

Summary
Viene presentato il confronto e l'ottimizzazione di due metodi di arricchimento organellare del DNA vegetale: centrifugazione differenziale tradizionale e frazionamento del gDNA totale sulla base dello stato di metilazione. Valutiamo la quantità e la qualità del DNA risultanti, dimostriamo le prestazioni in sequenza di prossima generazione e discutiamo il potenziale di utilizzo in sequenza a lunga lettura di singole molecole.
Abstract
I genomi organellari delle piante contengono grandi elementi ripetitivi che possono subire un accoppiamento o una ricombinazione per formare strutture complesse e / o frammenti subgenomici. Anche i genomi organistici esistono in aggiunte all'interno di una determinata cellula o tipo di tessuto (eteroplasma) e un'abbondanza di sottotipi può cambiare durante lo sviluppo o quando è sotto lo stress (spostamento sotto-stechiometrico). Sono necessarie tecnologie di sequenziamento di nuova generazione (NGS) per ottenere una comprensione più approfondita della struttura e della funzione del genoma organellare. Gli studi di sequenziamento tradizionali utilizzano diversi metodi per ottenere DNA organellare: (1) Se viene utilizzata una grande quantità di tessuto di partenza, viene omogeneizzata e sottoposta a centrifugazione differenziale e / o depurazione gradiente. (2) Se si utilizza una quantità minore di tessuti ( cioè, se i semi, il materiale o lo spazio sono limitati), lo stesso processo viene eseguito come in (1), seguito da un'amplificazione del genoma completa per ottenere un DNA sufficiente. (3) L'analisi di bioinformatica può essere usata per seqIl DNA totale genomico e per analizzare le letture organellari. Tutti questi metodi hanno sfide intrinseche e compromessi. In (1), può essere difficile ottenere una così grande quantità di tessuto di partenza; In (2), l'amplificazione del genoma intero potrebbe introdurre una bias di sequenziamento; E in (3), l'omologia tra genomi nucleari e organellari potrebbe interferire con l'assemblaggio e l'analisi. Nelle piante con grandi genomi nucleari è vantaggioso arricchire il DNA organellare per ridurre i costi di sequenza e la complessità delle sequenze per le analisi di bioinformatica. Qui confrontiamo un tradizionale metodo di centrifugazione differenziale con un quarto metodo, un approccio adattato CpG-metile, per separare il DNA genomico totale in frazioni nucleari e organellari. Entrambi i metodi forniscono un sufficiente DNA per NGS, DNA che è altamente arricchito per sequenze organellari, anche se a diversi rapporti nei mitocondri e nei cloroplasti. Presentiamo l'ottimizzazione di questi metodi per i fogli di frumento e discutere di importanti vantaggi e dI vantaggi di ogni approccio nel contesto dell'input di campionamento, della facilità di protocollo e dell'applicazione a valle.
Introduction
Il sequenziamento del genoma è un potente strumento per disseccare la base genetica sottostante di importanti tratti vegetali. La maggior parte degli studi di sequenziamento genomico si concentra sul contenuto del genoma nucleare, in quanto la maggioranza dei geni si trova nel nucleo. Tuttavia, i genomi organellari, inclusi i mitocondri (attraverso eucarioti) e plastidi (in piante, la forma specializzata, il cloroplasto, funzionano in fotosintesi) contribuiscono a fornire notevoli informazioni genetiche fondamentali per lo sviluppo organico, la risposta allo stress e la forma fisica1 . I genomi organellari sono tipicamente inclusi nelle estrazioni totali del DNA destinate al sequenziamento del genoma nucleare, anche se vengono impiegati anche metodi per ridurre i numeri degli organeli prima dell'estrazione del DNA 2 . Molti studi hanno utilizzato risultati di sequenziamento delle estrazioni totali di gDNA per assemblare genomi organistici 3 , 4 , 5 ,xref "> 6, 7. Tuttavia, quando l'obiettivo dello studio è di concentrarsi su genomi organellari, utilizzando il totale gDNA aumenta i costi di sequenziamento perché molti letture sono 'perso' per le sequenze di DNA nucleare, in particolare in impianti di grandi genomi nucleari Inoltre, a causa della duplicazione e del trasferimento di sequenze organellari nel genoma nucleare e tra gli organelli, risolvendo la corretta posizione di mappatura del sequenziamento legale al genoma corretto è bioinformatica impegnativa 2 , 8. La purificazione dei genomi organellari dal genoma nucleare è una Strategia per ridurre questi problemi.Per ulteriori strategie di bioinformatics possono essere utilizzati per separare le letture che mappano alle regioni di omologia tra i mitocondri ei cloroplasti.
Mentre i genomi organellari da molte specie vegetali sono stati sequenziati, poco si sa sulla larghezza della diversità genomica organellareDisponibile in popolazioni selvatiche o in piscine coltivate. I genomi organellari sono noti anche per essere molecole dinamiche che subiscono un significativo riarrangiamento strutturale a causa della ricombinazione tra sequenze di ripetizione 9 . Inoltre, molte copie del genoma organellare sono contenute all'interno di ciascuna organella e sono presenti più organelli all'interno di ciascuna cellula. Non tutte le copie di questi genomi sono identiche, che è conosciuto come eteroplasma. Contrariamente all'immagine canonica di "cerchi master", ora si registra una crescente evidenza per un quadro più complesso di strutture del genoma organellare, compresi cerchi subgenomiali, cromosomi lineari, concatamers lineari e strutture ramificate 10 . L'assemblaggio di genomi organellari vegetali è ulteriormente complicato dalle loro dimensioni relativamente grandi e dalle sostanziali ripetizioni invertite e dirette.
Protocolli tradizionali per l'isolamento organellare, la purificazione del DNA e il genoma successivo I sequenziamenti sono spesso ingombranti e richiedono grandi volumi di input tissutale, con diversi grammi in aumento di centinaia di grammi di tessuti fogliari giovani necessari come punto di partenza 11 , 12 , 13 , 14 , 15 , 16 , 17 . Questo rende il sequenziamento del genoma organellare inaccessibile quando il tessuto è limitato. In alcune situazioni, gli importi dei semi sono limitati, ad esempio quando è necessario sequenziare su base generazionale o in linee sterili maschili che devono essere mantenute attraverso l'attraversamento. In queste situazioni, il DNA organellare può essere purificato e quindi sottoposto ad amplificazione del genoma intero. Tuttavia, l'amplificazione del genoma intero può introdurre significative bias di sequenziamento, che costituisce un problema particolare nella valutazione della variazione strutturale, delle strutture subgenomiche e dei livelli di eteroplasmi> 18. I recenti progressi nella preparazione della biblioteca per le tecnologie di sequenza brevettate hanno superato le barriere a basso input per evitare l'amplificazione del genoma intero. Ad esempio, il kit di preparazione alla libreria Illumina Nextera XT consente di utilizzare fino a 1 ng di DNA come input 19 . Tuttavia, le preparazioni standard di libreria per applicazioni di sequenziamento a lungo lette, come le tecnologie di sequenziamento PacBio o Oxford Nanopore, richiedono ancora una quantità relativamente elevata di DNA di input, che può rappresentare una sfida per il sequenziamento del genoma organellare. Recentemente, sono stati sviluppati nuovi protocolli di sequencing realizzati a livello manuale per ridurre gli importi e contribuire a facilitare il sequenziamento dei genomi nei campioni in cui è difficile ottenere quantità di DNA di microgrammi 20 , 21 . Tuttavia, ottenendo frazioni organolari puri ad alto peso molecolare per alimentare questi preparati alla libreria rimane una sfida.
Abbiamo cercato tO confrontare e ottimizzare i metodi di arricchimento e isolamento del DNA organellare adatti a NGS senza la necessità di amplificazione del genoma intero. In particolare, il nostro obiettivo era quello di determinare le migliori pratiche per arricchire il DNA organellare a peso molecolare dai materiali di partenza limitati, ad esempio un sottoinsieme di foglia. Questo lavoro presenta un'analisi comparativa dei metodi da arricchire per il DNA organellare: (1) un protocollo di centrifugazione differenziale tradizionale modificato rispetto a (2) un protocollo di frazionamento del DNA basato sull'uso di un approccio pulldown di proteine del dominio di DNA di CpG 22 applicato al tessuto vegetale 23 . Raccomandiamo le migliori pratiche per l'isolamento del DNA organellare da tessuti di foglie di grano, che possono essere facilmente estesi ad altre piante e tipi di tessuti.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Generazione di materiali vegetali per l'isolamento organico e l'estrazione del DNA
- Crescita standard dei semenzali di frumento
- Sementi vegetali in vermiculite in piccoli vasi quadrati con 4 - 6 semi per angolo. Trasferire in serra o in camera di crescita con un ciclo leggero da 16 h, 23 ºC giorno / 18 ºC notte.
- Acqua le piante ogni giorno. Fertilizzare le piante con ¼ di cucchiaino di fertilizzante granulare 20-20-20 NPK alla germinazione ea 7 giorni dopo la germinazione.
- Eziolazione alternativa delle piantagioni di frumento
- Seguire il passo 1.1, ma mettere le pentole in una camera di crescita scura, 23 ° C per 16 h / 18 ºC per 8 ore. In alternativa, coprire le piante nella serra ( ad esempio, con un contenitore di stoccaggio, tuttavia, deve essere mantenuta una corretta ventilazione).
- Crescita e raccolta dei tessuti
- Coltivi le piante per 12 - 14 giorni. Per la maggior parte del genotipo del granoS, 75 - 100 piantine producono circa 10 - 12 g di tessuto, che è sufficiente per due estratti organellari utilizzando il metodo di centrifugazione differenziale (sezione 2); Solo una pianta è necessaria se si utilizza l'approccio pulldown a base di DNA CpG-metilazione per frazionare l'organellare dal DNA nucleare (sezione 3).
- Se si utilizza l'approccio di centrifugazione differenziale, raccogliere il tessuto fresco e procedere immediatamente alla lavorazione dei campioni, come descritto nella sezione 2.
- Se si utilizza l'approccio pulldown CpG-metil, raccogliere le sezioni di tessuto giovanile da 20 mg in tubi di microcentrifuga (utilizzare i tessuti standardizzati o etiolati, vedere i risultati rappresentativi ). Snap-freeze sull'azoto liquido e congelare a -80 ºC fino all'uso. Procedere alla frazionamento a distanza del DNA, come descritto nella sezione 3.
2. Metodo # 1: Estrazione del DNA mediante centrifugazione differenziale (DC)
NOTA: il diffIl protocollo di centrifugazione erenziale è stato modificato da due pubblicazioni che ottimizzano le condizioni per isolare entrambi gli organelli ma arricchire per i mitocondri 17 , 24 . Il protocollo risultante è meno intensivo e utilizza meno sostanze chimiche tossiche rispetto ai metodi precedenti. In particolare, abbiamo apportato modifiche ai buffer e ai passaggi di lavaggio, compreso l'aggiunta di polivinilpirrolidone (PVP) al tampone di estrazione STE e l'eliminazione del passaggio finale di lavaggio in buffer NETF, che contiene fluoruro di sodio (NaF).
Attenzione: La preparazione e l'uso del tampone STE deve essere eseguita sotto una cappa chimica chimica con adeguata attrezzatura per la protezione personale, in quanto questo tampone contiene 2-mercaptoetanolo (BME).
- Cose da fare prima di iniziare
- Assicurarsi che tutte le apparecchiature siano estremamente pulite e autoclave tutte le apparecchiature autoclavabili ( ad esempio, cilindri di macinazione , centri ad alta velocitàTubi flessibili, ecc .).
NOTA: Consigli di filtro sono consigliati per tutti i passaggi che richiedono pipettature per evitare contaminazioni incrociate. - Vedere l'elenco delle attrezzature e dei reagenti necessari e preparare i buffer e le scorte richieste per il metodo # 1 ( tabella 1 ). Far raffreddare i blocchi criogenici a -20 ºC ei rotori e tamponi a 4 ºC, impostare la microcentrifuga a 4 ºC e accendere un bagno d'acqua a 37 ºC.
- Assicurarsi che tutte le apparecchiature siano estremamente pulite e autoclave tutte le apparecchiature autoclavabili ( ad esempio, cilindri di macinazione , centri ad alta velocitàTubi flessibili, ecc .).
- Isolamento di organelli
- Raccogliete 5 g di tessuto fresco e sciacqualo in acqua fredda e sterile in un bicchiere raffreddato sul ghiaccio.
NOTA: Tenere sempre i campioni in ghiaccio durante tutte le operazioni e trasporti da e verso le centrifughe, le cappe di fumi, ecc. In alternativa, lavorare in una stanza fredda se è possibile accedere a sufficienti spazi e attrezzature per eseguire il protocollo. - Usando le forbici, tagliare il tessuto delle foglie in pezzi di ~ 1 cm direttamente in un tubo da 50 ml contenente due macinature ceramichecilindri.
NOTA: Pulire o cambiare le forbici tra i campioni per evitare contaminazioni incrociate. - Se non vi è alcun omogeneizzatore di tessuti, utilizzare un mortaio e un pestello e seguire per sostituire i punti 2.2.4 - 2.2.9.
- Tagliare il tessuto foglia in un mortaio pre-raffreddato sul ghiaccio. Mescolare i campioni per 2 - 3 minuti in 15 ml di STE (nella cappa).
- Versare il tampone (lasciare il tessuto nella malta) attraverso un imbuto contenente uno strato di panno di filtrazione pre-bagnato e sterile (~ 22 a 25 μm di pori, vedere il protocollo principale per i dettagli) in un altro tubo da 50 ml . Aggiungere un ulteriore 10 mL di STE alla mortaia e pestello e nuovamente omogeneizzare.
- Versare il tessuto omogeneizzato e il tampone nello stesso imbuto. Sciacquare il mortaio e pestarli con 10 ml di STE e versarlo nell'imbuto. Strizzare e strizzare il panno di filtrazione nell'imbuto per recuperare il più liquido possibile.
NOTA: Cambiare guanti tra i campioni per evitare contaminazioni incrociate. Continuare con il proTocol al punto 2.2.10.
- Aggiungere 20 ml di STE (nella cappa di fumo) ad ogni tubo da 50 ml.
- Posizionare i campioni in blocchi criogenici pre-refrigerati in un dispositivo di macinazione tessuto e macinare i campioni per 2 x 30 s a 1.750 giri / min. Ruotare le posizioni del campione e posizionare i campioni sul ghiaccio per circa 1 min tra le macine.
NOTA: in questo passaggio può essere utilizzato un mortaio e un pestello, un frullatore o un altro dispositivo di macinazione / omogeneizzazione dei tessuti. Tuttavia, ogni metodo influirà sulla qualità risultante del DNA a gradi diversi e pertanto la lunghezza e la qualità del DNA devono essere valutati prima di continuare con le applicazioni a valle. - Inserire un imbuto in un tubo pulito da 50 ml posto in ghiaccio. Posizionare uno strato di panno di filtrazione nell'imbuto e pre-bagnarlo con 5 ml di STE. Non scartare il flusso.
- Versare il tessuto omogeneizzato nell'imbuto. Sciacquare il tubo di macinazione con 15 ml di STE, raccogliere e invertire il tubo per sciacquare le pareti e il coperchio e versare nel funnEL.
- Rimuovere con cautela le pietre ceramiche e poi spremere e strizzare il panno di filtrazione nell'imbuto.
NOTA: Cambiare guanti tra i campioni per evitare contaminazioni incrociate. - Avvolgere i tappi del tubo con il parafilm per evitare lo spargimento. Centrifugare a 2000 xg per 10 min a 4 ºC.
- Aspirare con cautela il surnatante usando una pipetta serologica (evitare di disturbare il pellet) e collocarlo in un tubo centrifugo ad alta velocità da 50 ml (se i tubi non hanno guarnizioni di tenuta strette, avvolgere i tappi di tubo con parafilm per evitare lo spargimento). Scartare i pellet.
- Bilanciare i tubi fino a 0,1 g utilizzando STE e centrifugare il supernatante risultante per 20 minuti a 18,000 xg e 4 ºC. Per equilibrare i tubi, posizionare un piccolo bicchiere di ghiaccio sul bilanciere, tirare la bilancia e pesare i campioni sul ghiaccio per tenerli freddi. In alternativa, utilizzare un equilibrio e una cappa di fumo in una stanza fredda.
- Scartare il surnatante. Aggiungere 1 ml di ST alla pellicola e riposaldare delicatamenteUn pennello morbido. Aggiungere 24 ml di ST (volume finale di 25 ml) e mescolare / turbare ( cioè, premere il pennello sul lato del tubo per rimuovere tutto il liquido).
- Bilanciare i tubi fino a 0,1 g utilizzando ST. Centrifugare per 20 minuti a 18.000 xg e 4 ºC. Nel frattempo, preparate la soluzione DNaseI (vedere la Tabella 1 per le ricette di magazzino e di lavorazione). Per ogni campione, fare una aliquota da 200 μL in un tubo da 1,5 ml.
- Scartare il supernatante, sbucciare il tubo e riposizionare il pellet (ancora in un tubo centrifugo ad alta velocità) in 300 μL di ST utilizzando un pennello morbido. Posizionare il pennello nel tubo da 1,5 ml precedentemente preparato contenente 200 μL di soluzione DNaseI e avvolgere il pennello per rimuovere qualsiasi pellet residuo bloccato nella spazzola. Pipettare la soluzione DNaseI nel tubo centrifuga ad alta velocità e mescolare delicatamente per mescolare.
- Incubare a 37 ° C per 30 minuti in un bagno d'acqua (avvolgere il parafilm intorno alla parte superiore del tubo per evitare la formazione di condensaG nel cappuccio). Mescolare delicatamente scorrendo due volte durante l'incubazione.
- Pipettare delicatamente la miscela di pellet fuori dal tubo usando una punta pipetta con un ampio orifizio e collocarla in un tubo a bassa tenuta da 1,5 ml. Aggiungere 500 μl di 400 mM EDTA, pH 8,0, al tubo di centrifuga ad alta velocità e pipetta delicatamente per ottenere tutto il pellet residuo fuori dal tubo. Trasferire l'EDTA allo stesso tubo da 1,5 ml di bassa legatura come la miscela di pellet e mescolare delicatamente per inversione.
- Centrifugare a 18.000 xg per 20 minuti a 4 ºC. Scartare il supernatante, macchiare il tubo e utilizzare immediatamente per l'isolamento del DNA. Se necessario, congelare i pellet a -20 ºC, ma questo può comportare una riduzione del rendimento, in quanto il residuo DNaseI può degradare il DNA campione se non viene immediatamente elaborato.
- Raccogliete 5 g di tessuto fresco e sciacqualo in acqua fredda e sterile in un bicchiere raffreddato sul ghiaccio.
- Estrazione del DNA da organelli isolati utilizzando un approccio basato sulla colonna commerciale
NOTA: vedere il manuale del kit per il protocollo completo 25 e vedere di seguito le modifiche. PrÈ preferibile passare direttamente dall'isolamento organellare all'estrazione del DNA. Il congelamento e lo scongelamento ripetuti riducono le dimensioni del frammento del DNA e portano al degrado del DNA da DNaseI residuo. Limitare vortexing o pipettature vigorose, in quanto questo può tagliare il DNA. Si raccomanda l'uso di tubi di microcentrifuga a basso tenore per massimizzare il recupero del DNA.- Procedura di estrazione del DNA
NOTA: Leggere il protocollo commerciale dettagliato 25 prima di iniziare a garantire che i buffer siano correttamente realizzati / memorizzati e che le procedure di spin-colonna siano comprese.- Aggiungere 180 μL di Tampone ATL direttamente nel tubo con il pellet (scongelato se precedentemente congelato e equilibrato a temperatura ambiente sul banco).
- Procedere con la fase 3 del protocollo "Purificazione dei tessuti del DNA" nel manuale del kit, con le seguenti modifiche: una lisi di 30 minuti nel passaggio 3 comprende la digestione opzionale di RNasi A e eluisce in 3 x 200 μL di AE ( Ciascuno in un seTubo del parato e quindi combinare le eluzioni).
- Salvare una aliquota (almeno 20 μL) per qPCR (vedere la fase 4.1). Per quantificare prima di concentrarsi, salvare un ulteriore 1 μL per la quantificazione ad alta sensibilità.
- Se si desidera, procedere con la concentrazione del campione.
- Procedura di estrazione del DNA
- Concentrazione di campione con unità di filtraggio commerciale
NOTA: per maggiori dettagli vedere il protocollo commerciale 26 . A seconda dell'uso a valle, potrebbe non essere necessario eseguire la concentrazione del campione ( ad esempio, per applicazioni PCR e qPCR a fine punto). Tuttavia, per la costruzione della libreria NGS, sarà probabilmente necessario concentrare il DNA diluito organellare ottenuto dopo l'estrazione del DNA.- Procedura della colonna di concentrazione
- Pesare con precisione (vedi tabella 2 ) l'unità filtro vuota (senza tubo) su un pezzo pulito di carta di pesata su un bilanciamento analitico digitale. Registra il peso.
- PiPettinare le eluzioni combinate nell'unità filtrante e pesare nuovamente con attenzione.
NOTA: Il manuale commerciale 26 afferma che il volume massimo del filtro è di 500 μL, ma può essere aggiunto fino a 575 μL all'unità contemporaneamente senza overflow. - Posizionare con cautela l'unità filtrante riempita in un tubo (fornito con le colonne). Centrifugare a 500 xg per il tempo desiderato per ottenere il volume di concentrato richiesto. Per un volume di campione di ~ 575 μL, una spin di 20 minuti di solito provoca un volume di concentrato di 15 - 30 μL.
- Rimuovere l'unità filtrante dal tubo e pesare di nuovo. Utilizzare la tabella per determinare se il volume di concentrato desiderato è stato raggiunto. In caso contrario, centrifugare nuovamente a 500 xg per un tempo più breve e pesare di nuovo; Ripetere fino a raggiungere il volume concentrato desiderato.
- Inserire un nuovo tubo (fornito con le colonne) sopra la parte superiore dell'unità filtrante e invertire. Centrifugare per 3 minuti a 1.000 xg per trasferire il coCentrare il tubo.
- Determinare il volume recuperato. Questo sarà di solito ~ 3 - 5 μL di meno del volume calcolato, a causa della ritenzione del filtro. Se è troppo concentrato, diluire con acqua sterile o TE per ottenere il volume desiderato.
- Quantificare il DNA usando la quantificazione ad alta sensibilità (per istruzioni del produttore).
- Procedura della colonna di concentrazione
3. Metodo n ° 2: Metodo di frazionamento (MF) per arricchire il DNA organellare del DNA genomico totale
NOTA: Questo protocollo è stato modificato da un protocollo di estrazione del DNA Genomic Tip Kit sviluppato dall'utente per piante e funghi 27 e il protocollo commerciale del kit di arricchimento del DNA Microbiome 28 . In teoria, qualsiasi protocollo di isolamento del DNA che produce un DNA ad alto peso molecolare può essere utilizzato per il pulldown. Per la sequenza di brevi letture, qualsiasi estrazione che produce prevalentemente> frammenti di 15 kb è sufficiente per l'uso nel pulldown. Per loNg-read la sequenza, i frammenti più grandi possono essere desiderabili. Pertanto, abbiamo ottimizzato questo protocollo per ottenere DNA ad alto peso molecolare.
- Isolamento del DNA totale
NOTA: Vedere l'elenco delle attrezzature e dei reagenti necessari e preparare i buffer e le scorte richieste per il metodo # 2 ( Tabella 1 ). Aggiungere gli enzimi di lysing allo stock di tampone di lisi per rendere la soluzione di lavoro del tampone di lisi. Accendere il termometro e impostarlo a 37 ° C. Accendere il bagno d'acqua a 50 ° C e collocare il tampone QF nel bagno. Mettere il 70% di EtOH nel congelatore e impostare la microcentrifuga a 4 ° C.- Estrazione totale del DNA utilizzando colonne di estrazione del DNA commerciale
NOTA: Prima di iniziare, leggere il manuale commerciale 29 per informazioni dettagliate sull'utilizzo delle colonne di scambio anionico a flusso di gravità. Le colonne possono essere impostate utilizzando un rack specializzato o posizionato sopra i tubi utilizzando gli anelli di plastica forniti. Tutti i passaggi, inclusi i gSi dovrebbe permettere di procedere con il flusso di gravità e il liquido residuo NON deve essere forzato.- Mescolare 20 mg di tessuto congelato in azoto liquido in un tubo a basso rilascio di 2 mL utilizzando pestelli di macinazione a mano progettati per tubi da 2 ml.
- Aggiungere 2 ml di soluzione di lavoro per tampone di lisi (i tubi saranno molto pieni).
- Incubare in un termomexer a 37 ° C per 1 h con agitazione leggera a 300 giri / min. Se non è disponibile un termomixer, l'incubazione su un blocco termico e la miscelazione con una leggera scorrimento ogni 15 minuti è un'alternativa appropriata.
- Aggiungere 4 μL di RNasi A (100 mg / mL, concentrazione finale di 200 μg / mL). Inverti per mescolare e incubare in un termomixer per 30 minuti a 37 ° C, con agitazione leggera a 300 giri / min.
- Aggiungere 80 μl di proteinasi K (20 mg / ml, concentrazione finale di 0,8 mg / ml), invertire per mescolare e incubare in un termomexer per 2 ore a 50 ° C, con agitazione leggera a 300 giri / min.
- Centrifugare per 20 minuti a 4 ° C e 1 °5.000 xg per far pelliccare i detriti insolubili.
- Mentre i campioni vengono centrifugati, bilanciare le colonne con 1 mL di Tampone QBT e lasciare che la colonna si svuota dal flusso di gravità.
- Utilizzare una punta di pipetta largo per applicare tempestivamente il campione (evitare il pallone) alla colonna equilibrata e lasciarla passare completamente attraverso la colonna. Se il campione diventa nuvoloso, filtrare o centrifugare nuovamente prima dell'applicazione alla colonna (vedere il manuale commerciale per i dettagli 29 ).
- Una volta che il campione è entrato pienamente nella resina, lavare la colonna con 4 x 1 ml di Tampone QC.
- Sospendere la colonna sopra un tubo di microcentrifuga a bassa tenuta da 2 ml. Eluire il DNA genomico con 0,8 mL di Tampone QF preriscaldato a 50 ° C.
- Precipitare il DNA aggiungendo 0,56 mL (0,7 volumi di tampone di eluizione) di isopropanolo a temperatura ambiente al DNA eluito.
- Mescolare per inversione (10X) e centrifugare immediatamente per 20 minuti a 15.000 xg e 4 ° C. CuraRimuovere completamente il surnatante senza pregiudicare il pellet vetroso e leggermente attaccato.
- Lavare il pellet di DNA centrifugo con 1 ml di etanolo a freddo del 70%. Centrifugare per 10 minuti a 15.000 xg e 4 ° C.
- Rimuovere con cautela il surnatante (essere cauto anche con questo passaggio) senza disturbare il pellet. Asciugare in aria per 5-10 min e risospendere il DNA in 0,1 mL di tampone di eluizione (EB). Sciogliere il DNA per una notte a temperatura ambiente. Evitare di pipettare, che può tagliare il DNA.
- Quantificare i campioni usando un test di quantificazione del DNA ad alta sensibilità (per istruzioni del produttore).
- Estrazione totale del DNA utilizzando colonne di estrazione del DNA commerciale
- Frazionamento a base di branco di DNA metilato e non metilato
NOTA: Una recente pubblicazione ha dimostrato l'uso di un kit 28 commercialmente disponibile che sfrutta un approccio a distanza che utilizza una proteina del dominio di legame di metil-legame FP-fusione fusione con il frammento Fc umano IgG (proteina MBD2-Fc)Hanno mangiato genomi organellari vegetali (nonmetilati) dal contenuto del genoma nucleare (altamente metilato) 23 . L'efficienza di frazionamento nei campioni di grano non è stata precedentemente testata utilizzando questo kit commerciale MF 28 .- Cose da fare prima di iniziare
- Preparare di recente l'etanolo al 80% (almeno 800 μL per reazione). Impostare il tampone di legame / lavaggio 5x a scongelare sul ghiaccio e preparare 5 mL di tampone 1x per campione (diluire il tampone 5X con acqua sterile e priva di nucleasi e mantenerlo su ghiaccio durante il protocollo).
- Preparare MBD2-Fc perline magnetiche legate alla proteina
- Preparare il numero di set di branelli richiesti. Scale le reazioni per utilizzare tra 1 e 2 μg di DNA totale di input, richiedendo 160 - 320 μL di perline. Si noti che le reazioni elencate di seguito sono per 1 μg di DNA totale in ingresso, quindi richiedono 160 μL di perline. Scala le reazioni secondo le esigenze.
- Utilizzando punte larghe, pipettare delicatamente la Proteina A Magnetica BEad slurry su e giù per creare una sospensione omogenea. In alternativa, ruotare delicatamente il tubo di perline per 15 minuti a 4 ° C.
NOTA: Non vortexare le perle. - Procedere con le istruzioni per le istruzioni del produttore 28 .
- Cattura DNA nucleare metilato
- Per ogni campione individuale, aggiungere 1 μg di DNA in ingresso a un tubo contenente 160 μL di perline magnetiche MBD2-Fc.
- Aggiungere un tampone di legame / lavaggio da 5x a seconda del volume del campione di ingresso del DNA per una concentrazione finale di 1x (volume di legame di 5x / tampone di lavaggio per aggiungere (μL) = volume di DNA in ingresso (μL) / 4). Pipette il campione su e giù alcune volte per mescolare usando una punta pipetta largo-bore.
- Ruotare i tubi a temperatura ambiente per 15 min. Pipettate con delicatezza i campioni con una punta di pipetta largo e fate scorrere i campioni 2-3 volte per tutta l'incubazione per evitare che si blocchi il tallone.
NOTA: la pipettazione e il flickiNg è fondamentale per garantire un pulldown efficiente del DNA metilato.
- Raccogli il DNA organellare arricchito e non metilato
- Girare brevemente il tubo contenente la miscela magnetica del tallone magnetico del DNA e MBD2-Fc. Mettere il tubo su un rack magnetico per almeno 5 min per raccogliere le perle sul lato del tubo. La soluzione dovrebbe apparire chiara.
- Utilizzando punte a forare larghe, rimuovete con attenzione il supernatante eliminato senza disturbare le perle. Trasferire il surnatante (contiene DNA non arricchito, organizzato), in un tubo di microcentrifuga a basso tenore di 2 mL. Conservare questo campione a -20 o -80 ° C, oppure procedere direttamente alla fase 3.2.6 per la purificazione.
- Elute ha catturato il DNA nucleare dalle perle magnetiche MBD2-Fc
- Se si desidera anche la frazione nucleare, seguire le istruzioni del produttore 28 per eluire il DNA nucleare dalle perle magnetiche legate a MBD2-Fc; Purificare come descritto nel passaggio 3.2.7.
- Purificazione di acido nucleico a base di branelli
- Assicurarsi che le perline di purificazione siano a temperatura ambiente e siano accuratamente mescolate. Procedere con il protocollo per le istruzioni contenute nel manuale del kit MF 28 .
NOTA: il campione può ora essere utilizzato per la costruzione della libreria NGS o un'altra analisi a valle.
- Assicurarsi che le perline di purificazione siano a temperatura ambiente e siano accuratamente mescolate. Procedere con il protocollo per le istruzioni contenute nel manuale del kit MF 28 .
4. Quantificazione del campione e controllo della qualità
- QPCR per valutare l'arricchimento organellare
NOTA: i parametri di reazione e parametri del test qPCR elencati qui sono stati progettati per essere utilizzati su un Roche LightCycler 480 e potrebbero essere necessari aggiustamenti per apparecchiature e reagenti diversi. Se qPCR non è disponibile, la PCR finale e la visualizzazione su un gel agarosio possono essere utilizzati come misura qualitativa della purezza del campione, utilizzando gli stessi primer e le condizioni qui descritte. Le dimensioni Amplicon saranno ~ 150 bp per tutti i set di primer. Vedere la Tabella 3 per il sequente primerE le accoppiamenti.- Impostazione della reazione qPCR
- Per impostare una reazione individuale di 20 μL di qPCR, pipettare con attenzione quanto segue in un unico pozzetto di una piastra qPCR a 96 pozzetti: 10 μL di 2x SYBR Green I Master; 2 μl del mix di primer 10 μM in avanti e invertito (per una concentrazione finale di 0,5 μM); 2 μl di template (all'interno della gamma della curva standard); E 6 μl di H 2 O sterile senza nucleasi. Per ridurre gli errori di pipettazione, è preferibile effettuare un mix master con tutti i componenti di reazione eccetto il template. Aggiungere il master mix alla piastra qPCR e quindi aggiungere il modello di interesse a ciascun pozzetto. Tre repliche tecniche per ciascun campione devono essere eseguite per ridurre al minimo gli effetti dell'errore di pipettamento.
NOTA: In ultima analisi, il rapporto tra cicli di quantificazione nucleare e organellare viene confrontato tra i campioni, per cui sono accettabili minime differenze di concentrazione. Tuttavia, le concentrazioni di DNA dovrebbero essere approssimativamente nell'ambito della eaCh altro. - Sigillare la lastra con una pellicola di sigillatura qPCR di alta qualità. Vorticare delicatamente i campioni, facendo attenzione a evitare la creazione di bolle. Girare brevemente il piatto giù per 2 min a 4 ° C per raccogliere il campione e eliminare le piccole bolle.
- Caricare la piastra nella macchina. Eseguire il programma qPCR per le linee guida elencate di seguito.
- Per impostare una reazione individuale di 20 μL di qPCR, pipettare con attenzione quanto segue in un unico pozzetto di una piastra qPCR a 96 pozzetti: 10 μL di 2x SYBR Green I Master; 2 μl del mix di primer 10 μM in avanti e invertito (per una concentrazione finale di 0,5 μM); 2 μl di template (all'interno della gamma della curva standard); E 6 μl di H 2 O sterile senza nucleasi. Per ridurre gli errori di pipettazione, è preferibile effettuare un mix master con tutti i componenti di reazione eccetto il template. Aggiungere il master mix alla piastra qPCR e quindi aggiungere il modello di interesse a ciascun pozzetto. Tre repliche tecniche per ciascun campione devono essere eseguite per ridurre al minimo gli effetti dell'errore di pipettamento.
- QPCR parametri di reazione
NOTA: sono parametri predefiniti, ad eccezione del ciclo di ricottura della fase di amplificazione. Regolare questa impostazione per ospitare primer specifici se quelli utilizzati differiscono dai primer presentati in questo protocollo.- Pre-incubare a 95 ° C per 5 min, con una velocità di rampa di 4,4 ° C / s.
- Eseguire 45 cicli di amplificazione di (1) 95 ° C per 10 s, con una velocità di rampa di 4,4 ° C / s; (2) 60 ° C per 20 s, con una velocità di rampa di 2,2 ° C / s; E (3) 72 ° C per 10 s, con una velocità di rampa di 4,4 ° C / s (dati acquisiti durante (3)).
- Usare un'opzioneIl ciclo della curva di fusione di 95 ° C per 5 s con una velocità di rampa di 4,4 ° C / s; 65 ° C per 1 min, con una velocità di rampa di 2,2 ° C / s; E 97 ° C, con modalità di acquisizione continua.
- Utilizzare un ciclo di raffreddamento di 40 ° C per 30 s, con una velocità di rampa di 1,5 ° C / s.
- Parametri di analisi
- Selezionare il modello SYBR. Controllare i parametri del programma nel pulsante Esperimento. Una volta caricata la lastra, il saggio può essere avviato e le impostazioni possono essere regolate mentre il saggio è in esecuzione.
- Assegnare i campioni utilizzando l'editor di esempio. Seleziona Abs Quant come flusso di lavoro e designi i campioni come sconosciuti, standard o controlli negativi. Designare replica e compilare i nomi di esempio del primo di ogni replica. Aggiungere concentrazioni e unità alle norme.
- Impostazione di sottogruppi per l'analisi; Questi vengono assegnati nell'editor di sottoinsieme.
- Per l'analisi, selezionare Abs Quant / 2nd Derivative Max dall'elenco "create new analysis".Importa la curva standard salvata esternamente (se applicabile) e quindi colpire il calcolo; Il report contiene le informazioni selezionate.
- Per eseguire una precisa quantificazione assoluta per la determinazione del numero di copie o della concentrazione, utilizzare una curva standard rappresentativa del campione in esame ( ad esempio, DNA organellare isolato dai metodi di cui sopra). Poiché la quantità di DNA mitocondriale richiesto per la preparazione di una curva standard è troppo elevata per essere raggiunta con una quantità ragionevole di tessuti, non utilizzare i calcoli del numero di copie forniti dal software, ma invece esaminare i valori di punto di attraversamento (Cp) per determinare l'arricchimento relativo Di organellare rispetto al DNA nucleare nei campioni. Confrontare questi valori relativi a quelli del DNA totale genomico (vedere i risultati rappresentativi ). Applicare efficienze del primer di test su cinque diluizioni 1:10 di DNA genomico totale da seminativi di grano di due settimane (le efficienze rappresentative riportate inE leggenda della Figura 2 ).
- Impostazione della reazione qPCR
- Elettroforesi a gel pulsata (PFGE)
NOTA: Questo protocollo si basa sulle linee guida del produttore per eseguire PFGE per risolvere il DNA ad alto peso molecolare. Vedere la tabella dei materiali.- Preparazione del gel e dei campioni
- Seguire le linee guida per la preparazione del gel e del campione e adattarle al sistema disponibile.
- Eseguire i parametri
- Seguire le linee guida per l'impostazione del sistema di elettroforesi e utilizzare i seguenti parametri: tempo di commutazione iniziale di 2 s, intervallo finale di 13 s, durata di esercizio di 15 h e 16 min, V / cm di 6 e angolo incluso di 120 ° .
- Macchiare e immaginare il gel
- Macchiare il gel con un colorante di scelta ( ad esempio, bromuro di etidio o un'alternativa appropriata) e l'immagine con un idoneo sistema di documentazione del gel.
- Preparazione del gel e dei campioni
- Utilizzare 1 ng di DNA come input per il kit DNA Prep Kit, secondo le istruzioni del produttore.
- Barcode e pool i campioni per sequenziare in una singola corsa. Eseguire la sequenza secondo le linee guida del produttore.
NOTA: i parametri di raggruppamento e sequenza possono essere modificati a seconda della specie di interesse, del livello di copertura desiderato e della piattaforma utilizzata per sequire le librerie. Ad esempio, una corsia HiSeq ha una produzione sostanzialmente maggiore di una corsia MiSeq, in modo che molti altri campioni possono essere multipli. Sequenza di un sottogruppo più piccolo di campioni per determinare se i livelli di copertura dei genomi organellari sono adeguati per l'analisi a valle.- Esaminare la qualità di lettura usando FastQC 31 per determinare l'estensione del taglio e del filtraggio richiesti per i dati.
- Tagliare e filtrare le letture raw utilizzando Trimmomatic 32 o un altro programma comparabile. Utilizzare le seguenti impostazioni: ILLUMINACLIP 2:30:10 (per rimuovere gli adattatori), LEADING 3, TRAILING 3, SLIDINGWINDOW 4:10 e MINLEN 100.
- Carteggiare le coppie di terminali (PE) con filtri qualitativamente filtrati e con adattatore (NC) alla cifra mitocondriale cinese (NCBI Sequenza di riferimento NC_007579.1 33 ), cloroplasti (NCBI Sequenza di riferimento NC_002762.1 34 ) e nuclei di riferimento nucleari 35 usando Bowtie2 36 , Con le seguenti impostazioni: -I 0 -X 800 - sensibile.
- Convertire i file di allineamento sam in formato bam (samtools) e ordinare i file bam. Utilizzare i file bam per calcolare la copertura genoma-wide e la copertura per-base con le utensili da letto. Visualizzare i risultati con la funzione R-plot.
- Cose da fare prima di iniziare
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
I protocolli presentati in questo manoscritto descrivono due metodi distinti per arricchire il DNA organellare dai tessuti vegetali. Le condizioni qui presentate riflettono l'ottimizzazione del tessuto di grano. Un confronto tra i passaggi chiave nei protocolli, l'input richiesto e l'output del DNA sono descritti in Figura 1 . Le fasi del protocollo DC che abbiamo testato seguono condizioni simili a quelle descritte in precedenza ( Figura 1A ). I tessuti raccolti devono essere freschi e sottoposti a centrifugazione differenziata e / o gradienti per isolare organelli intatti. Il DNA nucleare viene eliminato prima che gli organelli siano lisi e, infine, il DNA viene estratto e utilizzato per applicazioni a valle. Al contrario, nel protocollo MF, i tessuti vegetali possono essere raccolti e conservati prima dell'uso, e non sono richiesti organelli intatti. Invece, il DNA nucleare e organellare è frazionato da gDNA totale basato su Lo stato di metilazione del DNA. Entrambi i protocolli forniscono circa uguali quantità di DNA organellare ( Figura 1C ). In termini di produzione totale del DNA organellare in relazione all'ingresso tissutale, il protocollo MF è vantaggioso quando il tessuto è limitato, in quanto può essere utilizzato un piccolo campione da una singola pianta e la pianta può essere lasciata crescere per ulteriori analisi. Tipicamente, nei protocolli DC, sono necessari tutti i tessuti aerei di molti piantine e queste piante vengono scartate. Tuttavia, il metodo DC può essere ottimizzato per arricchire in modo specifico per un tipo di organelle rispetto all'altro, che non è possibile con l'approccio MF. Vale la pena ricordare che il tempo totale per ciascun protocollo è approssimativamente equivalente, sebbene non ci sia meno tempo nell'approccio MF.
Entrambi i metodi arricchiscono il DNA organellare, anche se con proporzioni diverse di mitocondri e sequenze plastiche:
"> Molto basse quantità di DNA organellare purificato sono ottenute da entrambi i metodi (sull'ordine di ~ 50-100 ng, Figura 1C ). Per valutare i livelli di arricchimento del genoma organellare e contaminazione del genoma nucleare in DNA isolati da entrambi i DC e MF In questo saggio, le relative abbondanze di tre ampliconi ( cioè specifici nucleari, ACTIN , mitocondriali-specifici, NAD3 e Cloroplast specifici, PSBB ) sono stati valutati in DNA totale genomico e la frazione DNA organellare è stata ottenuta da entrambi i metodi ( Figura 2 ). I valori di ciclo di quantificazione (C q ) sono stati esaminati per ciascun campione ( Figura 2A ) e poiché il Cq è definito come il ciclo PCR in cui la fluorescenza dall'ampliamento di destinazione aumenta al di sopra del livello di fluorescenza di sfondo, C q e l'abbondanza di bersaglio hanno un relazione inversa. Inil campione DC, C q di NAD3 e PSBB sono, rispettivamente, 17 e ~ ~ 15 cicli prima del ACTIN (che ha un q C di ~ 36) (vedere la Figura 2B per i valori q C e livelli di arricchimento). Ciò equivale a arricchimenti teorici di 167.181 e 47.790 volte per NAD3 e PSBB , rispettivamente, Rispetto a ACTIN nel campione DC ( figura 2B , vedere la leggenda della Figura 2 per il calcolo). Nel campione totale del DNA genomico, gli arricchimenti piegati per NAD3 e PSBB rispetto all'ACTIN sono rispettivamente solo 158 e 10.701. Non è sorprendente trovare un'abbondanza maggiore degli ampliconi organellari rispetto all'amplicone nucleare nel DNA genomico totale, dato che i genomi organellari esistono in numeri di copia maggiori per cellula rispetto al genoma nucleare 37 e che il numero di organelle peLa cellula r può variare a seconda del tipo di tessuto o dello stadio di sviluppo 38 , 39 . Nel complesso, i dati indicano che il metodo DC richiede preferenzialmente arricchimenti per i mitocondri, che ci si aspetta, poiché le velocità di centrifugazione sono ottimizzate per isolare selettivamente i mitocondri e ridurre la contaminazione nucleare e cloroplastica.La frazione nonmetilizzata del gDNA totale di MF mostra anche un notevole arricchimento di entrambi gli ampliconi organellari e si prevede di mantenere le quantità relative di questi obiettivi nativi. Gli arricchimenti piegati per NAD3 e PSBB rispetto all'ACTIN nella frazione nonmetilata sono rispettivamente 20.551 e 1.703.253 ( Figura 2A e 2B ). Nella frazione metilata, gli arricchimenti piegati per NAD3 e PSBB rispetto all'ACTIN sono 31 e 823, rispettivamente, indiPoiché la proteina MBD2-Fc è altamente efficace al primo momento del DNA nucleare metilato. Poiché l'amplicone del cloroplasto ha una maggiore abbondanza rispetto all'amplicone mitocondriale nel DNA totale genomico (~ 6 Cq in precedenza), frazione metilata (~ 5 C q prima) e frazioni non metilate (~ 6 C q precedenti), questo suggerisce che il L'abbondanza nativa di questi ampliconi non è sostanzialmente modificata dal pulldown MDB2. Ci concentriamo qui sulla frazione nonmetilata (organellare) a causa dell'interesse per sequenziare in modo specifico questi genomi. Tuttavia, se il genoma nucleare è l'interesse primario, la MF e la successiva sequenza della frazione metilata forniranno una copertura del genoma nucleare molto più elevata rispetto al sequenziamento totale del DNA genomico, a causa della riduzione della contaminazione del DNA organellare.
Vale la pena notare che se qPCR non è disponibile, il PCR finale (utilizzando gli stessi primer come per qPCR) fornisce la qualitaValutazione della purezza organellare. In questo caso, i campioni di DNA pura organellare mostreranno amplificazione per gli ampi- noni mitocondriali e plastidi, ma nessuna amplificazione rilevabile dell'amplicone nucleare sul gel agarosio, mentre il DNA genomico totale mostra l'amplificazione per tutti e tre i set di primer, come dimostrato negli studi precedenti 11 , 12 .
DNA organellare isolato da entrambi i metodi è adatto per NGS:
Le sequenze di sequenza di PE (PE) sono state mappate con genomi di riferimento organellari di grano precedentemente pubblicati e la quantità di letture utilizzate per la mappatura di ciascun campione variava da ~ 800.000 a 1.100.000 letture ( Figura 3I ). I risultati della mappatura di de novo sequenza di Illumina legati ai genomi disponibili del cloroplasto di grano e dei mitocondri sono coerenti con la resistenza qPCRUlts, con il metodo DC che produce DNA che è più arricchito nel DNA mitocondriale ( Figura 3A e 3B , ~ 80% e ~ 10% di letture mappa rispettivamente ai genomi mitocondriali (mt) e cloroplasti (cp) e al metodo MF Che produce il DNA che riflette probabilmente l'abbondanza nativa dei due genomi organellari ( figure 3A e 3B , ~ 20% e ~ 80% di letture mappa rispettivamente per i genomi mt e cp). In entrambi i metodi, la copertura teorica (vedi la leggenda della Figura 3 per il calcolo) di entrambi i genomi dell'organellare del grano supera la copertura di 100X (e varia fino a una copertura di ~ 2,000X per il genoma del cloroplasto nella frazione nonmetilata del metodo MF) Quando 12 librerie sono multiplexizzate ( Figura 3C e 3D , le 6 biblioteche incluse in questa analisi sono state raggruppate con altre 6 librerie per un'analisi separata, per un totale di 12 librerieRiuniti in una singola corsia di sequenza). Una visione più dettagliata della copertura è stata raggiunta esaminando la frazione del genoma coperto in profondità specifica, nonché a livelli di copertura per-base ( Figura 3E- 3I ). Per il metodo MF, la copertura media per base è stata di ~ 300-450X per il genoma mt e 4.000-5.000X per il genoma cp. Per il metodo DC, la copertura media per base era rispettivamente di ~ 900 - 1.300 e ~ 500 - 700x per i genomi mt e cp. Tuttavia, c'era una piccola frazione di entrambi i genomi mt e cp che hanno una copertura estremamente bassa o alta, e questo è stato visto nel DNA organellare derivato da entrambi i metodi ( Figura 3I ). Le regioni con una copertura superiore alla media corrispondono probabilmente a regioni di omologia tra i genomi organellari e le regioni a bassa copertura possono indicare SNP o altre piccole varianti tra le cultivar sequenziate ei riferimenti pubblicati. A sostegno di questa nozione, questi picchiDi elevata copertura erano più pronunciate per il DNA mt derivato dal metodo MF ( figure 3E e 3I ), probabilmente a causa dell'elevata copertura del genoma cp in questo metodo. In modo inesplicabile, la copertura del genoma cp è più irregolare nel metodo MF rispetto al metodo DC ( Figura 3G e 3H ), che potrebbe essere dovuto a piccole polarizzazioni nel pulldown MBD2-Fc lungo il cp DNA. Saranno necessari ulteriori esperimenti per determinare il motivo per cui questo è il caso. Indipendentemente da ciò, i genomi mt e cp hanno una copertura relativamente uniforme con entrambi i metodi e senza grandi aree di copertura mancante, che può essere dimostrato dall'esame della frazione di genomi sequenziati ad una determinata profondità ( Figura 3E -3H ). Inoltre, i livelli di copertura per entrambi i genomi sono considerati sufficienti per l'analisi a valle, ad esempio l'analisi di varianti. Se ritenuto necessario per l'analisi di varianti rare, riducendo il numeroR di campioni pool avrebbe ottenuto una maggiore copertura. In alternativa, un gran numero di campioni può essere raggruppato su una corsia HiSeq, pur raggiungendo una profondità di sequenza ancora maggiore, sebbene ad un sacrificio a lunghezza di sequenza, poiché le librerie HiSeq sono attualmente limitate alla lunghezza PE150 in contrasto con le librerie PE300 MiSeq.
Per esaminare i livelli di contaminazione del genoma nucleare utilizzando un approccio di mappatura, sono state esaminate le categorie di mappatura PE. Le letture PE possono mappare un genoma di riferimento in una varietà di configurazioni. Quando si legge 1 e 2 si allinea al riferimento in modo capo a testa, con una certa distanza "prevista" tra i due compagni (in base alla dimensione media inserto della libreria e tipicamente specificata come parametro di ingresso nel software di mappatura ), Queste letture PE sono dette mappe "concordantemente". Al contrario, la mappatura "discordante" è la situazione in cui i mappati mappano con un disco meno o superiore a quello previstoAl genoma di riferimento o alla mappa in configurazioni alternative (testa a coda o coda a coda). Se solo un compagno si allinea al genoma di riferimento, allora si dice che la lettura PE non sia mappata in modo concordante o discordante al genoma di riferimento. In tutte e tre le categorie di mappatura di lettura, le letture PE possono allinearsi al genoma di riferimento uno o più volte.
Per entrambi i DNA organellare isolati da DC e MF, la lettura del mappaggio al genoma mitocondriale è stata prevalentemente nella categoria allineata concordantemente una volta ( Figura 4A ), mentre le letture sono mappate al genoma di cloroplasto in proporzioni relativamente uguali di concordanza una volta e concordemente più di Una volta ( Figura 4B ), probabilmente a causa delle grandi ripetizioni invertite presenti nel genoma del cloroplasto e anche ai livelli di copertura estremamente elevati. Tuttavia, meno PE legge mappato al genoma nucleare e ha mappato in gran parte più di una volta in unNé moda concordante né discordante ( cioè, solo un compagno è in grado di mappare). Questi sono probabilmente mappatura "fuori bersaglio" a sequenze nel genoma nucleare, che sono omologhe ai genomi organellari o alle regioni misassate. Solo una piccola quantità di letture (<5%) mappata al genoma nucleare in modo concordante, indicando bassi livelli di contaminazione del genoma nucleare nel DNA organellare isolato dal metodo DC o MF ( Figura 4C ), come risulta anche dai risultati qPCR ( Figura 2A ). La frazione nucleare dopo il pulldown MBD2-Fc da tessuti non etiliolati cinesi di primavera è stata anche sequenziata per determinare l'efficacia del pulldown alla rimozione del DNA nonmetilato. Meno dell'1% delle letture nella biblioteca derivata dalla frazione nucleare sono mappate ai genomi di riferimento organellare, mentre ~ 45% di tutte le letture mappate al genoma nucleare ( Figura 4 ). Tuttavia, la maggior parte delle letture sono mappate in modo discordante, wChe riflette probabilmente gli elevati livelli di misassembly e frammentazione nel genoma di riferimento nucleare del grano. Indipendentemente, i risultati suggeriscono che il pulldown MBD2-Fc è altamente efficace per la rimozione del DNA organellare nonmetilato da DNA nucleare metilato. Vale la pena notare che, poiché il DNA arricchito da organellari risultante da questi metodi contiene una miscela di sequenze di mitocondri e cloroplasti e perché le somiglianze di sequenza risultanti dall'antico trasferimento di generi tra questi organi rimangono nei loro genomi, l'assegnazione appropriata di letture allo specifico I genomi devono essere risolti in bioinformatica.
L'etichetta del fogliame non altera appieno le abbondanze organelle:
Tradizionalmente, i tessuti etiolati sono preferiti per l'isolamento del DNA mitocondriale vegetale al fine di diminuire i livelli di fenoli e di amidi, che possono interferire con gli estrattiN o applicazioni a valle 13 . Per determinare se i livelli di arricchimento del genoma organellare potrebbero essere alterati o migliorati dalle condizioni di crescita, i tessuti etiolati e non etiolati sono stati sottoposti al protocollo MF e alla sequenza. È interessante notare che la etiolazione non ha modificato notevolmente la percentuale di letture che hanno mappato i genomi di riferimento organellare ( figure 3A e 3B ) o la copertura per-base ( Figura 3I ) rispetto alle condizioni non etiliolate. Abbiamo anche isolato il DNA organellare utilizzando centrifugazione differenziale, con tessuti etiolati e non etiolati, e piccole differenze nell'arricchimento sono state trovate tra i diversi tessuti usando qPCR (dati non mostrati). Ciò suggerisce che più tessuti fisiologicamente rilevanti non etiliolati possono essere utilizzati per studi organistici di sequenziamento, senza alcun cambiamento apprezzabile dell'arricchimento.
Il controllo di qualità suggerisce cheIl DNA di MF è più adatto per sequenza a lunga lettura:
Poiché il sequenziamento a lunga lettura diventa più accessibile ai ricercatori, l'isolamento del DNA a peso molecolare sta diventando sempre più importante. Per valutare il DNA organellare isolato con entrambi i metodi per l'integrità e la qualità, è stato impiegato PFGE. Il DNA genomico complessivo tipicamente migra come uno strato diffuso in PFGE e il peso molecolare è determinato dal protocollo e come il DNA è stato conservato e trattato dopo l'estrazione. Il DNA totale genomico isolato con punte genomiche dovrebbe superare 50 kb, che è stato verificato usando PFGE ( Figura 5 , corsia 2). Il DNA genomico totale dai suggerimenti genomici viene utilizzato come input nel kit di arricchimento dei microbiomi per frazionare il nucleare dal DNA organellare. La frazione nucleare ottenuta dopo la frazionamento diminuisce nelle dimensioni, ma rimane centrata di circa 50 kb ( figura 5 , corsia 4). Questo non è suRivolgendosi, dato che la manipolazione relativamente più ruvida della frazione nucleare come eluizione da branchi legati a MBD2-Fc richiede la digestione di calore e proteinasi K. A causa della massa limitata, la frazione organellare non è stata eseguita su PFGE, ma l'analisi successiva con la TapeStation ha indicato il DNA> 50 kb (dati non mostrati). Il DNA organellare ottenuto con centrifugazione differenziale ha una massa media di ~ 20 kb, probabilmente causata dal protocollo di isolamento organellare esteso e dalla successiva estrazione e concentrazione del DNA a base di colonna. L'isolamento organellare basato su gradiente e metodi alternativi di estrazione del DNA possono mantenere dimensioni maggiori dei frammenti di DNA. Indipendentemente dal fatto che il DNA della dimensione ottenuta in questo protocollo può essere utilizzato per generare letture di sequenza di 10 o 15 kb, se si prende cura durante la preparazione della libreria.
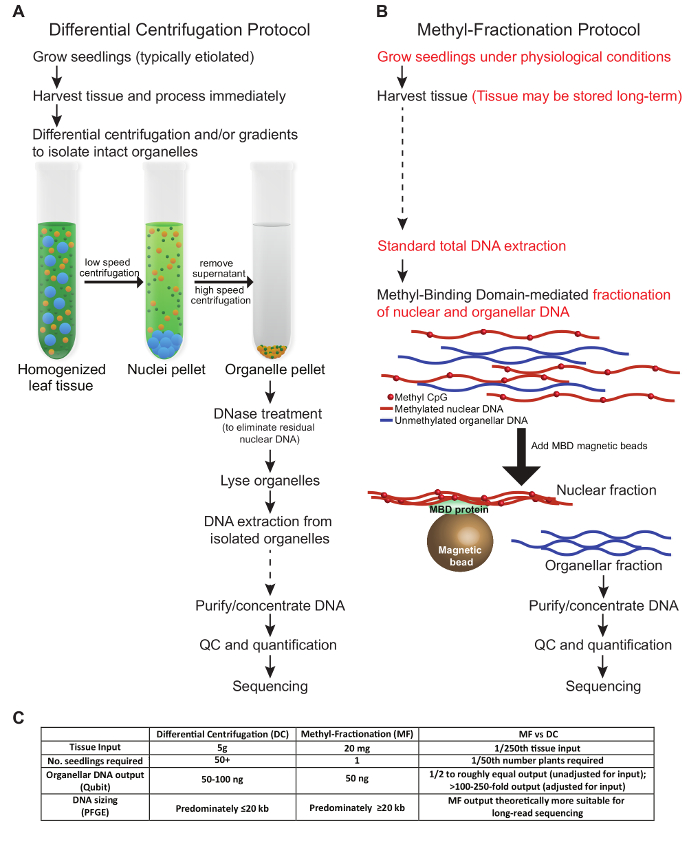
Figura 1: Una visione comparata di due metodiDs per arricchire il DNA del pianto organellare. Un protocollo DC tradizionale ( A ) è in contrasto con il protocollo MF ( B ). Si raccomanda di evitare di congelare e scongelare i campioni; Comunque, i punti in cui i campioni possono essere conservati a lungo termine sono indicati con frecce tratteggiate ( A e B ). Le differenze chiave tra i protocolli sono evidenziate in rosso ( B ). ( C ) La tabella confronta i metodi in termini di input tissutale, numero di piante richieste, produzione di DNA e dimensione del DNA risultante. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2: Valutazione della contaminazione del DNA nucleare nel DNA organellare isolato utilizzando due metodi. (
( B ) La tabella mostra i valori di C q , mostrati sul grafico in ( A ), e l'arricchimento pieghevole degli ampliconi organellari rispetto all'ACTIN . * Arricchire la piega = 2 (Cq ACTIN - Target Cq) . La formula assume un'efficienza perfetta di 2 per ogni set di primer, poiché il minore deviaIone di ciascuna serie di primer da 2 è trascurabile e avrebbe poco effetto sul calcolo e sulla tendenza generale ( ACTIN = 1.961, NAD3 = 1.95 e PSBB = 1.989). Le efficienze del primer sono state valutate realizzando una curva standard con una serie di cinque diluizioni 1:10 di DNA totale genomico. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3: Leggi la mappatura e la copertura teorica dei cloroplasti e dei genomi mitocondriali. Percentuale di letture mappate ai genomi di riferimento cinese di primavera mitocondriale ( A ) o cloroplasti ( B ). Correlazione di copertura teorica del geno di riferimento cinese di tipo mitocondriale ( C ) o cloroplasto ( D )Mes, assumendo rispettivamente le dimensioni del genoma di 450 e 135 kb, calcolate usando i numeri totali di lettura e la percentuale di letture mappate ai diversi genomi. Distribuzione genoma di copertura per DNA organellare dal metodo MF ( E e G ) o il metodo DC ( F e H ). I dati nei pannelli E - H sono dal campione culinario cinese esaminato, ma tutti gli altri campioni hanno mostrato una tendenza simile. ( I ) Copertura media, più bassa e più alta per tutti i campioni nei pannelli A - D. Le etichette di esempio compresi "E" indicano campioni etiocati, e "NE" indica campioni non etiliolati. DC indica il DNA isolato con il metodo di centrifugazione differenziale e Unmethylated indica il DNA che è nella frazione nonmetilata dopo il pulldown con MBD2-Fc (protocollo MF). I campioni contrassegnati con "Chris" indicano il grano Triticum aestivum'Chris.' CS designa campioni di primavera cinese di Triticum aestivum di grano. Nota: a causa dell'omologia di sequenza tra i cloroplasti, i mitocondri ei genomi nucleari derivanti da un trasferimento genico tra genomi organistici e tra genomi organellari e nucleari, una piccola percentuale di letture crude può mappare genomi multipli. Inoltre, le letture che non mappano a un genoma di riferimento organellare non sono rappresentate in questa figura. Quindi, le percentuali visualizzate qui ( A e B ) non sono totali del 100%. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 4: PE Leggi la mappatura al Genoma Nucleare del Grano. Percentuale di categorie di PE Leggere i tipi di mappatura ai genomi di riferimento di primavera mitocondriale (A) , chloroplast (B) o nucleare (C) . - E indica campioni etiolati e - NE indica campioni non etolati. DC indica il DNA isolato con il metodo di centrifugazione differenziale, Unmethylated indica il DNA che è nella frazione nonmetilata dopo il pulldown con MBD2-Fc nel protocollo MF e Methylated designa la frazione nucleare dopo pulldown MBD2-Fc. I campioni contrassegnati con "Chris" indicano il grano Triticum aestivum 'Chris'. CS designa campioni di primavera cinese di grano Triticum aestivum . Le letture non definite non vengono mostrate. Clicca qui per visualizzare una versione più grande di questa figura.
Oad / 55528 / 55528fig5.jpg "/>
Figura 5: Esame della qualità del DNA utilizzando PFGE. Il DNA genomico totale (corsia 2), il DNA organolitico del frumento ottenuto da centrifugazione differenziale (corsia 3) e la frazione nucleare dopo MF con l'approccio pulldown MBD2-Fc (corsia 4) sono stati sottoposti a PFGE su un gel agarosico del 1% con un 1 scala larga estesa utilizzata come marcatore (corsi 1 e 5). Clicca qui per visualizzare una versione più grande di questa figura.
| Nome del buffer | Ricetta | Gli appunti | Metodo |
| STE Buffer | 400 mM di saccarosio, 50 mM Tris pH 7,8, 20 mM EDTA pH 8,0, 0,6% (in peso) polivinilpirrolidone (PVP), 0,2% (w / v) albumina bovina serum (BSA), 0.1% (v / v) β-mercaptoetanolo (BME) | Il mix di bufere contenente solo saccarosio, Tris e EDTA può essere effettuato fino a un mese di anticipo e mantenuto a 4 ° C. PVP, BSA e BME devono essere aggiunti freschi ad una aliquota della quantità richiesta di tampone appena prima dell'uso. | Metodo # 1 |
| ST Buffer | 400 mM di saccarosio, 50 mM Tris pH 7,8, 0,6% (in peso) di polivinilpirrolidone (PVP), 0,1% (w / v) albumina bovina serum (BSA) | La miscela di bufere contenente solo saccarosio e Tris può essere effettuata fino a un mese in anticipo e mantenuta a 4 ° C. Si noti che il buffer ST non contiene EDTA o BME e contiene una concentrazione inferiore di BSA. | Metodo # 1 |
| DNase stock | 2 mg / ml di DNasi in 0.15 M NaCl ad una concentrazione di 2 mg / ml | Conservare le aliquote di 200 ul a -20 ° C. Per preparare la soluzione di lavoro DNase (200 μl di soluzione DNasi per campione) vedereTabella 1 di seguito. Vedere il protocollo completo qui sotto per i dettagli completi della digestione DNase. DNase soluzione di lavoro dovrebbe essere preparata fresca. Per fermare la reazione DNase è necessaria una soluzione di pH 400 di EDTA da 400 mM (la concentrazione finale necessaria per arrestare la reazione è 0,2 M EDTA, vedere protocollo completo per i dettagli). | Metodo # 1 |
| Soluzione di lavoro DNase | 0,25 mg / ml DNasi e 20 mM MgCl 2 a ST Buffer | Preparare fresco, 200 ul per campione. Le concentrazioni indicate sono per il volume finale di reazione, quindi mescolare: 62,5 μl 2 mg / ml DNasi (basata sul volume di reazione finale di 500 μl), 4 μl 1 M MgCl 2 (basata su 200 μl di soluzione della soluzione DNasi) e 133,5 μl di tampone ST Un volume finale di 200 μl. | Metodo # 1 |
| Tampone di lisi | 20 mM EDTA pH 8,0; 10 mM Tris pH 7,9; 500 mM guanidina-HCl; 200 mM NaCl; 1% Triton X-100; 0,5 mg / ml enzimi di lysazione daTrichoderma harzianum | Mescolare tutti gli ingredienti tranne gli enzimi di lysing e conservare a temperatura ambiente. Gli enzimi di lysing devono essere aggiunti freschi ad una piccola aliquota per un uso immediato. | Metodo # 2 |
Tabella 1: Ricette di tamponi fatti in casa e scorte di lavoro.
| Foglio di lavoro di concentrazione | |||||||
| SAMPLE NAME | Peso del dispositivo vuoto (g) | Peso del dispositivo riempito (g) | Volume pieno (ul, riempito meno pesi vuoti) | Peso dopo il primo spin (20 min *, g) | Volume dopo il primo spin (ul, riempitoMeno i pesi vuoti) | Peso dopo il secondo spin (X min *, g) | Volume dopo il secondo spin (ul, riempito meno i pesi vuoti) |
| Si noti che il volume effettivo di recupero sarà un po 'inferiore a quello calcolato. | |||||||
Tabella 2: Foglio di lavoro di concentrazione.
| Nome | Specificità del genoma | Fonte della sequenza di gene | Sequenza (5 '- 3') |
| Ta_ACTIN - F | Nucleare | Scaffold Gramene IWGSC_CSS_1AS_scaff_3272162: 10.663-12.557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | Nucleare | Come sopra | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | mitocondriale | L'adesione di NCBI EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | mitocondriale | Come sopra | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | cloroplasto | Entrata NCBI KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | cloroplasto | Come sopra | GCTGCCTGTTTCCTTGTAGTT |
Tabella 3: Elenco dei primer qPCR.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Ad oggi, la maggior parte degli studi organizzativi di sequenziamento si concentra sui metodi DC tradizionali per arricchire il DNA specifico. Sono stati descritti metodi per isolare gli organelli da diverse piante, tra cui il muschio 40 ; Monocots come il grano 15 e l'avena 11 ; E dicoti come arabidopsis 11 , girasole 17 e colza 14 . La maggior parte dei protocolli si concentra sui tessuti foglia 13 , 14 , 15 , 16 , 17 , alcuni che sono stati adattati per una varietà di tipi di tessuto, inclusi i semi 11 . È stato inoltre dimostrato l'isolamento degli organelli da protoplasti 41 . Tuttavia, questo non è adatto a tutti i sistemi, né è fattibile quando il tessuto di interesse è limitato. Molti di questi orgaI metodi di isolamento nellar sono stati progettati per recuperare organoli intatti per esperimenti specifici, come studi fisiologici. Questi protocolli sono ingombranti e in genere richiedono l'utilizzo di gradienti di densità, quali gradienti di saccarosio o di Percoll, che sono molto efficaci per isolare frazioni organiche specifiche, ma richiedono un ingente input tissutale ( cioè superiore a 5 g e in su di chilogrammi, a seconda Il tipo di tessuto). Tuttavia, il metodo DC può essere ottimizzato per arricchire per particolari frazioni cellulari, come i mitocondri o il cloroplasto, cambiando velocità di rotazione e gradienti di densità. Al contrario, l'approccio MF richiede molto meno materiale di partenza (20 mg), ma i DNA mitocondriali e plastidi saranno presenti per le loro abbondanze relative nel tessuto utilizzato per l'estrazione del DNA. Tuttavia, il protocollo MF offre un approccio alternativo per l'isolamento del DNA organellare misto ed è particolarmente utile per iniziare con piccole quantità di tessuto.
T O valutare la purezza del campione dopo l'isolamento dell'organone, la maggior parte degli studi fino ad oggi utilizza solo l'end-point PCR e l'elettroforesi gel 11 , 12 . Questo dà una giusta misura qualitativa della purezza del campione. Tuttavia, bassi livelli di amplificazione non possono essere visualizzati su un gel agarosio. Pochi rapporti comprendono misure quantitative di controllo qualità, come qPCR 14 . Per una valutazione quantitativa della purezza campionaria del DNA isolata da entrambi i metodi, abbiamo utilizzato qPCR e sequenziamento per determinare quanto il DNA nucleare rimane nel campione, così come le relative proporzioni di DNA mitocondriale contro il cloroplasto. Entrambi i metodi qui valutati sono efficaci nella rimozione del DNA nucleare. Entrambi i metodi producono un mix di DNA mitocondriale e cloroplastica, sebbene a diverse proporzioni.
Le piante crescenti nel buio (etiolazione) sono segnalate per facilitare l'isolamento organellare a causa di una riduzione delle sostanze fenolicheTuttavia, in questo confronto, non abbiamo trovato un notevole vantaggio per lavorare con tessuti etiolati su campioni coltivati a luce leggera. Anche se la percentuale di cloroplasti specializzati sarà probabilmente superiore a coltura leggera, il numero totale di plastidi, come Che si riflette nella proporzione di letture mappate al genoma di cloroplast, è invariata in condizioni di luce diverse. Pertanto, per le analisi funzionali a valle, come la valutazione dell'eteroplasma in diversi tessuti o sotto diversi stressori o per analisi di espressione, si consiglia di eseguire sequenziamenti genomici Piante coltivate in condizioni fisiologicamente rilevanti.
Per l'applicazione con tecnologie di sequenza brevettate, entrambe le tecniche qui comparate offrono una quantità e qualità adeguate del DNA. Tuttavia, per ottenere lunghe letture di> 20 kb per applicazioni di sequenziamento a singole molecole, è necessaria una maggiore quantità di DNA di elevata qualità. Per esempio, idealmente,> 1 μg di puro orgaDNA frumento nellar con un peso molecolare> 20 kb è necessario, protocolli interni basso input per 20 kb preparazioni libreria inserto 42. I nuovi protocolli a basso consumo sviluppati dall'utente possono ridurre i requisiti del DNA ( cioè a 50 ng o addirittura meno 20 ), ma la sfida continua ad avere un DNA di alta qualità e ad alto peso molecolare che entra nei preparativi della libreria. È essenziale che la maggior parte del DNA sia> 20 kb, poiché frammenti più piccoli saranno inseriti preferenzialmente nel SMRTbell e spegnere la distribuzione di dimensioni della libreria 43 . Abbiamo provato un certo numero di protocolli di estrazione del DNA casalinghe e un certo numero di protocolli commerciali per l'estrazione del DNA (non mostrati). Per il tessuto di foglie di frumento, il miglior equilibrio tra la quantità e la qualità del DNA, in particolare la lunghezza, è stato ottenuto utilizzando un kit commerciale 27 , 29 . A seconda delle specie vegetali e tessuti di interesse, alternatiI protocolli di estrazione possono essere ugualmente adatti o più fecondi. Tuttavia, concludiamo che l'estrazione totale di DNA genomico a peso molecolare elevato> 50 kb, seguita dalla frazionamento con l'approccio pulldown MBD2-Fc 28 , è suscettibile di sequenza a lungo letto da un materiale di partenza limitato. I lavori futuri dovrebbero verificare i limiti del materiale di partenza richiesto dopo la frazionamento per la preparazione della libreria a lungo inserire e successivi sequenziamenti a lungo leggere. Critico, questo approccio potrebbe fornire un metodo robusto per isolare il DNA da un substrato di una singola foglia che è adatta per sequenza a lungo leggere, senza amplificazione del genoma intero. Si prevede che questo approccio sarà facilmente adattabile a tipi di tessuti supplementari e generalmente applicabile ad altre specie vegetali. Sarà particolarmente utile in situazioni in cui gli importi dei tessuti stanno limitando, come ad esempio sequenziamento a generazioni individuali in uno schema di attraversamento o in tipi di tessuti più rari.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori dichiarano di non avere interessi in gioco.
La menzione dei nomi commerciali o dei prodotti commerciali della presente pubblicazione è esclusivamente ai fini della fornitura di informazioni specifiche e non implica raccomandazione o approvazione da parte del Dipartimento dell'agricoltura USA. USDA è un fornitore di pari opportunità e datore di lavoro.
Acknowledgments
Vorremmo riconoscere i finanziamenti del Dipartimento per l'Agricoltura-Agricoltura del Dipartimento degli Stati Uniti e della National Science Foundation (IOS 1025881 e IOS 1361554). Ringraziamo R. Caspers per la manutenzione delle serre e la cura delle piante. Ringraziamo anche l'Università del Minnesota Genomics Center, dove sono stati eseguiti i preparativi e la sequenza delle biblioteche Illumina. Siamo anche grati per i commenti dei redattori e dei quattro recensori anonimi che hanno ulteriormente rafforzato il nostro manoscritto. Ringraziamo anche l'OCSE per una borsa di studio alla SK per integrare questi protocolli per progetti di collaborazione con i colleghi in Giappone.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
