gDNA total com base no estado de metilação. Nós avaliamos a quantidade e a qualidade de DNA resultantes, demonstram desempenho em seqüenciamento de próxima geração de leitura curta e discutimos o potencial para uso em sequenciação de moléculas únicas de leitura longa.
Abstract
Os genomas orgânicos de plantas contêm elementos grandes e repetitivos que podem sofrer combinação ou recombinação para formar estruturas complexas e / ou fragmentos sub-genômicos. Os genomas de Organellar também existem em misturas dentro de uma determinada célula ou tipo de tecido (heteroplasmia), e uma abundância de subtipos pode mudar ao longo do desenvolvimento ou quando sob estresse (deslocamento sub-estequiométrico). As tecnologias de sequenciação de próxima geração (NGS) são necessárias para obter uma compreensão mais profunda da estrutura e da função do genoma organelar. Estudos de seqüenciamento tradicionais usam vários métodos para obter DNA organelar: (1) Se uma grande quantidade de tecido inicial é usada, é homogeneizada e submetida a centrifugação diferencial e / ou a depuração gradiente. (2) Se uma quantidade menor de tecido é usada ( ou seja, se sementes, material ou espaço é limitado), o mesmo processo é realizado como em (1), seguido de amplificação de todo o genoma para obter DNA suficiente. (3) A análise de bioinformática pode ser usada para seqUence o DNA genômico total e analisar as leituras orgânicas. Todos esses métodos têm desafios e compromissos inerentes. Em (1), pode ser difícil obter uma quantidade tão grande de tecido inicial; Em (2), a amplificação do genoma inteiro poderia introduzir um viés de seqüenciamento; E em (3), a homologia entre genomas nucleares e organelares poderia interferir na montagem e análise. Em plantas com grandes genomas nucleares, é vantajoso enriquecer o DNA organelar para reduzir os custos de sequenciação e a complexidade da sequência para análises de bioinformática. Aqui, comparamos um método de centrifugação diferencial tradicional com um quarto método, uma abordagem adaptada de CpG-methyl pulldown, para separar o DNA genômico total em frações nucleares e orgânicas. Ambos os métodos produzem DNA suficiente para NGS, DNA que é altamente enriquecido para seqüências organelares, embora em diferentes proporções nas mitocôndrias e nos cloroplastos. Apresentamos a otimização desses métodos para o tecido das folhas de trigo e discutimos grandes vantagens e dSão vantagens de cada abordagem no contexto de entrada de amostra, facilidade de protocolo e aplicação a jusante.
Introduction
O seqüenciamento do genoma é uma ferramenta poderosa para dissecar a base genética subjacente de traços importantes da planta. A maioria dos estudos de sequenciação do genoma se concentra no conteúdo do genoma nuclear, já que a maioria dos genes está localizada no núcleo. No entanto, os genomas organelares, incluindo as mitocôndrias (através de eucariotas) e os plastidios (nas plantas, a forma especializada, o cloroplasto, funciona na fotossíntese) contribuem com informações genéticas significativas essenciais ao desenvolvimento organizacional, à resposta ao estresse e à aptidão geral 1 . Os genomas organelares são tipicamente incluídos em extrações de DNA total destinadas ao seqüenciamento do genoma nuclear, embora também sejam empregados métodos para reduzir o número de organelas antes da extração de DNA 2 . Muitos estudos usaram resultados de seqüenciamento das extrações totais de gDNA para reunir genomas organelares 3 , 4 , 5 ,xref "> 6, 7. No entanto, quando o alvo do estudo é se concentrar em genomas organelares, usando o gADN total aumenta os custos de seqüenciamento porque muitas leituras são 'perdidos' para as seqüências de DNA nuclear, particularmente em plantas com grandes genomas nucleares Além disso, devido à duplicação e transferência de seqüências organelares para o genoma nuclear e entre organelas, a resolução da correta posição de mapeamento das leituras de seqüência para o genoma apropriado é um desafio bioinformático 2 , 8. A purificação de genomas organelares do genoma nuclear é uma Estratégia para reduzir esses problemas. Outras estratégias de bioinformática podem ser usadas para separar as leituras desse mapa para regiões de homologia entre mitocôndrias e cloroplastos.
Enquanto os genomas organelares de muitas espécies de plantas foram sequenciados, pouco se sabe sobre a amplitude da diversidade organometrica do genomaDisponíveis em populações selvagens ou em piscinas cultivadas. Os genomas orgânicos também são conhecidos por moléculas dinâmicas que sofrem rearranjo estrutural significativo devido à recombinação entre as seqüências repetidas 9 . Além disso, várias cópias do genoma orgânico estão contidas dentro de cada organela e várias organelas estão contidas em cada célula. Nem todas as cópias desses genomas são idênticas, o que é conhecido como heteroplasmia. Em contraste com a imagem canônica de "círculos mestres", há agora evidências crescentes de uma imagem mais complexa das estruturas do genoma organelar, incluindo círculos sub-genômicos, cromossomos lineares, concatomas lineares e estruturas ramificadas 10 . A montagem dos genomas orgânicos de plantas é ainda mais complicada por seus tamanhos relativamente grandes e repetições substanciais inversas e diretas.
Protocolos tradicionais para isolamento organelar, purificação de DNA e genoma subseqüente A seqüenciamento é muitas vezes pesada e requer grandes volumes de entrada de tecido, com vários gramas para cima de centenas de gramas de tecido de folhas jovens necessárias como ponto de partida 11 , 12 , 13 , 14 , 15 , 16 , 17 . Isso torna o seqüenciamento genômico organelar inacessível quando o tecido é limitado. Em algumas situações, as quantidades de sementes são limitadas, como quando é necessário seqüenciar em uma base geracional ou em linhas estéreis masculinas que devem ser mantidas por cruzamento. Nessas situações, o DNA organelar pode ser purificado e depois submetido a amplificação do genoma total. No entanto, a amplificação do genoma total pode introduzir um viés de sequenciação significativo, o que é um problema particular ao avaliar variações estruturais, estruturas sub-genômicas e níveis de heteroplastia> 18. Os avanços recentes na preparação da biblioteca para tecnologias de seqüência de leitura curta superaram barreiras de baixa entrada para evitar a amplificação do genoma total. Por exemplo, o kit de preparação da biblioteca Illumina Nextera XT permite que apenas 1 ng de DNA seja usado como entrada 19 . No entanto, as preparações de bibliotecas padrão para aplicações de seqüência de leitura longa, como as tecnologias de sequenciação PacBio ou Oxford Nanopore, ainda requerem uma quantidade relativamente alta de DNA de entrada, o que pode representar um desafio para o seqüenciamento genômico organelar. Recentemente, foram desenvolvidos novos protocolos de seqüência de leitura longa, elaborados para reduzir as quantidades de insumos e ajudar a facilitar o seqüenciamento do genoma em amostras onde a obtenção de microgramas de quantidades de DNA é difícil 20 , 21 . No entanto, a obtenção de fracções orgânicas puras de peso molecular elevado para alimentar estas preparações de biblioteca continua a ser um desafio.
Buscamos tO comparar e otimizar métodos de enriquecimento e isolamento de DNA organelar adequados para NGS sem a necessidade de amplificação do genoma total. Especificamente, nosso objetivo era determinar as melhores práticas para enriquecer o DNA organelar de alto peso molecular de materiais de partida limitados, como uma subconjunto de uma folha. Este trabalho apresenta uma análise comparativa de métodos para enriquecer para o DNA organelar: (1) um protocolo de centrifugação diferencial modificado e tradicional versus (2) um protocolo de fracionamento de DNA com base no uso de uma abordagem comercial de DNA CpG-ligação de metilação 22 aplicado ao tecido vegetal 23 . Recomendamos melhores práticas para o isolamento de DNA organelar a partir de tecido de folha de trigo, que pode ser facilmente estendido para outras plantas e tipos de tecido.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Geração de materiais vegetais para isolamento organológico e extração de DNA
- Crescimento padrão de mudas de trigo
- Plante as sementes em vermiculita em pequenos vasos quadrados com 4 a 6 sementes por canto. Transferir para uma estufa ou câmara de crescimento com ciclo leve de 16 h, dia 23 ºC / noite 18 ºC.
- Água as plantas todos os dias. Fertilize as plantas com ¼ de colher de chá de fertilizante granuloso 20-20-20 NPK após germinação e 7 dias após a germinação.
- Etiolação alternativa de mudas de trigo
- Siga o passo 1.1, mas coloque as panelas numa câmara de crescimento escuro, 23 ° C durante 16 h / 18 ºC durante 8 h. Alternativamente, cubra as plantas na estufa ( por exemplo, com um recipiente de armazenamento, no entanto, a ventilação adequada deve ser mantida).
- Crescimento e coleta de tecidos
- Cresça as plantas por 12 a 14 dias. Para a maioria dos genótipos de trigoS, 75 - 100 mudas produzem cerca de 10 a 12 g de tecido, o que é suficiente para duas extrações orgânicas utilizando o método de centrifugação diferencial (seção 2); Apenas uma planta é necessária se estiver usando a abordagem de desdobramento baseada em metilação de CpG de DNA para fragmentar organelar a partir de DNA nuclear (seção 3).
- Se utilizando a abordagem de centrifugação diferencial, colete o tecido fresco e proceda imediatamente ao processamento das amostras, conforme descrito na seção 2.
- Se utilizando a abordagem CpG-methyl pulldown, colhe 20 mg de seções de tecido de folha jovem em tubos de microcentrífuga (use tecido padronizado ou etiolado, veja os Resultados Representativos ). Snap-freez em nitrogênio líquido e congelar a -80 ºC até o uso. Proceda ao fraccionamento do DNA, conforme descrito na seção 3.
2. Método # 1: Extração de DNA Usando Centrifugação Diferencial (DC)
NOTA: O diffO protocolo de centrifugação erétil foi modificado a partir de duas publicações que otimizaram as condições para isolar ambas as organelas, mas enriquecem as mitocôndrias 17 , 24 . O protocolo resultante é menos intensivo em tempo e usa menos produtos químicos tóxicos do que os métodos anteriores. Especificamente, realizamos modificações nos passos de lavagem e lavagem, incluindo a adição de polivinilpirrolidona (PVP) ao tampão de extração STE e a eliminação do passo de lavagem final no tampão NETF, que contém fluoreto de sódio (NaF).
Cuidado: a preparação e o uso do tampão STE devem ser realizados sob um exaustor químico com equipamento de proteção pessoal apropriado, pois este tampão contém 2-mercaptoetanol (BME).
- Coisas para fazer antes de começar
- Certifique-se de que todo o equipamento é extremamente limpo e autoclave qualquer equipamento que possa ser autoclavado ( por exemplo, cilindros de moagem, centímetro de alta velocidadeTubos de fuge, etc. ).
NOTA: As dicas de filtro são recomendadas para todas as etapas que requerem pipetagem para evitar a contaminação cruzada. - Consulte a lista de equipamentos e reagentes necessários e prepare os buffers e estoques necessários para o Método nº 1 ( Tabela 1 ). Chill os blocos de moagem criogênica para -20 ºC e os rotores e buffers para 4 ºC, ajuste a microcentrífuga para 4 ºC e ative um banho-maria a 37 ºC.
- Certifique-se de que todo o equipamento é extremamente limpo e autoclave qualquer equipamento que possa ser autoclavado ( por exemplo, cilindros de moagem, centímetro de alta velocidadeTubos de fuge, etc. ).
- Isolamento de organelas
- Colher 5 g de tecido fresco e enxaguá-lo em água fria e estéril em um copo gelado no gelo.
NOTA: Mantenha sempre as amostras em gelo durante todas as operações e transportes de e para as centrífugas, exaustores, etc. Em alternativa, trabalhe em uma sala fria se houver acesso a espaço e equipamentos suficientes para realizar o protocolo. - Usando tesouras, corte o tecido foliar em pedaços de ~ 1 cm diretamente em um tubo de 50 mL contendo duas moagens cerâmicasCilindros.
NOTA: Limpe ou altere as tesouras entre as amostras para evitar a contaminação cruzada. - Se não houver homogeneizador de tecido, use uma argamassa e pilão e siga para substituir as etapas 2.2.4 - 2.2.9.
- Corte o tecido das folhas em uma argamassa pré-congelada no gelo. Moer as amostras por 2 a 3 min em 15 mL de STE (na exaustão).
- Despeje o tampão (deixe o tecido na argamassa) através de um funil contendo uma camada de pano de filtração pré-úmido e estéril (tamanho de poro de 22 a 25 μm, veja o protocolo principal para detalhes) em outro tubo de 50 mL . Adicione mais 10 mL de STE à argamassa e pilão e homogeneize novamente.
- Despeje o tecido homogeneizado e o tampão no mesmo funil. Enxaguar a argamassa e pilão com 10 mL de STE e despejá-la no funil. Aperte e remova o pano de filtração no funil para recuperar o máximo de líquido possível.
NOTA: Mude as luvas entre as amostras para evitar a contaminação cruzada. Continue com o profissionalTocol no passo 2.2.10.
- Adicione 20 mL de STE (na exaustão) a cada tubo de 50 mL.
- Coloque as amostras em blocos de moagem criogênicos pré-refrigerados em um aparelho de moagem de tecido e moer as amostras por 2 x 30 s a 1.750 rpm. Gire as posições da amostra e coloque as amostras no gelo durante ~ 1 min entre as moagens.
NOTA: Um adereço e pilão, liquidificador ou outro dispositivo de moagem / homogeneização de tecido pode ser usado nesta etapa. No entanto, cada método afetará a qualidade do DNA resultante em diferentes graus e, portanto, o comprimento e a qualidade do DNA devem ser avaliados antes de continuar com as aplicações a jusante. - Insira um funil em um tubo limpo de 50 mL colocado em gelo. Coloque uma camada de pano de filtração no funil e molhe-o previamente com 5 mL de STE. Não descarte o fluxo.
- Despeje o tecido homogeneizado no funil. Enxaguar o tubo de moagem com 15 mL de STE, recapitular e inverter o tubo para enxaguar as paredes e a tampa, e despejar no funnEl.
- Retire cuidadosamente as pedras de cerâmica e, em seguida, aperte e remova o pano de filtração no funil.
NOTA: Mude as luvas entre as amostras para evitar a contaminação cruzada. - Enrole as tampas do tubo com parafilme para evitar derrames. Centrifugar a 2.000 xg por 10 min a 4 ºC.
- Elimine cuidadosamente o sobrenadante usando uma pipeta serológica (evite perturbar a pelotização) e coloque-a em um tubo de centrífuga de alta velocidade de 50 mL (se os tubos não tiverem juntas de vedação apertadas, enrole as tampas do tubo com parafilme para evitar derrames). Descarte as pastilhas.
- Balance os tubos até 0,1 g usando STE e centrifugar o sobrenadante resultante durante 20 min a 18,000 xg e 4 ºC. Para equilibrar os tubos, coloque uma pequena taça de gelo no balanço, estude a balança e pesa as amostras no gelo para mantê-las frias. Alternativamente, use um balanço e uma aspiradora em uma sala fria.
- Descarte o sobrenadante. Adicione 1 mL de ST à pastilha e volte a suspender-nos suavemente.Com um pincel macio. Adicione 24 mL de ST (volume final de 25 mL) e misture / redemoinho ( ou seja, pressione o pincel do lado do tubo para remover todo o líquido).
- Balance os tubos para dentro de 0,1 g usando ST. Centrifugar por 20 min a 18,000 xg e 4 ºC. Enquanto isso, prepare a solução DNaseI (veja a Tabela 1 para receitas de estoque e solução de trabalho). Para cada amostra, faça uma alíquota de 200 μL em um tubo de 1,5 mL.
- Descarte o sobrenadante, retire o tubo e volte a suspender o sedimento (ainda em um tubo de centrífuga de alta velocidade) em 300 μL de ST usando um pincel macio. Coloque o pincel no tubo de 1,5 mL previamente preparado contendo 200 μL de solução de DNaseI e arraste o pincel para remover qualquer pellet residual preso na escova. Pipetar a solução DNaseI de volta ao tubo de centrífuga de alta velocidade e girar suavemente para misturar.
- Incube a 37 ° C durante 30 min em banho-maria (envolva parafilme em torno da parte superior do tubo para evitar vazamento de condensaçãoG na tampa). Misture gentilmente rodando 2 vezes durante a incubação.
- Pipie suavemente a mistura de pelotização do tubo usando uma ponta de pipeta com um orifício largo e coloque-a em um tubo de 1,5 mL de baixa ligação. Adicione 500 μL de EDTA 400 mM, pH 8,0, ao tubo de centrífuga de alta velocidade e uma pipeta suavemente para remover todo o sedimento residual do tubo. Transfira o EDTA para o mesmo tubo de 1,5 mL, de baixa ligação, como a mistura de pelotização e misture suavemente por inversão.
- Centrifugar a 18,000 xg por 20 min a 4 ºC. Descarte o sobrenadante, apague o tubo e use imediatamente para isolar o DNA. Se necessário, congelar pellets a -20 ºC, mas isso pode resultar em uma redução de rendimento, uma vez que a DNase residual pode degradar o DNA da amostra se não for processado imediatamente.
- Colher 5 g de tecido fresco e enxaguá-lo em água fria e estéril em um copo gelado no gelo.
- Extração de DNA de organelas isoladas usando uma abordagem comercial baseada em coluna
NOTA: Consulte o manual do kit para o protocolo completo 25 e veja abaixo as modificações. PrA partir do isolamento organelar, a extração de DNA é preferida. O congelamento e descongelamento repetidos reduzirão o tamanho dos fragmentos de DNA e levará à degradação do DNA pela DNase I residual. Limite o vortex ou a pipetagem vigorosa, pois isso pode cortar o DNA. Recomenda-se o uso de tubos de microcentrífuga de baixa ligação para maximizar a recuperação do DNA.- Procedimento de extração de DNA
NOTA: Leia o protocolo comercial detalhado 25 antes de começar a garantir que os buffers sejam devidamente criados / armazenados e que os procedimentos de spin-column sejam entendidos.- Adicione 180 μL de tampão ATL diretamente no tubo com o sedimento (descongelado se previamente congelado e equilibrado à temperatura ambiente na bancada).
- Proceda com o passo 3 no protocolo para "Purificações de DNA de tecidos" no manual do kit, com as seguintes modificações: uma lise de 30 minutos no passo 3, inclui a digestão opcional com RNase A e elute em 3 x 200 μL de AE ( Cada um em um seParate tube e depois combine as eluções).
- Salve uma alíquota (pelo menos 20 μL) para qPCR (veja o passo 4.1). Para quantificar antes da concentração, salve 1 μL adicional para quantificação de alta sensibilidade.
- Se desejar, prossiga com a concentração da amostra.
- Procedimento de extração de DNA
- Concentração de amostras com unidades comerciais de filtro
NOTA: Consulte o protocolo comercial 26 para obter mais detalhes. Dependendo do uso a jusante, pode não ser necessário realizar a concentração da amostra ( por exemplo, para PCR de ponto final e aplicações qPCR). No entanto, para a construção da biblioteca NGS, provavelmente será necessário concentrar o DNA organelar diluído obtido após a extração do DNA.- Procedimento da coluna de concentração
- Pré-pesa cuidadosamente (veja a Tabela 2 ) a unidade de filtro vazia (sem tubo) em um pedaço limpo de papel de pesagem em um balanço analítico digital. Registre o peso.
- PiPique as eluções combinadas na unidade de filtro e pesa cuidadosamente novamente.
NOTA: O manual comercial 26 diz que o volume máximo da unidade de filtro é de 500 μL, mas até 575 μL podem ser adicionados à unidade de uma só vez sem excesso. - Coloque cuidadosamente a unidade de filtro preenchida em um tubo (fornecido com as colunas). Centrifugar a 500 xg durante o tempo desejado para atingir o volume de concentrado necessário. Para um volume de amostra de ~ 575 μL, uma rotação de 20 minutos geralmente resultará em um volume de concentrado de 15-30 μL.
- Remova a unidade de filtro do tubo e pesa novamente. Use a tabela para determinar se o volume de concentrado desejado foi alcançado. Caso contrário, centrifugue novamente a 500 xg por um período mais curto e pesa novamente; Repita até atingir o volume de concentrado desejado.
- Coloque um novo tubo (fornecido com as colunas) na parte superior da unidade de filtro e inverta. Centrifugar por 3 min a 1.000 xg para transferir o coConcentre-se no tubo.
- Determine o volume recuperado. Isso geralmente será ~ 3 - 5 μL inferior ao volume calculado, devido à retenção do filtro. Se excessivamente concentrado, diluir com água estéril ou TE para atingir o volume desejado.
- Quantifique o DNA usando quantificação de alta sensibilidade (por instruções do fabricante).
- Procedimento da coluna de concentração
3. Método n. ° 2: Abordagem de metil-fraccionamento (MF) para enriquecer para o DNA organelole do DNA Genômico Total
NOTA: Este protocolo foi modificado a partir de um protocolo de extração de DNA Genomic Tip Kit desenvolvido pelo usuário para plantas e fungos 27 e o protocolo comercial Microbiome DNA Enrichment Kit 28 . Em teoria, qualquer protocolo de isolamento de DNA que produz DNA de alto peso molecular pode ser usado para o pulldown. Para uma seqüência de leitura curta, qualquer extração que provoca predominantemente fragmentos de 15 kb é adequada para uso no pulldown. Para oNg-read seqüenciamento, fragmentos maiores podem ser desejáveis. Portanto, otimizamos este protocolo para produzir DNA de alto peso molecular.
- Isolamento do DNA total
NOTA: Consulte a lista de equipamentos e reagentes necessários e prepare os buffers e estoques necessários para o Método # 2 ( Tabela 1 ). Adicione enzimas de lise ao estoque de tampão de lise para fazer a solução de trabalho de tampão de lise. Ligue o termomixer e ajuste-o para 37 ° C. Ligue o banho de água a 50 ° C e coloque o tampão QF no banho. Coloque 70% de EtOH no congelador e ajuste a microcentrífuga para 4 ° C.- Extração total de DNA utilizando colunas comerciais de extração de DNA
NOTA: Antes de iniciar, leia o manual comercial 29 para informações detalhadas sobre o uso das colunas de permuta aniónica de fluxo gravitacional. As colunas podem ser configuradas usando um rack especializado ou colocadas sobre os tubos usando os anéis de plástico fornecidos. Todas as etapas, incluindo o gDicas enológicas, devem ser permitidas para o fluxo gravitacional, e o líquido residual NÃO deve ser forçado.- Moer 20 mg de tecido congelado em nitrogênio líquido em um tubo de baixa ligação de 2 mL usando pilões de moagem de mão projetados para tubos de 2 mL.
- Adicione 2 mL de solução de solução de tampão de lise (os tubos estarão muito cheios).
- Incubar em termomixer a 37 ° C durante 1 h com agitação suave a 300 rpm. Se um termomixer não estiver disponível, a incubação em um bloco de calor e a mistura por um suave deslizamento a cada 15 minutos é uma alternativa adequada.
- Adicionar 4 μL de RNase A (100 mg / mL, concentração final de 200 μg / mL). Inverter para misturar e incubar em termomixer durante 30 min a 37 ° C, com agitação suave a 300 rpm.
- Adicionar 80 μL de proteinase K (20 mg / mL, concentração final de 0,8 mg / mL), inverter para misturar e incubar em termomixer durante 2 h a 50 ° C, com agitação suave a 300 rpm.
- Centrifugar durante 20 min a 4 ° C e 15.000 xg para sedimentar os detritos insolúveis.
- Enquanto as amostras são centrifugadas, equilibre as colunas com 1 mL de Buffer QBT e permita que a coluna seja esvaziada pelo fluxo gravitacional.
- Use uma ponta de pipeta de diâmetro largo para aplicar prontamente a amostra (evitar a pelotização) na coluna equilibrada e permitir que ela flua completamente pela coluna. Se a amostra ficar turva, filtre ou centrifugue novamente antes da aplicação na coluna (consulte o manual comercial para detalhes 29 ).
- Uma vez que a amostra tenha entrado completamente na resina, lave a coluna com 4 x 1 mL de Buffer QC.
- Suspenda a coluna por um tubo de microcentrífuga limpo, de 2 mL e de baixa ligação. Eluir o DNA genômico com 0,8 mL de tampão QF pré-aquecido a 50 ° C.
- Precipitar o ADN por adição de 0,56 mL (0,7 volumes de tampão de eluição) de isopropanol à temperatura ambiente ao ADN eluído.
- Misture por inversão (10X) e centrifugue imediatamente por 20 min a 15.000 xg e 4 ° C. CuidadoRemova completamente o sobrenadante sem perturbar o grânulo vidrado e solto.
- Lavar a pastilha de DNA centrifugada com 1 mL de etanol a 70% frio. Centrifugar por 10 min a 15,000 xg e 4 ° C.
- Remova cuidadosamente o sobrenadante (seja cauteloso com este passo também) sem perturbar a pelotização. Secar ao ar por 5-10 min e ressuspender o DNA em 0,1 mL de tampão de eluição (EB). Dissolver o DNA durante a noite à temperatura ambiente. Evite a pipetagem, que pode cortar o DNA.
- Quantifique as amostras usando um ensaio de quantificação de DNA de alta sensibilidade (por instruções do fabricante).
- Extração total de DNA utilizando colunas comerciais de extração de DNA
- Fracionamento à base de grânulos de DNA metilado e não metilado
NOTA: Uma publicação recente demonstrou o uso de um kit comercialmente disponível 28 que aproveita uma abordagem de pulldown utilizando uma proteína de domínio de ligação de metilma CpG específica fundida ao fragmento de IgG Fc humano (proteína MBD2-Fc) a fraçãoComeu genomas orgânicos de plantas (não metilados) a partir do conteúdo do genoma nuclear (altamente metilado) 23 . A eficiência de fracionamento em amostras de trigo não foi previamente testada usando este kit MF comercial 28 .- Coisas para fazer antes de começar
- Prepare recentemente 80% de etanol (pelo menos 800 μL por reação). Coloque 5x de ligação / tampão de lavagem para descongelar em gelo e prepare 5 mL de 1x tampão por amostra (diluir o tampão 5X com água estéril, livre de nuclease e manter o gelo durante o protocolo).
- Prepare bolinhas magnéticas ligadas a proteínas MBD2-Fc
- Prepare o número necessário de conjuntos de contas. Reduza as reações para usar entre 1 e 2 μg de DNA de entrada total, exigindo 160 - 320 μL de esferas. Observe que as reações listadas abaixo são para 1 μg de DNA de entrada total, então eles requerem 160 μL de esferas. Reduza as reações de acordo com as necessidades.
- Usando pontas de diâmetro largo, pipetar suavemente a Proteína A Magnética BAlmofada de gordura para cima e para baixo para criar uma suspensão homogênea. Como alternativa, gire suavemente o tubo de pérolas por 15 minutos a 4 ° C.
NOTA: Não vorteie as contas. - Proceda com as instruções de acordo com as instruções do fabricante 28 .
- Capturar DNA nuclear metilado
- Para cada amostra individual, adicione 1 μg de DNA de entrada a um tubo contendo 160 μL de pérolas magnéticas ligadas a MBD2-Fc.
- Adicionar 5x tampão de ligação / lavagem, conforme apropriado, dado o volume da amostra de entrada de DNA para uma concentração final de 1x (volume de 5x bind / buffer de lavagem para adicionar (μL) = volume de DNA de entrada (μL) / 4). Pipete a amostra para cima e para baixo algumas vezes para misturar usando uma ponta de pipeta de diâmetro largo.
- Gire os tubos à temperatura ambiente durante 15 min. Elimine delicadamente as amostras com uma ponta de pipeta de diâmetro largo e mova as amostras 2 a 3 vezes ao longo da incubação para evitar a acumulação de grânulos.
NOTA: o pipetting e flickiNg é fundamental para assegurar o pulldown eficiente do DNA metilado.
- Coletar DNA orgânico enriquecido e não metilado
- Gire rapidamente o tubo contendo o DNA e a mistura de grânulos magnéticos ligados a MBD2-Fc. Coloque o tubo em uma prateleira magnética por pelo menos 5 min para coletar as contas ao lado do tubo. A solução deve parecer clara.
- Usando pontas de diâmetro largo, remova cuidadosamente o sobrenadante limpo sem perturbar as contas. Transfira o sobrenadante (contém DNA não metilado, enriquecido organelarmente) para um tubo de microcentrífuga de 2 mL limpo e de baixa ligação. Armazene esta amostra a -20 ou -80 ° C, ou vá diretamente para o passo 3.2.6 para a purificação.
- Elute capturou DNA nuclear das esferas magnéticas ligadas a MBD2-Fc
- Se a fração nuclear também for desejada, siga as instruções do fabricante 28 para eluir o DNA nuclear das esferas magnéticas ligadas a MBD2-Fc; Purificar como descrito no passo 3.2.7.
- Purificação de ácido nucleico à base de talão
- Certifique-se de que as esferas de purificação estão à temperatura ambiente e estão completamente misturadas. Proceda com o protocolo de acordo com as instruções do manual MF kit 28 .
NOTA: A amostra agora pode ser usada para a construção da biblioteca NGS ou outra análise a jusante.
- Certifique-se de que as esferas de purificação estão à temperatura ambiente e estão completamente misturadas. Proceda com o protocolo de acordo com as instruções do manual MF kit 28 .
4. Quantificação de Amostras e Controle de Qualidade
- Ensaio de qPCR para avaliar o enriquecimento orgânico
NOTA: Os parâmetros de reação e análise de qPCR listados aqui foram projetados para uso em Roche LightCycler 480 e podem precisar ser ajustados para diferentes equipamentos e reagentes. Se qPCR não estiver disponível, a PCR de ponto final e a visualização em um gel de agarose podem ser usadas como uma medida qualitativa da pureza da amostra, usando os mesmos primers e condições aqui descritos. Os tamanhos de amplicão serão ~ 150 pb para todos os conjuntos de iniciadores. Veja a Tabela 3 para a sequência inicialPares e emparelhamentos.- Configuração de reação do qPCR
- Para configurar uma reação individual de 20 μL de qPCR, pipeteie cuidadosamente o seguinte em um único poço de uma placa qPCR de 96 poços: 10 μL de 2x SYBR Green I Master; 2 μL da mistura de iniciador direta e reversa de 10 μM (para uma concentração final de 0,5 μM); 2 μL de modelo (dentro do intervalo da curva padrão); E 6 μL de H 2 O estéril e livre de nucleases. Para reduzir os erros de pipetagem, é preferível fazer uma mistura principal com todos os componentes de reação, exceto o modelo. Adicione a mistura principal à placa qPCR e, em seguida, adicione o modelo de interesse para cada poço. Três repetições técnicas para cada amostra devem ser realizadas para minimizar os efeitos do erro de pipetagem.
NOTA: Em última análise, a relação entre os ciclos de quantificação nuclear e organelar é comparada entre as amostras, portanto pequenas diferenças de concentração são aceitáveis. No entanto, as concentrações de DNA devem estar aproximadamente na faixa deOutros. - Selar a placa com uma película de vedação qPCR de alta qualidade. Suavemente vorteie as amostras, tomando cuidado para evitar a criação de bolhas. Gire rapidamente a placa durante 2 minutos a 4 ° C para coletar a amostra e eliminar pequenas bolhas.
- Coloque a placa na máquina. Execute o programa qPCR de acordo com as diretrizes listadas abaixo.
- Para configurar uma reação individual de 20 μL de qPCR, pipeteie cuidadosamente o seguinte em um único poço de uma placa qPCR de 96 poços: 10 μL de 2x SYBR Green I Master; 2 μL da mistura de iniciador direta e reversa de 10 μM (para uma concentração final de 0,5 μM); 2 μL de modelo (dentro do intervalo da curva padrão); E 6 μL de H 2 O estéril e livre de nucleases. Para reduzir os erros de pipetagem, é preferível fazer uma mistura principal com todos os componentes de reação, exceto o modelo. Adicione a mistura principal à placa qPCR e, em seguida, adicione o modelo de interesse para cada poço. Três repetições técnicas para cada amostra devem ser realizadas para minimizar os efeitos do erro de pipetagem.
- Parâmetros de reação de qPCR
NOTA: Estes são parâmetros padrão, exceto para o ciclo de recozimento do estágio de amplificação. Ajuste esta configuração para acomodar primers específicos se os usados diferirem dos primers apresentados neste protocolo.- Pré-incubar a 95 ° C durante 5 minutos, com uma taxa de rampa de 4,4 ° C / s.
- Execute 45 ciclos de amplificação de (1) 95 ° C durante 10 s, com uma taxa de rampa de 4,4 ° C / s; (2) 60 ° C durante 20 s, com uma taxa de rampa de 2,2 ° C / s; E (3) 72 ° C por 10 s, com uma taxa de rampa de 4.4 ° C / s (dados adquiridos durante (3)).
- Use uma optioCiclo de curva de fusão nal de 95 ° C durante 5 s, com uma taxa de rampa de 4,4 ° C / s; 65 ° C durante 1 min, com uma taxa de rampa de 2,2 ° C / s; E 97 ° C, com um modo de aquisição contínuo.
- Use um ciclo de resfriamento de 40 ° C durante 30 s, com uma taxa de rampa de 1,5 ° C / s.
- Parâmetros de ensaio
- Selecione o modelo SYBR. Verifique os parâmetros do programa no botão Experiência. Uma vez que a placa é carregada, o ensaio pode ser iniciado e as configurações podem ser ajustadas enquanto o ensaio está funcionando.
- Atribua amostras usando o editor de amostra. Selecione Abs Quant como fluxo de trabalho e designe as amostras como padrões desconhecidos, padrões ou negativos. Designe as réplicas e preencha os nomes das amostras da primeira de cada repetição. Adicione concentrações e unidades aos padrões.
- Configurar subconjuntos para análise; Estes são atribuídos no editor do subconjunto.
- Para análise, selecione Abs Quant / 2nd Derivative Max na lista "criar nova análise".Importe a curva padrão guardada externamente (se aplicável) e, em seguida, clique em calcular; O relatório conterá a informação selecionada.
- Para realizar uma quantificação absoluta precisa para a determinação do número ou concentração da cópia, use uma curva padrão representativa da amostra testada ( por exemplo, DNA organelar isolado dos métodos acima). Uma vez que a quantidade de ADN mitocondrial necessária para preparar uma curva padrão é muito alta para ser alcançada com uma quantidade razoável de tecido, não utilize os cálculos do número de cópias fornecidos pelo software, mas analise os valores do ponto de passagem (Cp) para determinar o enriquecimento relativo De organelar em comparação com o DNA nuclear nas amostras. Compare estas quantidades relativas às do DNA genômico total (ver os Resultados Representativos ). Teste as eficiências dos iniciadores em cinco diluições de 1:10 de DNA genômico total de mudas de trigo de duas semanas de idade totalmente cultivadas (eficiências representativas relatadas naE legenda da Figura 2 ).
- Configuração de reação do qPCR
- Electroforese em gel de campo pulsado (PFGE)
NOTA: Este protocolo é baseado em diretrizes do fabricante para executar o PFGE para resolver o DNA de alto peso molecular. Veja a Tabela de Materiais.- Preparando o gel e amostras
- Siga as diretrizes para preparação de gel e amostras e adapte-as ao sistema disponível.
- Execute os parâmetros
- Siga as diretrizes para a instalação do sistema de eletroforese e use os seguintes parâmetros: tempo de comutação inicial de 2 s, tempo de troca final de 13 s, tempo de execução de 15 h e 16 min, V / cm de 6 e ângulo de 120 ° incluído .
- Mancha e imagem do gel
- Manchar o gel com um corante de escolha ( por exemplo, brometo de etidio ou uma alternativa adequada) e imagem com um sistema de documentação de gel adequado.
- Preparando o gel e amostras
- Use 1 ng de DNA como entrada para o Kit de Preparação da Biblioteca de DNA, de acordo com as instruções do fabricante.
- Código de barras e agrupe as amostras para seqüenciamento em uma única execução. Execute a seqüência de acordo com as diretrizes do fabricante.
NOTA: Os parâmetros de agrupamento e seqüenciamento podem ser alterados dependendo das espécies de interesse, do nível de cobertura desejado e da plataforma usada para seqüenciar as bibliotecas. Por exemplo, uma linha HiSeq tem substancialmente mais saída do que uma faixa MiSeq, muitas outras amostras podem ser multiplexadas. Seqüência um subconjunto menor de amostras para determinar se os níveis de cobertura dos genomas orgânicos são adequados para a análise a jusante.- Examine a qualidade de leitura usando FastQC 31 para determinar a extensão do corte e filtragem necessários para os dados.
- Corte e filtre as leituras brutas usando Trimmomatic 32 ou outro programa comparável. Use as seguintes configurações: ILLUMINACLIP 2:30:10 (para remover adaptadores), LEADING 3, TRAILING 3, SLIDINGWINDOW 4:10 e MINLEN 100.
- Mapear as leituras filtradas filtradas por qualidade e adaptadas ao adaptador (PE) para a mitocôndria da Primavera Chinesa (NCBI Reference Sequence NC_007579.1 33 ), cloroplasto (NCBI Reference Sequence NC_002762.1 34 ) e genomas de referência nucleares 35 usando Bowtie2 36 , Com as seguintes configurações: -I 0 -X 800 - sensível.
- Converta os arquivos de alinhamento de sam para o formato bam (samtools) e classifique os arquivos bam. Use os arquivos bam para calcular a cobertura do genoma e a cobertura por base com toalhas de cama. Visualize os resultados com a função R-plot.
- Coisas para fazer antes de começar
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Os protocolos apresentados neste manuscrito descrevem dois métodos distintos para enriquecer o DNA organelar do tecido vegetal. As condições aqui apresentadas refletem a otimização do tecido do trigo. Uma comparação de etapas-chave nos protocolos, entrada de tecido requerida e saída de DNA são descritas na Figura 1 . As etapas do protocolo CC que testamos seguem condições semelhantes às descritas anteriormente ( Figura 1A ). O tecido colhido deve ser processado recentemente e submetido a centrifugação diferencial e / ou gradientes para isolar organelas intactas. O DNA nuclear é eliminado antes que as organelas sejam lisadas e, finalmente, o DNA é extraído e usado para aplicações a jusante. Em contraste, no protocolo MF, o tecido vegetal pode ser colhido e armazenado antes do uso, e não são necessárias organelas intactas. Em vez disso, o DNA nuclear e organelar é fracionado do gDNA total com base em O estado de metilação do DNA. Ambos os protocolos produzem quantidades aproximadamente iguais de DNA organelar ( Figura 1C ). Em termos de produção total de DNA organelar em relação à entrada de tecido, o protocolo MF é vantajoso quando o tecido é limitado, uma vez que uma amostra pequena de uma única planta pode ser usada e a planta pode ser cultivada para análise posterior. Normalmente, em protocolos DC, todos os tecidos aéreos de muitas mudas são necessários e essas plantas são descartadas. No entanto, o método DC pode ser otimizado para enriquecer especificamente um tipo organelle sobre o outro, o que não é possível com a abordagem MF. Vale ressaltar que o tempo total para cada protocolo é aproximadamente equivalente, embora haja menos tempo de uso na abordagem MF.
Ambos os métodos enriquecem o DNA organelar, embora com diferentes proporções de mitocôndrias e seqüências de plastidios:
"> As quantidades muito baixas de DNA organelar purificado são obtidas a partir de qualquer método (na ordem de ~ 50 - 100 ng, Figura 1C ). Para avaliar os níveis de enriquecimento do genoma orgânico e a contaminação do genoma nuclear no DNA isolada tanto da DC quanto do MF Métodos, utilizou-se um ensaio de qPCR. Neste ensaio, as abundâncias relativas de três amplicões ( ie, nuclear-específico, ACTIN , mitocondrial-específico, NAD3 e Cloroplasto específico, PSBB ) foram avaliados em DNA genômico total, e a fração de DNA organelar foi obtida de ambos os métodos ( Figura 2 ). Os valores do ciclo de quantificação (C q ) foram examinados para cada amostra ( Figura 2A ), e porque o C q é definido como o ciclo de PCR em que a fluorescência da ampliação alvo aumenta acima do nível de fluorescência de fundo, C q e abundância de alvo têm um relação inversa. Dentroa amostra de DC, a C Q de NAD3 e PSBB são, respectivamente, ~ 17 e ~ 15 ciclos mais cedo do que ACTINA (que tem um Q C de ~ 36) (ver Figura 2B para valores q C e níveis de enriquecimento). Isso equivale a enriquecimentos teóricos 167,181 e 47,790 vezes para NAD3 e PSBB , respectivamente, Em relação ao ACTIN na amostra DC ( Figura 2B , veja a legenda da Figura 2 para o cálculo). Na amostra total de DNA genômico, os enxames de enxofre para NAD3 e PSBB em relação ao ACTIN são apenas 158 e 10.701, respectivamente. Não é surpreendente encontrar uma maior abundância de amplicões organelares em relação ao amplicão nuclear no DNA genômico total, dado que os genomas organelares existem em maiores números de cópias por célula do que o genoma nuclear 37 e que o número de organelos peAs células r podem diferir dependendo do tipo de tecido ou do estágio de desenvolvimento 38 , 39 . Em geral, os dados indicam que o método DC preferencialmente enriquece para as mitocôndrias, o que é esperado, uma vez que as velocidades de centrifugação são otimizadas para isolar seletivamente as mitocôndrias e reduzir a "contaminação" nuclear e de cloroplasto.A fração não metilada do gDNA total MF também mostra enriquecimento substancial de ambos os amplicões organelares e espera-se reter as quantidades relativas nativas desses alvos. Os enxames de enxofre para NAD3 e PSBB em relação à ACTIN na fração não metilada são 20,551 e 1,703,253, respectivamente ( Figura 2A e 2B ). Na fração metilada, os enxames de enxofre para NAD3 e PSBB em relação ao ACTIN são 31 e 823, respectivamente, indiIndicando que a proteína MBD2-Fc é altamente eficiente no pulldown do DNA nuclear metilado. Como o amplicão de cloroplasto tem maior abundância do que o amplicão mitocondrial em amostras de DNA genômico total (~ 6 C q mais cedo), fração metilada (~ 5 C q anterior) e fração não metilada (~ 6 C q anterior), isso sugere que a A abundância nativa desses amplicões não é substancialmente alterada pelo desdobramento MDB2. Focamos aqui a fração não metilada (organelar) devido ao interesse em seqüenciar esses genomas especificamente. No entanto, se o genoma nuclear for o principal interesse, o MF e o subsequente seqüenciamento da fração metilada renderiam uma cobertura muito maior do genoma nuclear do que o seqüenciamento total de DNA genômico, devido à redução na "contaminação" do DNA organelar.
Vale ressaltar que, se qPCR não estiver disponível, o PCR de ponto final (usando os mesmos primers como para qPCR) fornece a qualitaAvaliação da pureza organelar. Neste caso, as amostras de DNA organelar puro mostrarão amplificação para os amplicões mitocondriais e plástidos, mas não há amplificação detectável do amplicão nuclear no gel de agarose, enquanto que o DNA genômico total mostra amplificação para os três conjuntos de iniciadores, como demonstrado em estudos anteriores 11 , 12 .
O DNA Organellar Isolado de Ambos os Métodos é adequado para NGS:
As leituras de sequenciação de PE aparadas e limpas (ver passo 4.3) foram mapeadas para genomas de referência orgânicos de trigo previamente publicados e a quantidade de leituras usadas para mapear cada amostra variou de ~ 800,000 a 1,100,000 leituras ( Figura 3I ). Os resultados do mapeamento de novos níveis de sequenciação de Illumina para o cloroplasto de trigo disponível e os genomas das mitocôndrias são consistentes com o res qPCRCom o método DC que produz DNA que é mais enriquecido no DNA mitocondrial ( Figura 3A e 3B , ~ 80% e ~ 10% do mapa de leituras para os genomas mitocondriais (mt) e cloroplasto (cp), respectivamente) e o método MF Produzindo DNA que provavelmente reflete a abundância nativa dos dois genomas organelares ( Figuras 3A e 3B , ~ 20% e ~ 80% do mapa de leituras para os genomas mt e cp, respectivamente). Em ambos os métodos, a cobertura teórica (ver a legenda da Figura 3 para o cálculo) de ambos os genomas orgânicos de trigo excede a cobertura de 100X (e varia até uma cobertura de ~ 2,000X para o genoma do cloroplasto na fração não metilada do método MF), mesmo Quando 12 bibliotecas são multiplexadas ( Figura 3C e 3D ; as 6 bibliotecas incluídas nesta análise foram agrupadas com 6 bibliotecas adicionais para uma análise separada, para um total de 12 bibliotecasAgrupados em uma única pista de seqüenciamento). Uma visão mais detalhada da cobertura foi alcançada examinando a fração do genoma coberto em profundidades específicas, bem como nos níveis de cobertura por base ( Figura 3E- 3 ). Para o método MF, a cobertura média por base foi de ~ 300 - 450X para o genoma de mt e 4,000 - 5,000X para o genoma de cp. Para o método DC, a cobertura média por base foi de ~ 900 - 1.300 e ~ 500 - 700X para os genomas mt e cp, respectivamente. No entanto, houve uma pequena fração de ambos os genomas de mt e cp que apresentaram cobertura extremamente baixa ou alta, e isso foi observado em DNA organelar derivado de qualquer dos métodos ( Figura 3I ). As regiões de cobertura superior à média provavelmente correspondem a regiões de homologia entre os genomas organelares e regiões com baixa cobertura podem indicar SNPs ou outras pequenas variantes entre as cultivares que sequenciamos e as referências publicadas. Em apoio desta noção, esses picosDe alta cobertura foram mais pronunciadas para o ADN mt derivado do método MF ( Figuras 3E e 3I ), provavelmente devido à alta cobertura do genoma cp neste método. Inexplicável, a cobertura do genoma do cp é mais desigual no método MF do que o método DC ( Figura 3G e 3H ), o que pode ser devido a ligeiros distúrbios no pulverizador MBD2-Fc ao longo do DNA cp. Outras experiências serão necessárias para determinar por que esse é o caso. Independentemente disso, os genomas mt e cp tiveram uma cobertura relativamente uniforme com ambos os métodos e sem grandes áreas de cobertura faltante, o que pode ser demonstrado pelo exame da fração de genomas seqüenciados em uma determinada profundidade ( Figura 3E -3H ). Além disso, os níveis de cobertura para ambos os genomas são considerados suficientes para a análise a jusante, como a análise variante. Se for considerado necessário para a análise de variantes raras, reduzindo o númeroR das amostras agrupadas alcançarão uma maior cobertura. Alternativamente, um número muito maior de amostras pode ser agrupado em uma linha HiSeq, enquanto obtém ainda maior profundidade de seqüenciamento, embora seja um sacrifício para o comprimento da seqüência, pois as bibliotecas HiSeq são atualmente limitadas no comprimento do PE150 em contraste com as bibliotecas PE300 MiSeq.
Para examinar os níveis de contaminação do genoma nuclear usando uma abordagem de mapeamento, foram examinadas as categorias de mapeamento de leitura PE. As leituras PE podem ser mapeadas para um genoma de referência em uma variedade de configurações. Quando lê 1 e 2 alinharem a referência de uma maneira direta, com uma certa distância "esperada" entre os dois companheiros (com base no tamanho médio de inserção da biblioteca e tipicamente especificado como um parâmetro de entrada no software de mapeamento ), Essas leituras de PE são ditas para mapear "concordantly". Em contraste, o mapeamento "discordante" é a situação em que os mates mapeiam com um menor ou maior do que o esperado.Para o genoma de referência ou o mapa em configurações alternativas (de cabeça para cauda ou de cauda a cauda). Se apenas um companheiro se alinha ao genoma de referência, então o PE lido diz que não mapeia nem concordante nem discordante com o genoma de referência. Nas três categorias de mapeamento de leitura, as leituras PE podem se alinhar ao genoma de referência uma ou várias vezes.
Para o DNA organelar isolado de DC e MF, o mapeamento de leitura para o genoma mitocondrial foi predominantemente na categoria concordante concordante de uma única vez ( Figura 4A ), enquanto as leituras são mapeadas para o genoma do cloroplasto em proporções relativamente iguais de uma forma concordante e concordantemente mais do que Uma vez ( Figura 4B ), provavelmente devido às grandes repetições invertidas presentes no genoma do cloroplasto e também aos níveis de cobertura extremamente altos. No entanto, menos leituras de PE mapeadas para o genoma nuclear e, em grande parte, mapeadas mais de uma vez em umNem uma maneira concordante nem discordante ( ou seja, apenas um parceiro pode mapear). Estes são, provavelmente, mapeando "off-target" para seqüências no genoma nuclear, que são homólogas aos genomas organelares ou regiões mal montadas. Apenas uma pequena quantidade de leituras (<5%) mapeadas para o genoma nuclear de forma concordante, indicando níveis baixos de contaminação do genoma nuclear em DNA organelar isolado do método DC ou MF ( Figura 4C ), como também é refletido pelos resultados do qPCR ( Figura 2A ). A fração nuclear após o pulldown MBD2-Fc dos tecidos não-etiolados da Primavera Chinesa também foi sequenciada para determinar a eficiência do pulldown na remoção do DNA não metilado. Menos de 1% das leituras na biblioteca derivada da fração nuclear mapeada para genomas de referência organelar, enquanto que 45% de todas as leituras mapeadas para o genoma nuclear ( Figura 4 ). No entanto, a maioria lê mapeada de forma discordante, wQue provavelmente reflete os altos níveis de misassembly e fragmentação no genoma de referência nuclear do trigo. Independentemente disso, os resultados sugerem que o pulverizador MBD2-Fc é altamente eficiente na remoção de DNA organelar não metilado a partir de DNA nuclear metilado. Vale ressaltar que, porque o DNA enriquecido organelar resultante desses métodos contém uma mistura de mitocôndrias e sequências de cloroplasto, e porque as semelhanças de seqüência resultantes da transferência de genes antigos entre essas organelas permanecem em seus genomas, a atribuição adequada das leituras ao específico Os genomas devem ser resolvidos bioinformática.
A Etiolação do Tecido da Folha Não é Admitida Alterar as Abundâncias de Organelle:
Tradicionalmente, os tecidos etiolados são preferidos para o isolamento do DNA mitocondrial da planta, a fim de diminuir os níveis de fenólicos e amidos, o que pode interferir com a extraçãoN ou aplicações a jusante 13 . Para determinar se os níveis de enriquecimento do genoma orgânico podem ser alterados ou melhorados por condições de crescimento, ambos os tecidos etiolados e não etiolados foram submetidos ao protocolo MF e à seqüenciamento. Curiosamente, a etiolação não alterou de forma apreciável a porcentagem de leituras mapeadas para os genomas de referência organelares ( Figuras 3A e 3B ) ou a cobertura por base ( Figura 3I ) em comparação com condições não etioladas. Nós também isolamos DNA organelar usando centrifugação diferencial, com tecidos etiolados e não etiolados, e pouca diferença no enriquecimento foi encontrada entre os diferentes tecidos usando qPCR (dados não mostrados). Isto sugere que podem ser utilizados mais tecidos não etiolados fisiologicamente relevantes para estudos de sequenciação organelar, sem alteração apreciável de enriquecimento.
Controle de qualidade sugere queMF DNA é mais adequado para seqüenciamento de leitura longa:
Como a seqüência de leitura longa torna-se mais acessível aos pesquisadores, o isolamento do DNA de alto peso molecular está se tornando cada vez mais importante. Para avaliar DNA organelar isolado com qualquer método de intacto e qualidade, o PFGE foi empregado. O DNA genômico total geralmente migra como um esfregaço difuso em PFGE e o peso molecular é determinado pelo protocolo e como o DNA foi armazenado e manuseado após a extração. O DNA genômico total isolado com dicas genômicas deve exceder 50 kb, o que foi verificado usando PFGE ( Figura 5 , pista 2). O DNA genômico total das dicas genômicas é usado como entrada no Kit de Enriquecimento de Microbioma para fracionar o DNA nuclear a partir do DNA organelar. A fração nuclear obtida após o fraccionamento diminui de tamanho, mas permanece centrada em cerca de 50 kb ( Figura 5 , pista 4). Isso não é suO levantamento, dado que o tratamento relativamente mais áspero da fração nuclear como eluição de pérolas ligadas a MBD2-Fc requer a digestão por calor e proteinase K. Devido à massa limitada, a fração organelar não foi executada no PFGE, mas a análise subseqüente com o TapeStation indicou DNA> 50 kb (dados não mostrados). O DNA organelar obtido com centrifugação diferencial tem uma massa média de ~ 20 kb, provavelmente causada pelo protocolo de isolamento organelar estendido e a posterior extração e concentração de DNA à base de coluna. O isolamento organelar baseado em gradiente e os métodos alternativos de extração de DNA podem manter tamanhos maiores de fragmentos de DNA. Independentemente disso, o DNA do tamanho obtido neste protocolo pode ser usado para gerar leituras de sequenciação de 10 ou 15 kb se for tomado cuidado durante a preparação da biblioteca.
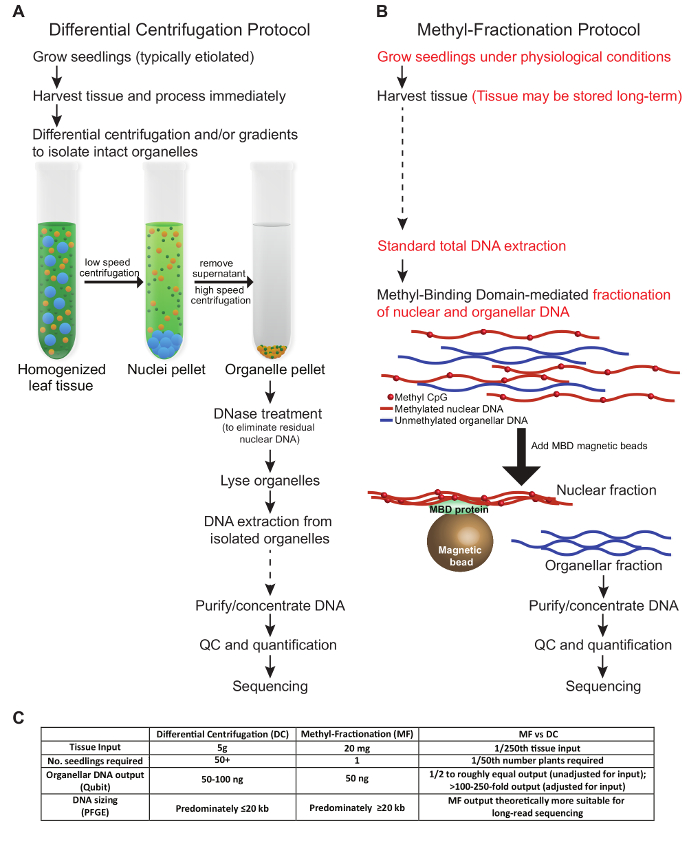
Figura 1: Uma visão comparativa de dois MethoDs para enriquecer para DNA de plantas orgânicas. Um protocolo DC tradicional ( A ) é contrastado com o protocolo MF ( B ). Recomenda-se evitar o congelamento e descongelamento das amostras; No entanto, as etapas nas quais as amostras podem ser armazenadas a longo prazo são indicadas com setas tracejadas ( A e B ). As principais diferenças entre os protocolos são realçadas em vermelho ( B ). ( C ) A tabela compara os métodos em termos de entrada de tecido, número de plantas necessárias, saída de DNA e tamanho de DNA resultante. Clique aqui para ver uma versão maior desta figura.

Figura 2: Avaliação da Contaminação de DNA Nuclear em DNA Organelar Isolado Usando dois Métodos. (
( B ) A tabela mostra os valores de C q , que são mostrados no gráfico em ( A ), e o enriquecimento de dobra dos amplicões organelares em relação ao ACTIN . * Fold enriquecimento = 2 (Cq ACTIN - Cq Target) . A fórmula assume uma eficiência perfeita de 2 para cada conjunto de iniciadores, uma vez que o menor deviatO íon de cada conjunto de iniciadores de 2 é insignificante e teria pouco efeito no cálculo e na tendência geral ( ACTIN = 1.961, NAD3 = 1.95 e PSBB = 1.989). As eficiências dos iniciadores foram avaliadas fazendo uma curva padrão com uma série de cinco diluições 1:10 de DNA genômico total. Clique aqui para ver uma versão maior desta figura.

Figura 3: Mapeamento de leitura e cobertura teórica de cloroplasto e genomas mitocondriais. Porcentagem de leituras mapeadas para os genomas de referência de mitochondrial ( A ) ou de cloroplasto ( B ) Chinese Spring. Cobertura teórica correspondente do geno da referência mitocondrial ( C ) ou de cloroplasto ( D ) da Primavera ChinesaMes, assumindo tamanhos de genoma de 450 e 135 kb, respectivamente, calculados usando o total de números de leitura e a porcentagem de mapeamento de leituras para os diferentes genomas. Distribuição de cobertura genômica para o DNA organelar do método MF ( E e G ) ou do método DC ( F e H ). Os dados nos painéis E - H são da amostra etiolada da Primavera chinesa, mas todas as outras amostras apresentaram uma tendência similar. ( I ) Cobertura média, menor e maior por base para todas as amostras nos painéis A - D. Os rótulos de amostra, incluindo "E", designam amostras etioladas e "NE" designa amostras não etioladas. DC indica DNA isolado com o método de centrifugação diferencial e Unmethylated indica DNA que está na fração não metilada após pulldown com MBD2-Fc (protocolo MF). Amostras rotuladas como "Chris" designam trigo Triticum aestivum'Chris'. CS designa amostras de trigo Triticum aestivum 'Chinese Spring. Nota: Devido à homologia de sequência entre o cloroplasto, as mitocôndrias e os genomas nucleares resultantes da antiga transferência de genes entre os genomas organelares, bem como entre os genomas orgânicos e orgânicos, uma pequena porcentagem de leituras cruas pode ser mapeada para múltiplos genomas. Além disso, as leituras que não se mapeiam para o genoma de referência organelar não estão representadas nesta figura. Assim, as percentagens apresentadas aqui ( A e B ) não totalizam 100%. Clique aqui para ver uma versão maior desta figura.

Figura 4: Mapeamento de leitura de PE para o genoma nuclear de trigo. Porcentagem de categorias de PE Leia os tipos de mapeamento para os genomas de referência de mitochondrial (A) , cloroplasto (B) ou nuclear (C) chinesa. - E designa amostras etioladas e - NE designa amostras não etioladas. DC indica DNA isolado com o método de centrifugação diferencial, Unmethylated indica DNA que está na fração não metilada após pulldown com MBD2-Fc no protocolo MF, e Methylated designa a fração nuclear após o pulverizador MBD2-Fc. As amostras rotuladas como "Chris" designam trigo Triticum aestivum 'Chris'. CS designa amostras de trigo Triticum aestivum 'Spring chinês'. Não são mostradas leituras não mapeadas. Clique aqui para ver uma versão maior desta figura.
Oad / 55528 / 55528fig5.jpg "/>
Figura 5: Exame da qualidade do DNA usando PFGE. O DNA genômico total de trigo (pista 2), o DNA organelar de trigo obtido a partir de centrifugação diferencial (pista 3) e a fração nuclear após MF com a abordagem pulverizada MBD2-Fc (pista 4) foram submetidos a PFGE em um gel de agarose a 1% com uma Escala extensa de 1 kb utilizada como marcador (pistas 1 e 5). Clique aqui para ver uma versão maior desta figura.
| Nome do Buffer | Receita | Notas | Método |
| Tampão de STE | Sacarose 400 mM, Tris 50 mM pH 7,8, EDTA 20 mM pH 8,0, polivinilpirrolidona a 0,6% (p / v) (PVP), albumina de soro bovino a 0,2% (p / v) (BSA), 0.1% (v / v) de p-mercaptoetanol (BME) | A mistura tampão contendo apenas sacarose, Tris e EDTA pode ser feita com um mês de antecedência e mantida a 4 ° C. PVP, BSA e BME devem ser adicionados frescos a uma alíquota da quantidade necessária de buffer imediatamente antes da utilização. | Método # 1 |
| ST Buffer | Sacarose 400 mM, Tris 50 mM pH 7,8, polivinilpirrolidona (PVP) a 0,6% (p / v), albumina de soro bovino a 0,1% (p / v) (BSA) | A mistura tampão contendo apenas sacarose e Tris pode ser feita com um mês de antecedência e mantida a 4 ° C. Observe que o buffer ST não contém EDTA ou BME, e contém uma menor concentração de BSA. | Método # 1 |
| Estoque DNase | DNase 2 mg / ml em NaCl 0,15 M até uma concentração de reserva de 2 mg / ml | Armazene alíquotas de 200 ul a -20 ° C. Para preparar a solução de trabalho DNase (200 μl de solução de DNase por amostra) verTabela 1 abaixo. Veja o protocolo completo abaixo para detalhes completos da digestão com DNase. A solução de trabalho DNase deve ser preparada de forma fresca. Para parar a reação DNase é necessária uma solução 400 mM EDTA pH 8.0 (a concentração final necessária para parar a reação é 0,2 M EDTA, consulte o protocolo completo para detalhes). | Método # 1 |
| Solução de trabalho DNase | 0,25 mg / ml de DNase e 20 mM MgCl2 em tampão de ST | Prepare fresco, 200 ul por amostra. As concentrações são indicados para o volume de reacção final, assim misturar: 62,5? L de 2 mg / ml de DNase (com base no volume de reaco final de 500), 4 ul de 1M MgCl 2 (com base em 200 ul de volume da solução de ADNase), e 133,5 de tamp de ST para Um volume final de 200 μl. | Método # 1 |
| Tampão de lise | EDTA 20 mM pH 8,0; Tris 10 mM pH 7,9; 500 mM de Guanidina-HCl; NaCl 200 mM; 1% Triton X-100; Enzimas de lise de 0,5 mg / ml deTrichoderma harzianum | Misture todos os ingredientes, exceto lise enzimas e guarde-os à temperatura ambiente. As enzimas Lysing devem ser adicionadas frescas a uma pequena alíquota para uso imediato. | Método # 2 |
Tabela 1: Receitas de buffers caseiros e estoques de trabalho.
| Ficha de Concentração | |||||||
| SAMPLE NAME | Peso vazio do dispositivo (g) | Peso do dispositivo preenchido (g) | Volume cheio (ul, cheio de pesos vazios) | Peso após a primeira rodada (20 min *, g) | Volume após a primeira rodada (ul, cheioMenos pesos vazios) | Peso após a segunda rotação (X min *, g) | Volume após a segunda rotação (ul, cheio menos pesos vazios) |
| Observe que o volume recuperado real será de pouco mais do que o volume calculado. | |||||||
Tabela 2: Planilha de Concentração.
| Nome | Especificidade do genoma | Fonte de sequência de genes | Sequência (5 '- 3') |
| Ta_ACTIN - F | Nuclear | Gramene Scaffold IWGSC_CSS_1AS_scaff_3272162: 10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | Nuclear | O mesmo que acima | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | Mitocondrial | Adesão NCBI EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | Mitocondrial | O mesmo que acima | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | Cloroplasto | Acesso NCBI KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | Cloroplasto | O mesmo que acima | GCTGCCTGTTTCCTTGTAGTT |
Tabela 3: Lista de primers qPCR.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Até à data, a maioria dos estudos de sequenciação organelar se centra em métodos tradicionais de DC para enriquecer para DNA específico. Foram descritos métodos para isolar organelas de diversas plantas, incluindo o musgo 40 ; Monocotiledose, como o trigo 15 e a aveia 11 ; E dicotiledóneas, como arabidopsis 11 , girassol 17 e colza 14 . A maioria dos protocolos concentra-se no tecido foliar 13 , 14 , 15 , 16 , 17 , tendo alguns sido adaptados para uma variedade de tipos de tecido, incluindo sementes 11 . O isolamento de organelas dos protoplastos também foi demonstrado 41 . No entanto, isso não é passível de todos os sistemas, nem é viável quando o tecido de interesse é limitado. Muitos desses orgaOs métodos de isolamento nellar foram concebidos para recuperar organelas intactas para experiências específicas, tais como estudos fisiológicos. Esses protocolos são pesados e tipicamente requerem o uso de gradientes de densidade, como gradientes de sacarose ou Percoll, que são muito eficientes para isolar frações organelares específicas, mas requerem uma grande entrada de tecido ( ou seja, em excesso de 5 g e mais de quilogramas, dependendo de O tipo de tecido). No entanto, o método DC pode ser otimizado para enriquecer para frações celulares específicas, como a mitocôndria ou o cloroplasto, alterando as velocidades de rotação e os gradientes de densidade. Em contrapartida, a abordagem MF requer muito menos material inicial (20 mg), mas os DNA mitocondriais e plástidos estarão presentes por suas abundâncias relativas no tecido utilizado para extração de DNA. No entanto, o protocolo MF oferece uma abordagem alternativa para isolar DNA organelole mixto e é particularmente benéfico para começar com pequenas quantidades de tecido.
T O avaliar a pureza da amostra após o isolamento de organelas, a maioria dos estudos até o momento apenas usa PCR de ponto final e eletroforese em gel 11 , 12 . Isso dá uma medida qualitativa justa da pureza da amostra. No entanto, baixos níveis de amplificação podem não ser visualizados em um gel de agarose. Poucos relatórios incluem mais medidas quantitativas de controle de qualidade, como qPCR 14 . Para uma avaliação quantitativa da pureza da amostra de DNA isolada de ambos os métodos, utilizamos qPCR e seqüenciamento para determinar quanto DNA nuclear permanece na amostra, bem como as proporções relativas do DNA mitocondrial versus cloroplasto. Ambos os métodos aqui avaliados são eficientes na remoção de DNA nuclear. Ambos os métodos produzem uma mistura de DNA mitocondrial e cloroplasto, embora em proporções diferentes.
As plantas crescentes no escuro (etiolação) são relatadas para ajudar a facilitar o isolamento organológico devido a uma redução de fenólicosNo entanto, nesta comparação, não encontramos uma vantagem apreciável para trabalhar com tecido etiolado em amostras cultivadas de forma leve. Embora a proporção de cloroplastos especializados seja provavelmente maior quando cultivada de forma leve, o número total de plastidios, como Refletido na proporção de mapeamento de leituras para o genoma do cloroplasto, é inalterado sob diferentes condições de luz. Portanto, para análises funcionais a jusante, como a avaliação de heteroplasmia em diferentes tecidos ou sob diferentes estressores ou para análises de expressão, recomendamos a realização de seqüenciamento genômico em Plantas cultivadas sob condições fisiologicamente relevantes.
Para aplicação com tecnologias de sequenciação de leitura curta, ambas as técnicas aqui comparadas produzem quantidade e qualidade adequadas de DNA. No entanto, para obter longas leituras de> 20 kb para aplicações de sequenciação de moléculas únicas, é necessária uma maior quantidade de DNA de qualidade superior. Por exemplo, idealmente,> 1 μg de orga puraO DNA de trigo nelar com um peso molecular> 20 kb é necessário para protocolos internos e de baixa entrada para preparações de biblioteca de inserção de 20 kb 42 . Novos protocolos desenvolvidos pelo usuário e de baixa entrada podem reduzir os requisitos de DNA ( isto é, 50 ng ou menos 20 ), mas o desafio continua a ter DNA de alta qualidade, de alto peso molecular, para os preparativos da biblioteca. É essencial que a maioria do DNA seja> 20 kb, uma vez que fragmentos menores serão inseridos preferencialmente no SMRTbell e descartarão a distribuição de tamanho da biblioteca 43 . Tentamos vários protocolos de extração de DNA caseiros e vários protocolos comerciais para extração de DNA (não mostrados). Para o tecido das folhas de trigo, obteve-se o melhor equilíbrio entre a quantidade e a qualidade do DNA, particularmente o comprimento, utilizando um kit comercial 27 , 29 . Dependendo das espécies de plantas e do tecido de interesse, alternatiOs protocolos de extração podem ser igualmente adequados ou mais frutíferos. No entanto, concluímos que a extração total de DNA genômico de alto peso molecular> 50 kb de tamanho, seguida do fraccionamento com a abordagem pulverizada MBD2-Fc 28 , é passível de sequenciação de leitura longa a partir de material de partida limitado. O trabalho futuro deve testar os limites do material de partida necessário após o fraccionamento para a preparação da biblioteca de longa inserção e subseqüente seqüência de leitura longa. Criticamente, essa abordagem poderia fornecer um método robusto para isolar o DNA de uma sub-amostra de uma única folha que é adequada para a seqüência de leitura longa, sem amplificação de todo o genoma. Prevemos que essa abordagem será facilmente adaptável a tipos de tecido adicionais e amplamente aplicável a outras espécies de plantas. Será particularmente útil em situações em que as quantidades de tecido são limitantes, como seqüenciamento em gerações individuais em um esquema de cruzamento ou em tipos de tecido mais raros.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores declaram que não têm interesses concorrentes.
A menção de nomes comerciais ou produtos comerciais nesta publicação é unicamente com o objetivo de fornecer informações específicas e não implica recomendação ou aprovação pelo Departamento de Agricultura dos EUA. O USDA é um fornecedor de oportunidades de igualdade e empregador.
Acknowledgments
Gostaríamos de reconhecer financiamento do Departamento de Agricultura dos Estados Unidos - Serviço de Pesquisa Agrícola e da National Science Foundation (IOS 1025881 e IOS 1361554). Agradecemos a R. Caspers pela manutenção de estufas e cuidados com plantas. Agradecemos também ao Centro de Genômica da Universidade de Minnesota, onde foram realizados os preparativos e sequenciações da biblioteca Illumina. Também agradecemos os comentários dos editores da revista e quatro revisores anônimos que reforçaram ainda mais o nosso manuscrito. Também agradecemos à OCDE uma bolsa de estudos para a SK para integrar esses protocolos para projetos colaborativos com colegas no Japão.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
