

Summary
提出了两种植物细菌DNA富集方法的比较和优化:传统差速离心和基于甲基化状态的总gDNA分馏。我们评估产生的DNA数量和质量,证明在短读下一代测序中的性能,并讨论长读单分子测序中使用的潜力。
Abstract
植物细胞器基因组包含大的重复元件,其可能经历配对或重组以形成复合结构和/或亚基因组片段。细胞器基因组也存在于给定细胞或组织类型(异质性)内的混合物中,并且丰富的亚型可能在整个发育过程中或当处于应力(亚化学计量转移)时)发生变化。需要下一代测序(NGS)技术来更深入地了解细胞器基因组结构和功能。传统的测序研究使用几种方法来获得细胞器DNA:(1)如果使用大量的起始组织,将其均质化并进行差速离心和/或梯度纯化。 (2)如果使用较少量的组织( 即如果种子,材料或空间有限),则按照(1)所述进行相同的处理,然后进行全基因组扩增以获得足够的DNA。 (3)生物信息学分析可用于分析使用总基因组DNA并解析出细胞器读数。所有这些方法都有固有的挑战和权衡。在(1)中,可能难以获得如此大量的起始组织;在(2)中,全基因组扩增可能引入测序偏倚;在(3)中,核和细胞器基因组之间的同源性可能会干扰组装和分析。在具有大核基因组的植物中,富集细菌DNA以降低生物信息学分析的测序成本和序列复杂度是有利的。在这里,我们将传统的差速离心法与第四种方法(适应性CpG-甲基下拉法)进行比较,将总基因组DNA分离成核和细胞器级分。两种方法为NGS产生足够的DNA,对于细胞器序列高度富集的DNA,尽管在线粒体和叶绿体中具有不同的比例。我们介绍了这些方法对小麦叶组织的优化,并讨论了主要优点和d在样本输入,协议易用性和下游应用的上下文中的每种方法的优点。
Introduction
基因组测序是解析重要植物性状的潜在遗传基础的有力工具。大多数基因组测序研究集中于核基因组内容,因为大多数基因位于细胞核中。然而,细胞器基因组,包括线粒体(跨真核生物)和质(植物;在特殊形式,叶绿体,工作在光合作用)促进生物发育,应激反应和整体健康至关重要的1显著的遗传信息。细胞器基因组通常包括在用于核基因组测序的总DNA的提取,虽然方法,以减少前,DNA提取的细胞器号码也采用2。许多研究已经使用测序结果从总的gDNA提取组装细胞器基因组3,4,5,外部参照“> 6,7。然而,当研究的目标是把重点放在细胞器基因组,使用总的gDNA增加了测序成本,因为很多的读取,但‘丢失’的核的DNA序列,特别是在植物具有大核基因组此外,由于重复和传输细胞器序列的进核基因组和细胞器之间,解决测序的正确的映射位置读取到适当的基因组是生物信息学有挑战性2,8。细胞器基因组的从核基因组的纯化是一个减少这些问题的策略,进一步的生物信息学策略可用于分离映射到线粒体和叶绿体之间同源区域的读数。
虽然来自许多植物物种的细胞器基因组已被测序,但是对于细胞器基因组多样性的广度几乎不了解可用于野生种群或栽培育种池。还知道细胞器基因组是由于重复序列9之间的重组而经历显着的结构重排的动态分子。此外,每个细胞器中含有多个拷贝的细胞器基因组,并且每个细胞内都含有多个细胞器。并非所有这些基因组的拷贝都是相同的,这被称为异质性。与“主圈”的规范图相比,现在越来越多的证据表明,细胞基因组结构更复杂,包括亚基因组圆,线性染色体,线性连接子和支链结构10 。植物细胞器基因组的组装由于其相对较大的尺寸和大量的反向和直接重复而进一步复杂化。
用于细胞器分离,DNA纯化和随后基因组的传统方案 È测序往往笨重且需要大体积的组织输入的,具有几克到几百个向上必要为起点11,12,13,14,15,16,17克幼叶组织的组成。当组织受限时,这使得细胞器基因组测序不可及。在某些情况下,种子数量有限,例如当需要在代际基础上进行序列或在必须通过杂交维持的雄性不育系中时。在这些情况下,可以纯化细胞器DNA,然后进行全基因组扩增。然而,全基因组扩增可以引入显着的测序偏差,这在评估结构变异,亚基因组结构和异质性水平时是一个特别的问题> 18。用于短读序列技术的图书馆准备工作的最新进展克服了低输入障碍,以避免全基因组扩增。例如,Illumina Nextera XT库制备试剂盒允许使用少至1ng的DNA作为输入19 。然而,用于长读取测序应用(如PacBio或Oxford Nanopore测序技术)的标准库制备仍然需要相对较高量的输入DNA,这可能对组织基因组测序构成挑战。最近,新的用户制作,长读测序的协议已被开发,以减少输入量和帮助促进在获得的DNA微克,批量样品的基因组测序是困难的20,21。然而,获得高分子量纯的细胞器级分以进入这些文库制剂仍是一个挑战。
我们寻求o比较和优化不需要全基因组扩增的适用于NGS的细胞器DNA富集和分离方法。具体来说,我们的目标是确定从有限起始材料(如叶子子样品)中丰富高分子量细胞器DNA的最佳实践。这项工作提供了丰富细胞DNA的方法的比较分析:(1)修改的传统差速离心方案与(2)基于使用市售DNA CpG-甲基结合结构域蛋白质下拉法的DNA分级方案22施用于植物组织23 。我们建议从小麦叶组织中分离细胞叶DNA的最佳实践,这可能容易地扩展到其他植物和组织类型。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1.生成用于细胞分离和DNA提取的植物材料
- 小麦幼苗的标准生长
- 在每个角落有4-6个种子的小方形花盆中种植蛭石。转移到温室或生长室,16小时光周期,23ºC/ 18ºC夜间。
- 每天浇灌植物。在萌发和发芽后7天,用¼茶匙颗粒状20-20-20 NPK肥料施肥。
- 小麦幼苗的替代品种
- 按照步骤1.1,但将盆置于暗生长室中,23℃16小时/ 18℃8小时。或者,覆盖温室中的植物( 例如,使用储存容器;但必须保持适当的通风)。
- 生长和组织收集
- 种植植物12-14天。对于大多数小麦基因型s,75-100个幼苗产生约10-12g的组织,这对于使用差速离心法的两种细胞器提取是足够的(第2节);如果利用基于DNA CpG-甲基化的下拉方法从核DNA中分离细胞器,则仅需要一种植物(第3节)。
- 如果使用差速离心法,请将新鲜的组织收集,并立即进行处理样品,如第2节所述。
- 如果利用CpG甲基下拉法,将20毫克的年轻叶组织切片收集到微量离心管中(使用标准培养的或蚀刻的组织,见代表性的结果 )。在液氮上快速冷冻并在-80℃下冷冻直至使用。按照第3节所述进行DNA的下拉分离。
2.方法1:使用差速离心(DC)的DNA提取
注意:erential离心方案从该优化条件来隔离两个细胞器但富集线粒体17,24两个发布修改。所得到的方案比以前的方法更少时间密集,使用更少的有毒化学品。具体来说,我们对缓冲液和洗涤步骤进行了修改,包括向STE提取缓冲液中加入聚乙烯吡咯烷酮(PVP),并消除了含有氟化钠(NaF)的NETF缓冲液中的最终洗涤步骤。
注意:STE缓冲液的制备和使用应在化学通风橱下进行,并配有适当的个人防护设备,因为该缓冲液含有2-巯基乙醇(BME)。
- 开始之前要做的事情
- 确保所有设备都非常干净,并对任何可以进行高压灭菌的设备( 例如,研磨气瓶,高速离心机)进行高压灭菌护眼管等 )。
注意:推荐使用过滤嘴,适用于所有需要移液的步骤,以避免交叉污染。 - 查看所需设备和试剂的清单,并准备方法#1( 表1 )所需的缓冲液和工作库存。将低温研磨块冷却至-20℃,转子和缓冲液至4℃,将微量离心机置于4℃,然后打开37℃水浴。
- 确保所有设备都非常干净,并对任何可以进行高压灭菌的设备( 例如,研磨气瓶,高速离心机)进行高压灭菌护眼管等 )。
- 细胞器的分离
- 收获5克新鲜的纸巾,并在冰冷的无菌水中,在冰上的冷冻烧杯中冲洗。
注意:在进行离心机,通风橱等的所有操作和运输过程中,始终将样品保持在冰上。或者,如果有足够的空间和设备来执行协议,则可以在冷藏室工作。 - 使用剪刀将叶组织切成约1厘米的片,直接装入含有两个陶瓷研磨的50mL管中缸。
注意:清洁或更换样品之间的剪刀,以避免交叉污染。 - 如果没有组织匀浆器,请使用砂浆和杵,然后按照替代步骤2.2.4 - 2.2.9。
- 将叶组织切成冰块中的预冷砂浆。在15 mL STE(通风柜)中研磨样品2 - 3 min。
- 将缓冲液(留在研钵中的组织)通过一个含有一层预湿无菌滤布(约22至25μm孔径的漏斗)的漏斗倒入另一个50mL管中。向研钵和杵中加入另外10mL的STE,再次匀浆。
- 将匀浆的组织和缓冲液倒入同一漏斗。用10mL STE冲洗研钵和杵,倒入漏斗中。将滤布挤压并拧入漏斗,以尽可能多地回收液体。
注意:在样品之间更换手套以避免交叉污染。继续亲在步骤2.2.10。
- 在每个50mL管中加入20mL STE(在通风橱中)。
- 将样品放入组织研磨设备中的预冷冻低温研磨块中,并以1,750rpm研磨样品2×30秒。旋转样品位置,将样品放在冰上约1分钟之间。
注意:在此步骤中可以使用砂浆和研杵,搅拌机或其他组织研磨/均质装置。然而,每种方法都会对不同程度的DNA质量产生影响,因此在继续下游应用前应对DNA长度和质量进行评估。 - 将漏斗插入放置在冰中的干净的50mL管。将一层滤布放入漏斗中,并用5 mL STE进行预湿。不要丢弃流通。
- 将匀浆的组织倒入漏斗。用15 mL STE冲洗研磨管,重新整理并翻转管子以冲洗墙壁和盖子,并倒入机体埃尔。
- 仔细取出陶瓷石头,然后将滤布挤压并拧入漏斗。
注意:在样品之间更换手套以避免交叉污染。 - 用石蜡膜包裹管帽以避免溢出。在4℃下以2,000×g离心10分钟。
- 使用血清移液管小心地吸出上清液(避免干扰沉淀),并将其置于50mL高速离心管中(如果管没有紧密的密封垫圈,用石蜡膜包裹管帽以避免溢出)。丢弃颗粒
- 使用STET将管平衡至0.1g,并将得到的上清液以18,000xg和4℃离心20分钟。为了平衡管,将一小杯冰块放在天平上,称重秤,并在冰上称重样品以保持冷。或者,在冷藏室使用天平和通风橱。
- 丢弃上清液。将1mL的ST加入沉淀中并轻轻重新悬浮一个软的画笔。加入24 mL ST(最终体积为25 mL),混匀/旋转( 即按下管子侧的油漆刷除去所有液体)。
- 使用ST将管平衡至0.1g以内。以18,000 xg和4℃离心20分钟。同时,准备DNaseI溶液( 参见表1的库存和工作溶液配方)。对于每个样品,将一个200μL等分试样置于1.5 mL管中。
- 丢弃上清液,吸干管,并使用柔软的画笔将沉淀物(仍然在高速离心管中)重新悬浮在300μL的ST中。将画笔放在预先制备的含有200μLDNA酶溶液的1.5mL管中,并旋转刷子以除去卡在刷子中的残留丸粒。将DNaseI溶液吸入高速离心管中,轻轻旋转混匀。
- 在水浴中在37℃下孵育30分钟(在管的顶部包裹石蜡膜以防止冷凝泄漏g进入帽子)。在孵育过程中轻轻搅拌2次。
- 使用具有宽孔口的移液管尖端将小球混合物轻轻移出管内,并将其置于1.5 mL低粘合管中。向高速离心管中加入500μL400 mM EDTA,pH 8.0,轻轻吸管,将所有剩余的颗粒从管中取出。将EDTA转移到与沉淀混合物相同的1.5-mL低粘合管,并通过反转轻轻混合。
- 在4℃下以18,000xg离心20分钟。丢弃上清液,吸管,并立即使用DNA分离。如果需要,在-20℃冷冻丸粒,但这可能导致产量降低,因为如果不立即处理,残留的DNA酶I可能降解样品DNA。
- 收获5克新鲜的纸巾,并在冰冷的无菌水中,在冰上的冷冻烧杯中冲洗。
- 使用商业色谱柱方法从分离的细胞器中提取DNA
注意:有关完整协议25 ,请参阅套件手册,有关修改,请参见下文。镨优选直接从细胞器分离到DNA提取。反复冷冻和解冻将减少DNA片段大小,并导致残留DNA酶I的DNA降解。限制涡旋或剧烈移液,因为它可以剪切DNA。建议使用低结合微量离心管,以最大限度地提高DNA的回收率。- DNA提取程序
注意:在开始之前,请阅读详细的商业协议25 ,以确保缓冲区已正确生成/存储,并且了解自旋柱程序。- 将缓冲液ATL直接加入到管中,加入沉淀(如果先前冷冻并在台式平台上平衡至室温)。
- 在试剂盒手册中的“组织DNA纯化”方案中的步骤3进行,具有以下修改:步骤3中30分钟裂解包括任选的RNA酶A消化,并在3×200μLAE中洗脱每一个成为一个然后组合洗脱液)。
- 保存qPCR的等分试样(至少20μL)(参见步骤4.1)。在浓缩前进行定量,另外还需要1μL进行高灵敏度定量。
- 如果需要,进行样品浓度。
- DNA提取程序
- 采用商用过滤器的样品浓度
注意:有关详细信息,请参阅商业协议26 。根据下游用途,可能无需进行样品浓度( 例如,对于终点PCR和qPCR应用)。然而,对于NGS图书馆建设,可能需要浓缩DNA提取后获得的稀释的细胞器DNA。- 浓缩柱程序
- 在数字分析天平上的干净的称重纸张上仔细预先称重( 见表2 )空滤芯(无管)。记录重量。
- 皮将组合洗液转移到过滤器单元中,并重新称重。
注意:商业手册26说,过滤器最大体积为500μL,但是一次即可添加高达575μL,无溢出。 - 小心地将填充的过滤器单元放入管中(随柱提供)。以500 xg离心达到所需时间,达到所需浓缩液体积。对于约575μL的样品体积,20分钟的旋转通常会导致浓缩液体积为15 - 30μL。
- 从管中取出过滤器,重新称重。使用表格确定是否已经达到所需的浓缩物体积。如果没有,再次以500 xg的速度离心一段较短的时间并再次称重;重复直到达到所需的浓缩体积。
- 将新管(随柱提供)放在过滤器单元的顶部并进行反转。以1,000 xg离心3分钟,以转移浓缩至管。
- 确定回收的体积。由于过滤器保留,这通常比计算的体积小约3-5微升。如果过度浓缩,用无菌水或TE稀释以达到所需体积。
- 使用高灵敏度定量(根据制造商的说明书)量化DNA。
- 浓缩柱程序
3.方法2:甲基分馏(MF)从总基因组DNA中丰富细胞DNA的方法
注意:该协议是从用户开发的Genomic Tip Kit DNA提取方案修改植物和真菌27以及商业的Microbiome DNA Enrichment Kit方案28 。在理论上,可以使用产生高分子量DNA的任何DNA分离方案进行下拉。对于短读序列,产生主要> 15 kb片段的任何提取都足以用于下拉。对于洛ng读取测序,较大的片段可能是期望的。因此,我们优化了该方案以产生高分子量DNA。
- 总DNA的分离
注意:请参阅所需的设备和试剂清单,并准备方法#2所需的缓冲液和工作库存( 表1 )。向裂解缓冲液中加入裂解酶以制备裂解缓冲液工作溶液。打开热敏混合器并将其设置为37°C。将水浴打开至50°C,并将QF缓冲液放入浴中。将70%EtOH放入冷冻器中,并将微量离心机置于4℃。- 使用商业DNA提取柱进行总DNA提取
注意:在开始之前,请阅读商业手册29了解有关使用重力流阴离子交换柱的详细信息。柱可以使用专门的机架设置,或者使用提供的塑料环放置在管上。所有步骤,包括g应该通过重力流动来允许肠系统提示,残留液体不应该被强制通过。- 使用专为2 mL管设计的手持式研杵,在2 mL低粘合管中研磨20 mg液氮中的冷冻组织。
- 加入2 mL裂解缓冲液工作溶液(管将非常满)。
- 在37℃的温热混合器中孵育1小时,同时以300rpm的温和搅拌。如果不能使用热混合器,则每隔15分钟通过温和的轻拂孵育,并加热混合是一种合适的选择。
- 加入4μLRNA酶A(100 mg / mL,终浓度200μg/ mL)。在37℃下,在温热混合器中反复混合并孵育30分钟,在300rpm下轻轻搅拌。
- 加入80μL蛋白酶K(20mg / mL,终浓度0.8mg / mL),反转混合,并在50℃下在温热混合器中孵育2小时,在300rpm下轻轻搅拌。
- 在4℃和1℃离心20分钟5,000 xg,以沉淀不溶性碎屑。
- 当样品离心时,用1 mL缓冲液QBT平衡色谱柱,并通过重力流将色谱柱排空。
- 使用大孔移液管尖端将样品(避免沉淀)迅速施加到平衡的柱上,并使其完全流过柱。如果样品变得多云,请在应用于色谱柱之前再次过滤或离心(详见商业手册29 )。
- 样品完全进入树脂后,用4 x 1 mL Buffer QC洗涤柱。
- 将柱悬挂在干净的2 mL低结合微量离心管上。用0.8 mL预热温度为50°C的缓冲液QF洗脱基因组DNA。
- 通过向洗脱的DNA中加入0.56mL(0.7体积的洗脱缓冲液)的室温异丙醇来沉淀DNA。
- 通过倒置(10X)混合,并在15,000 xg和4℃下立即离心20分钟。关心充分去除上清液,不会干扰玻璃状,松散附着的颗粒。
- 用1 mL冷的70%乙醇洗涤离心的DNA沉淀。在15,000 xg和4℃下离心10分钟。
- 仔细清除上清液(同样要谨慎),而不会干扰沉淀。空气干燥5-10分钟,并将DNA重悬于0.1mL洗脱缓冲液(EB)中。在室温下将DNA溶解过夜。避免移液,这可能剪切DNA。
- 使用高灵敏度DNA定量测定(根据制造商说明书)量化样品。
- 使用商业DNA提取柱进行总DNA提取
- 甲基化和非甲基化DNA的基于珠的分馏
注意:最近的出版物证明了使用市售的试剂盒28 ,其利用使用与人IgG Fc片段(MBD2-Fc蛋白)融合的CpG特异性甲基结合结构域蛋白的下拉方法来分级从核基因组(高甲基化)含量的植物细胞器基因组(未甲基化) 23 。以前没有使用这种商业MF试剂盒28测试小麦样品的分馏效率。- 开始之前要做的事情
- 新鲜制备80%乙醇(每次反应至少800μL)。设置5x结合/洗涤缓冲液在冰上解冻,并为每个样品(稀释5X缓冲液,无菌,无核酸酶水)准备5 mL 1x缓冲液,并在方案中保持在冰上。
- 制备MBD2-Fc蛋白结合磁珠
- 准备所需数量的珠套。缩放反应,使用1到2μg的总输入DNA,需要160 - 320μL的珠子。请注意,以下列出的反应用于1μg总输入DNA,因此需要160μL珠粒。根据需要量化反应。
- 使用宽孔提示,轻轻吸取蛋白A磁性Bead浆料上下形成均匀的悬浮液。作为替代方案,在4℃下轻轻旋转珠子管15分钟。
注意:不要涡旋珠。 - 按照制造商的说明进行操作28 。
- 捕获甲基化核DNA
- 对于每个单独的样品,将1μg输入DNA加入到含有160μLMBD2-Fc结合的磁珠的管中。
- 加入5x结合/洗涤缓冲液,给定DNA输入样品的体积,最终浓度为1x(体积为5x的结合/洗涤缓冲液以加入(μL)=输入DNA的体积(μL)/ 4)。将样品上下移动几次,使用宽孔移液器吸头进行混合。
- 将管在室温下旋转15分钟。用大口径移液管尖端轻轻吸取样品,并在整个孵化过程中轻拍2-3次,以防止珠粒团聚。
注意:移液和flicking对于确保甲基化DNA的有效下拉是至关重要的。
- 收集富集的,未甲基化的细胞器DNA
- 简单旋转含有DNA和MBD2-Fc结合的磁珠混合物的管。将管放置在磁性架上至少5分钟以将珠收集到管侧。解决方案应该清楚。
- 使用大口径提示,小心地清除清除的上清液,而不打扰珠子。将上清液(含有未甲基化的富含细胞的DNA)转移到干净,低结合的2 mL微量离心管中。将此样品储存在-20或-80°C,或直接进入步骤3.2.6进行纯化。
- 洗脱液从MBD2-Fc结合的磁珠中捕获核DNA
- 如果还需要核级分,按照制造商说明书28从MBD2-Fc结合的磁珠中洗脱核DNA;如步骤3.2.7所述进行纯化。
- 基于珠的核酸纯化
- 确保净化珠在室温下充分混合。按照MF套件手册28中的说明继续执行协议。
注意:该样本现在可用于NGS库构建或另一个下游分析。
- 确保净化珠在室温下充分混合。按照MF套件手册28中的说明继续执行协议。
4.样品定量和质量控制
- qPCR测定以评估细胞器富集
注意:此处列出的qPCR反应和测定参数设计用于Roche LightCycler 480,可能需要针对不同的设备和试剂进行调整。如果qPCR不可用,则使用本文所述的相同引物和条件,可以使用琼脂糖凝胶上的端点PCR和可视化作为样品纯度的定性测量。所有引物组的扩增子大小将为〜150 bp。引物序列见表3ces和配对。- qPCR反应设置
- 要建立一个单独的20μLqPCR反应,请小心地将以下物质移至96孔qPCR板的一个孔中:10μL2x SYBR Green I Master; 2μL10μM正向和反向引物混合物(最终浓度为0.5μM); 2μL模板(标准曲线范围内);和6μL无菌,无核酸酶的H 2 O.为了减少移液误差,优选与除模板之外的所有反应组分一起制备主混合物。将主混合物添加到qPCR板中,然后将感兴趣的模板添加到每个孔中。应对每个样品进行三次技术重复,以尽量减少移液误差的影响。
注意:最终,样品之间比较核与细胞定量循环的比例,因此浓度的微小差异是可以接受的。然而,DNA浓度应大致在ea的范围内ch其他。 - 用高质量的qPCR密封膜密封板。轻轻旋转样品,注意避免产生任何气泡。在4℃下将板短暂旋转2分钟以收集样品并消除任何小的气泡。
- 将板装入机器。按照下列准则运行qPCR程序。
- 要建立一个单独的20μLqPCR反应,请小心地将以下物质移至96孔qPCR板的一个孔中:10μL2x SYBR Green I Master; 2μL10μM正向和反向引物混合物(最终浓度为0.5μM); 2μL模板(标准曲线范围内);和6μL无菌,无核酸酶的H 2 O.为了减少移液误差,优选与除模板之外的所有反应组分一起制备主混合物。将主混合物添加到qPCR板中,然后将感兴趣的模板添加到每个孔中。应对每个样品进行三次技术重复,以尽量减少移液误差的影响。
- qPCR反应参数
注意:这些是默认参数,除了放大级的退火循环。调整此设置以适应特定的引物,如果使用的不同于本协议中引用的引物。- 在95℃预孵育5分钟,升温速率为4.4℃/ s。
- 进行(1)95℃10秒的45个放大循环,斜率为4.4℃/ s; (2)60℃20s,升温速率为2.2℃/ s;和(3)72℃10秒,斜坡率为4.4℃/ s(在(3)期间获得的数据)。
- 使用optio正常熔融曲线循环95°C 5 s,斜坡速率为4.4°C / s; 65℃1分钟,升温速率为2.2℃/ s;和97°C,具有连续采集模式。
- 使用40°C的冷却循环30 s,斜率为1.5°C / s。
- 测定参数
- 选择SYBR模板。检查实验按钮中的程序参数。一旦装载板,可以开始测定,并且可以在测定运行时调整设置。
- 使用示例编辑器分配样本。选择Abs Quant作为工作流,并将样品指定为未知,标准或阴性对照。指定复制并填写每个重复的第一个的样本名称。添加浓度和单位到标准。
- 设置子集进行分析;这些在子集编辑器中分配。
- 要进行分析,请从“创建新分析”列表中选择Abs Quant / 2nd Derivative Max。导入外部保存的标准曲线(如果适用),然后按计算;该报告将包含所选信息。
- 要进行准确的绝对定量以确定拷贝数或浓度,请使用代表被检样品的标准曲线( 例如,从上述方法分离的细菌DNA)。由于准备标准曲线所需的线粒体DNA的量太高,无法用合理数量的组织达到,所以不要使用软件提供的拷贝数计算,而是检查交叉点(Cp)值以确定相对浓度的细胞器与样品中的核DNA相比。将这些相对量与总基因组DNA相比较(见代表性结果 )。测试来自完全光生长的两周龄小麦幼苗的总基因组DNA的5×10稀释的引物效率(代表性效率报告在th图2的传说)。
- qPCR反应设置
- 脉冲场凝胶电泳(PFGE)
注意:该协议基于制造商的指导方针,以执行PFGE来解决高分子量DNA。见材料表。- 准备凝胶和样品
- 遵循凝胶和样品制备准则,并适应可用的系统。
- 运行参数
- 按照设置电泳系统的指导原则,使用以下参数:初始切换时间为2秒,最终切换时间为13秒,运行时间为15小时,16分钟,V / cm为6,夹角为120度。
- 污渍和成像凝胶
- 用选择的染料( 例如溴化乙锭或合适的替代品)染色凝胶,并使用合适的凝胶文件系统进行成像。
- 准备凝胶和样品
- 根据制造商的说明书,使用1ng DNA作为DNA Library Prep Kit的输入。
- 条码并汇总样品进行排序,一次运行。按照制造商的指导进行排序。
注意:可以根据感兴趣的种类,期望的覆盖水平和用于对文库进行排序的平台来更改池和排序参数。例如,HiSeq通道具有比MiSeq通道更多的输出,因此可以复用更多的样本。序列较小的样本子集,以确定细胞器基因组的覆盖水平是否足以用于下游分析。- 使用FastQC 31检查读取质量,以确定数据所需的修剪和过滤的程度。
- 使用Trimmomatic 32或另一个可比较的程序修剪和过滤原始读取。使用以下设置:ILLUMINACLIP 2:30:10(删除适配器),LEADING 3,TRAILING 3,SLIDINGWINDOW 4:10和MINLEN 100。
- 映射质量过滤并适配器修剪配对末端(PE)读取到中国春线粒体(NCBI参考序列NC_007579.1 33),叶绿体(NCBI参考序列NC_002762.1 34),以及使用36 Bowtie2核35个参考基因组,具有以下设置:-I 0 -X 800 - 敏感。
- 将sam对齐文件转换为bam格式(samtools)并对bam文件进行排序。使用bam文件来计算基因组范围的覆盖率和床单的覆盖面积。用R绘图功能可视化结果。
- 开始之前要做的事情
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
本手册中提出的方案描述了从植物组织中丰富细胞器DNA的两种不同的方法。这里提出的条件反映了小麦组织的优化。 图1中描述了协议,组织输入和DNA输出中的关键步骤的比较。我们测试的DC协议的步骤遵循与先前描述的相似的条件( 图1A )。收获的组织必须新鲜处理,并进行差速离心和/或梯度以分离完整的细胞器。在细胞器被裂解之前,核DNA被去除,最后提取DNA并用于下游应用。相比之下,在MF方案中,植物组织可以在使用前收获和储存,并且不需要完整的细胞器。相反,核和细胞器DNA基于总DNA进行分馏 DNA的甲基化状态。两种方案产生大致相等量的细胞器DNA( 图1C )。关于组织输入的总体细胞器DNA输出,当组织受限时,MF方案是有利的,因为可以使用来自单一植物的小样品,并且可允许植物生长用于进一步分析。通常,在DC协议中,需要许多幼苗的所有空中组织,并且丢弃这些植物。然而,可以优化DC方法以特异性地丰富一种细胞器类型,而MF方法是不可能的。值得一提的是,每个协议的总时间大致相当,尽管MF方法中的实际操作时间较短。
这两种方法丰富了细胞质DNA,尽管线粒体和质体序列的比例不同:
“从两种方法(约50-100ng; 图1C )获得非常少量的纯化的细胞器DNA,以评估从DC和MF分离的DNA中的细胞器基因组富集和核基因组污染的水平在该测定中,三个扩增子( 即核特异性, ACTIN ,线粒体特异性, NAD3和相对丰度)的相对丰度 叶绿体特异性, PSBB )在总基因组DNA中进行评估,并从两种方法获得细胞器DNA级分( 图2 )。检测每个样品的定量循环(C q )值( 图2A ),并且由于C q被定义为来自靶扩增的荧光增加到背景荧光水平以上的PCR循环,C q和目标丰度具有反向关系。在直流样品,NAD3和PSBB的C- q是分别比〜ACTIN(其具有一个C Q〜36)前面17首〜15个循环(参见图2B对C的Q值和富集水平)。这相当于分别NAD3和PSBB,理论167,181-和47790倍富集, 相对于DC样品中的ACTIN ( 图2B ,参见图2的图例进行计算)。在总基因组DNA样品中, NAD3和PSBB相对于ACTIN的折叠浓缩分别仅为158和10,701。相对于核基因组DNA中的核扩增子,发现更丰富的细胞器扩增子并不奇怪,鉴于细胞器基因组以比核基因组37更多的拷贝数存在每个细胞,而细胞器数目peř小区可以根据组织类型或发育阶段38,39不同。总体而言,数据表明DC方法优先丰富线粒体,这是预期的,因为离心速度被优化以选择性分离线粒体并减少核和叶绿体“污染”。MF总gDNA的未甲基化部分还显示两种细胞扩增子的实质富集,并且预期保留这些靶的天然相对量。未甲基化部分中NAD3和PSBB相对于ACTIN的折叠富集分别为20,551和1,703,253 ( 图2A和2B )。在甲基化部分中, NAD3和PSBB相对于ACTIN的折叠浓缩分别为31和823,indi说明MBD2-Fc蛋白在甲基化核DNA的下拉是高效的。由于叶绿体扩增子在总基因组DNA(〜6 C q较早),甲基化级分(〜5 C q较早)和未甲基化级分(〜6 C q较早)样品中的线粒体扩增子丰度较高,这表明这些扩增子的天然丰度没有通过MDB2下拉基本上改变。由于对这些基因组进行了特异性测序的兴趣,我们重点关注非甲基化(细胞器)部分。然而,如果核基因组是主要兴趣,由于细胞器DNA“污染”的减少,MF和随后的甲基化部分的测序将产生比全基因组DNA测序更高的核基因组覆盖。
值得注意的是,如果qPCR不可用,则终点PCR(使用与qPCR相同的引物)提供质量细胞器纯度的评估。在这种情况下,纯的细胞器DNA样品将显示线粒体和质体扩增子的扩增,但在琼脂糖凝胶上没有检测到扩增核扩增子,而总基因组DNA显示所有三个引物组的扩增,如先前的研究11所示 , 12 。
从两种方法分离的细菌DNA适用于NGS:
修剪和清洁的PE测序读数(见步骤4.3)被映射到先前发表的小麦细胞参考基因组,并且用于绘制每个样品的读数量范围为约80万到1,100,000读数( 图3I )。从映射从头 Illumina测序结果读取到可用小麦叶绿体和线粒体基因组与qPCR的RES一致通过DC方法产生更丰富线粒体DNA的DNA( 图3A和3B ,分别为线粒体(mt)和叶绿体(cp)基因组的读数的〜80%和〜10%)和MF方法产生可能反映两种细胞器基因组的天然丰度的DNA( 图3A和3B ,分别为mt和cp基因组的读数的〜20%和〜80%)。在两种方法中,两种小麦细胞器基因组的理论覆盖率(参见图3的传奇计算)超过了100X的覆盖范围(对于来自MF方法的未甲基化部分中的叶绿体基因组的覆盖范围高达〜2000X),甚至当12个文库被复用时( 图3C和3D ;将该分析中包括的6个文库与另外6个文库进行汇总以进行单独的分析,共有12个文库汇集在单个测序通道中)。通过检查在特定深度覆盖的基因组以及每基础覆盖水平( 图3E- 3I ), 可以获得覆盖率的更详细的观点。对于MF方法,mt基因组的平均每基因覆盖率为〜300-450倍,cp基因组的平均每基因覆盖率为4000〜5,000倍。对于DC方法,mt和cp基因组的平均每基因覆盖率分别为〜900 - 1,300和〜500 - 700X。然而,mt和cp基因组中有一小部分具有极低或较高的覆盖率,而且在从任一种方法得到的细胞器DNA中都可以看到( 图3I )。高于平均覆盖率的区域可能对应于细胞器基因组之间的同源区域,低覆盖率的区域可能表示SNP或我们测序的品种之间的其他小变体和已发表的参考文献。为了支持这个概念,这些尖峰对于来自MF方法的mt DNA( 图3E和3I ),高覆盖率最为显着,这可能是由于该方法中cp基因组的高覆盖率。不可思议的是,在MF方法中,cP基因组的覆盖率比DC方法( 图3G和3H )更加不均匀,这可能是由于沿着cp DNA的MBD2-Fc下拉的轻微偏差。将需要进一步的实验来确定为什么会这样。无论如何,mt和cp基因组具有两种方法相对均匀的覆盖率,没有大面积的缺失覆盖,这可以通过检查在给定深度测序的基因组的分数来证明( 图3E -3H )。此外,两种基因组的覆盖程度被认为对于下游分析是足够的,如变体分析。如果认为有必要分析稀有变体,减少数量汇集的样本将达到更大的覆盖率。或者,在HiSeq通道上可以汇集更多数量的样品,同时获得甚至更高的测序深度,尽管牺牲序列长度,因为与PE300 MiSeq文库相反,HiSeq文库目前受限于PE150长度。
使用映射方法检查核基因组污染的水平,检查了PE读取映射类别。 PE读取可以以各种配置映射到参考基因组。当读取1和2以头对头的方式与参考对齐时,在两个配对之间具有一定的“预期”距离(基于库的平均插入大小,并且通常在映射软件中被指定为输入参数) ),这些PE读取据说是“一致地”映射的。相比之下,“不一致”的映射是伴侣以较小或超过预期的映射映射的情况参考基因组或替代配置中的图(头对尾或尾对尾)。如果只有一个配偶符合参考基因组,那么该PE读取被认为既不一致地或不一致地映射到参考基因组。在所有三种读取映射类别中,PE读数可以与参考基因组一致或多次。
对于DC和MF分离的细胞器DNA,读取到线粒体基因组的映射主要在一致的一次类别中( 图4A ),而以相对相等比例的读数映射到叶绿体基因组,一致地一次,并且一致地超过一次( 图4B ),可能是由于存在于叶绿体基因组中的大的反向重复以及极高的覆盖水平。然而,较少的PE读取映射到核基因组,并且在一个时间内主要映射了多于一次既不一致又不一致的时尚( 即只有一个伴侣能够映射)。这些最有可能将“脱靶”映射到核基因组中与细胞器基因组或错配组织区域同源的序列。只有少量的读数(<5%)一致地映射到核基因组,表明从DC或MF方法分离的细胞器DNA中核基因组污染水平较低( 图4C ),qPCR结果也反映了( 图2A )。来自中国春季非蛋白质组织的MBD2-Fc下拉后的核分数也进行了测序,以确定下拉在去除未甲基化DNA方面的效率。少于1%的核分馏衍生文库中的读数映射到细胞器参考基因组,而〜45%的读数映射到核基因组( 图4 )。然而,大多数读取以不和谐的方式映射,w这可能反映了小麦核参考基因组中错配和分裂的高水平。无论如何,结果表明,MBD2-Fc下拉在从甲基化核DNA中去除未甲基化的细胞器DNA方面是高效的。值得注意的是,由于这些方法产生的富集细胞的DNA含有线粒体和叶绿体序列的混合物,并且由于这些细胞器之间的古代基因转移产生的序列相似性保留在其基因组中,因此将读取的适当分配到具体生物信息学必须解决基因组。
叶组织病变不明显改变细胞器丰度:
传统上,为了降低酚类和淀粉的水平,植物线粒体DNA分离是优选的,这可能会干扰提取物n或下游应用13 。为了确定细胞器基因组富集水平是否可以通过生长条件改变或改善,两种病原体和非病原体组织都经过MF方案和测序。有趣的是,etiolation没有明显改变映射到细胞器参考基因组( 图3A和3B )或每个基础覆盖( 图3I )与非病态条件相比的读数百分比。我们还使用差异离心法分离细胞器DNA,同时用etiolated和non-etiolated组织,并且使用qPCR在不同组织之间发现很少的富集差异(数据未显示)。这表明更多的生理相关的非病原体组织可以用于细胞器测序研究,没有明显的富集变化。
质量控制建议MF DNA最适合长读序列:
随着长期阅读测序变得更易于研究人员访问,高分子量DNA的分离变得越来越重要。为了评估用任一种方法分离的完整性和质量的细胞器DNA,使用PFGE。总基因组DNA通常在PFGE中作为弥漫性涂片迁移,分子量由方案确定,DNA如何在提取后储存和处理。用基因组尖端分离的总基因组DNA应该超过50kb,这是使用PFGE进行验证的( 图5 ,泳道2)。来自基因组尖端的总基因组DNA用作微生物组合富集试剂盒的输入,以从细胞器DNA中分离核。分馏后获得的核级分确实在尺寸上减小,但保持在50kb左右( 图5 ,泳道4)。这不是苏令人惊奇的是,鉴于从MBD2-Fc结合珠洗脱的核级分比较粗糙的处理需要加热和蛋白酶K消化。由于质量有限,细胞器级分不在PFGE上运行,但随后用TapeStation分析显示DNA> 50 kb(数据未显示)。通过差速离心获得的细胞器DNA具有约20kb的平均质量,可能由延长的细胞器分离方案和随后的基于柱的DNA提取和浓缩引起。基于梯度的细胞器分离和替代的DNA提取方法可以保持较大的DNA片段大小。无论如何,如果在文库制备过程中注意,在本协议中获得的大小的DNA可用于产生10或15-kb测序读数。
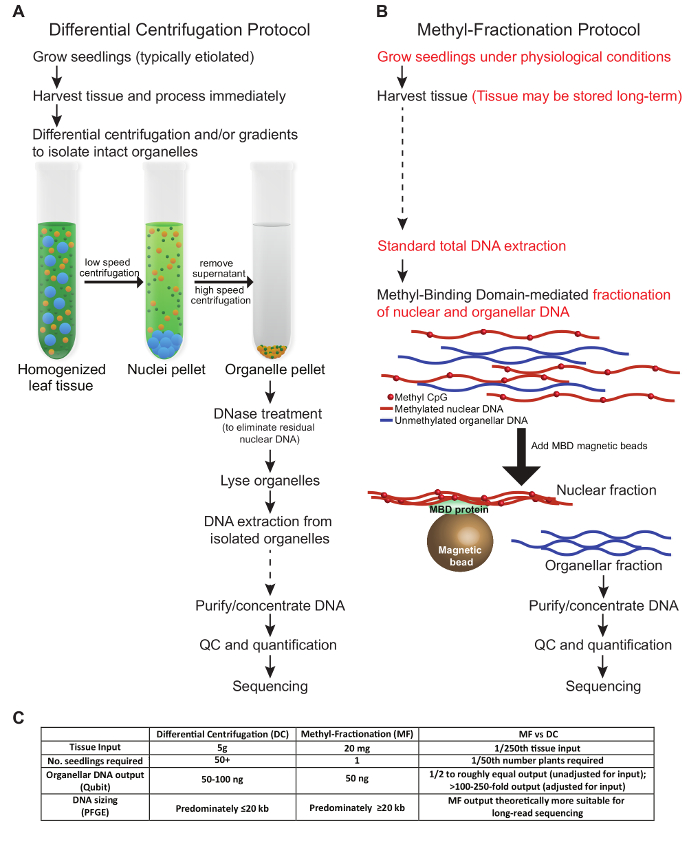
图1:两个Metho的比较视图ds浓缩植物细胞DNA。传统的DC协议( A )与MF协议( B )形成对照。建议避免冻结和解冻样品;然而,用虚线箭头( A和B )表示长期存储样品的步骤。协议之间的主要区别以红色( B )突出显示。 ( C )该表比较了组织输入,所需植物数,DNA输出和所得DNA大小的方法。 请点击此处查看此图的较大版本。

图2:使用两种方法分离的细菌DNA中核DNA污染的评估。 (
( B )该表显示了在( A )的图表中显示的C q值和相对于ACTIN的细胞器扩增子的折叠富集。 *浓缩浓度= 2 (Cq ACTIN - Cq Target) 。该公式假定每个引物组的完美效率为2,因为次要偏差每个引物组的离子从2可忽略不计,对计算和总体趋势几乎没有影响( ACTIN = 1.961, NAD3 = 1.95, PSBB = 1.989)。通过制备具有5个1:10稀释基因组DNA的标准曲线来评价引物效率。 请点击此处查看此图的较大版本。

图3:叶绿体和线粒体基因组的读取和理论覆盖。映射到线粒体( A )或叶绿体的读数百分比( B )中国春季参考基因组。中国春季线粒体( C )或叶绿体( D )参考基因的理论覆盖mes,假设基因组大小分别为450和135 kb,使用总读数和映射到不同基因组的读取百分比计算。来自MF方法( E和G )或DC法( F和H )的细胞器DNA覆盖的基因组范围分布。 E - H图中的数据来自中国春季样品,但所有其他样品均呈现类似趋势。 ( I )面板A - D中所有样品的平均,最低和最高基数覆盖。包括“E”的样品标记指定了样品,“NE”表示非质子样品。 DC表示用差速离心法分离的DNA,Unmethylated表示在用MBD2-Fc(MF方案)下拉之后在未甲基化部分中的DNA。标记为“克里斯”的样品表示小麦小麦'克里斯。' CS表示小麦小麦 “春天”的样品。注意:由于叶绿体,线粒体和细胞器基因组之间以及细胞器和核基因组之间的古代基因转移产生的核基因组之间的序列同源性,原始读数的一小部分可能映射到多个基因组。此外,该图中没有表示不映射到任何一个细胞器参考基因组的读数。因此,这里显示的百分比( A和B )不能达到100%。 请点击此处查看此图的较大版本。

图4:PE读取对小麦核基因组的映射。 PE类别百分比读取线粒体(A) ,叶绿体(B)或核(C)中国春季参考基因组的映射类型。 - E表示质子样品, - NE表示非质子样品。 DC表示用差示离心法分离的DNA,未甲基化表示在MF方案中与MBD2-Fc下拉之后的未甲基化部分中的DNA,并且甲基化指定在MBD2-Fc下拉之后的核级分。标记为“克里斯”的样品表示小麦小麦 '克里斯'。 CS表示小麦小麦 “中国春季”样品。未显示未映射的读取。 请点击此处查看此图的较大版本。
oad / 55528 / 55528fig5.jpg“/>
图5:使用PFGE检查DNA质量。将小麦总基因组DNA(泳道2),从差速离心(泳道3)获得的小麦细胞DNA和具有MBD2-Fc下拉方法(泳道4)的MF后的核部分在1%琼脂糖凝胶上进行PFGE, 1kb扩展梯用作标记(泳道1和5)。 请点击此处查看此图的较大版本。
| 缓冲区名称 | 食谱 | 笔记 | 方法 |
| STE缓冲液 | 400mM蔗糖,50mM Tris pH 7.8,20mM EDTA pH 8.0,0.6%(w / v)聚乙烯吡咯烷酮(PVP),0.2%(w / v)牛血清白蛋白(BSA),0.1%(v / v)β-巯基乙醇(BME) | 仅含蔗糖,Tris和EDTA的缓冲液混合物可以提前一个月提供并保持在4℃。在使用之前,PVP,BSA和BME应该新鲜加入所需量的缓冲液的等分试样中。 | 方法#1 |
| ST缓冲液 | 400mM蔗糖,50mM Tris pH 7.8,0.6%(w / v)聚乙烯吡咯烷酮(PVP),0.1%(w / v)牛血清白蛋白(BSA) | 仅含蔗糖和Tris的缓冲液混合物可以提前一个月提供并保持在4℃。请注意,ST缓冲液不含EDTA或BME,含有较低浓度的BSA。 | 方法#1 |
| DNase库存 | 2mg / ml DNase在0.15M NaCl中,浓度为2mg / ml | 在-20℃下储存200ul等分试样。制备DNase工作溶液(每个样品200μlDNA酶溶液)下表1。有关DNA酶消化的详细信息,请参阅下面的完整协议。 DNase工作液应准备新鲜。要停止DNase反应,需要400 mM EDTA pH 8.0溶液(终止反应所需的终浓度为0.2 M EDTA,详见完整方案)。 | 方法#1 |
| DNase工作解决方案 | 在ST缓冲液中0.25mg / ml DNase和20mM MgCl 2 | 准备新鲜,每个样品200ul。所显示的浓度用于最终反应体积,因此混合:62.5μl2mg / ml DNase(基于最终500μl反应体积),4μl1M MgCl 2 (基于200μlDNase溶液体积)和133.5μlST缓冲液最终体积为200μl。 | 方法#1 |
| 裂解缓冲液 | 20mM EDTA pH8.0; 10mM Tris pH 7.9; 500mM胍盐酸盐; 200mM NaCl; 1%Triton X-100; 0.5mg / ml溶解酶哈茨木霉 | 混合所有成分,除了裂解酶,并在室温下储存。应将新鲜的裂解酶加入到少量的等分试样中即可使用。 | 方法#2 |
表1:自制缓冲区和工作种群的配方。
| 集中工作表 | |||||||
| 样品名称 | 空装重量(g) | 填充装置重量(g) | 填充体积(ul,填充减空重量) | 第一次旋转后重量(20分钟*,g) | 体积第一次旋转(ul,充满减去空重量) | 第二次旋转后重量(X min *,g) | 第二次旋转后体积(ul,填充减空重量) |
| 请注意,实际回收的体积将比计算体积少几ul。 | |||||||
表2:浓缩工作表。
| 名称 | 基因组特异性 | 基因序列来源 | 序列(5' - 3') |
| Ta_ACTIN - F | 核 | Gramene脚手架IWGSC_CSS_1AS_scaff_3272162:10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | 核 | 与上述相同 | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | 线粒体 | NCBI登录号EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | 线粒体 | 与上述相同 | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | 叶绿体 | NCBI登录号KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | 叶绿体 | 与上述相同 | GCTGCCTGTTTCCTTGTAGTT |
表3:qPCR引物列表。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
迄今为止,大多数细胞器测序研究集中于传统的DC方法,以丰富特定的DNA。已经描述了从不同植物中分离细胞器的方法,包括苔藓40 ;单子叶植物如小麦15和燕麦11 ;和双子叶植物如拟南芥11 ,向日葵17和油菜籽14 。大多数协议集中在叶组织13,14,15,16,17,与一些已适用于多种组织类型,包括种子11。细胞器从原生质体的分离也已被证明41 。然而,这不适用于所有系统,当感兴趣的组织有限时也不可行。许多这些orga设计了Nellar分离方法以回收完整的细胞器,用于特定实验,如生理研究。这些方案是麻烦的,并且通常需要使用密度梯度,例如蔗糖或Percoll梯度,其在分离特定细胞器级分方面非常有效,但是需要大的组织输入( 即,超过5g和高于千克,取决于组织类型)。然而,可以通过改变旋转速度和密度梯度来优化DC方法以丰富特定细胞部分,例如线粒体或叶绿体。相比之下,MF方法需要的起始物质(20 mg)远少于线粒体和质体DNA,因为它们用于DNA提取的组织的相对丰度。然而,MF方案提供了用于分离混合的细胞器DNA的替代方法,并且特别有利于从少量的组织开始。
Ť o评估下列细胞器隔离样品纯度,大多数研究迄今为止只使用终点PCR和凝胶电泳11,12。这样可以对样品纯度进行公正的定性测量。然而,在琼脂糖凝胶上可能不可见低水平的扩增。很少的报告包括更多的质量控制量化措施,如qPCR 14 。为了对从两种方法分离的DNA样品纯度的定量评估,我们使用qPCR和测序来确定样品中残留的核DNA数量以及线粒体与叶绿体DNA的相对比例。在这里评估的两种方法都有效地去除核DNA。两种方法都产生线粒体和叶绿体DNA的混合物,尽管它们的比例不同。
据报道,生长在黑暗中的植物(etiolation)有助于促进由于酚类化合物的减少而进行的细胞分离然而,在这个比较中,我们没有发现在光生长样品上使用蚀刻组织的明显优势,虽然光生长时专业叶绿体的比例可能会更高,但总质量数反映在映射到叶绿体基因组的读数的比例在不同的光条件下是不变的,因此,对于下游功能分析,例如不同组织或不同胁迫下的异质性评估或表达分析,我们建议对在生理相关条件下生长的植物。
对于使用短读序列技术的应用,两种比较技术均可获得足够的DNA数量和质量。然而,为了实现单分子测序应用的> 20kb的长读数,需要更多量的更高质量的DNA。例如,理想情况下,>1μg纯粹的对于20-kb插入文库制备的内部低输入方案42 ,分子量> 20kb的中性小麦DNA是必需的。新用户开发的,低输入协议可以减少DNA的要求( 即,至50毫微克或甚至更少20),但挑战仍然具有高品质,高分子量DNA进入库制剂。大多数DNA大于20kb是重要的,因为较小的片段将被优先插入SMRTbell中,并且丢弃了文库43的大小分布。我们尝试了一些自制DNA提取方案和许多DNA提取的商业方案(未显示)。对于小麦叶组织,用市售试剂盒27,29得到的DNA的数量和质量,特别是长度之间的最佳平衡。取决于植物的种类和组织的兴趣,交替ve提取方案可能同样适合或更有成果。尽管如此,我们得出结论,大规模> 50kb的高分子量基因组DNA的总提取,然后用MBD2-Fc下拉法28分离 ,可以从有限的起始材料中长期读取测序。未来的工作应该测试用于长插入文库制备和随后长时间读取测序的分离后所需的起始材料的限制。最重要的是,这种方法可以提供一种强有力的方法来分离来自适合于长读取测序的单个叶子的子样本的DNA,而不需要全基因组扩增。我们预计这种方法将很容易适应于额外的组织类型,并广泛适用于其他植物物种。在组织量是限制性的情况下,例如在交叉方案中或在稀有组织类型中在各代进行测序的情况下,这将是特别有用的。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
作者宣称他们没有竞争的利益。
本出版物中提及的商品名称或商业产品仅用于提供具体信息,并不意味着美国农业部的建议或认可。 USDA是一个平等的机会提供者和雇主。
Acknowledgments
我们要承认美国农业部农业研究服务处和国家科学基金会(IOS 1025881和IOS 1361554)的资助。感谢R. Caspers进行温室维护和植物保养。我们还感谢明尼苏达大学基因组学中心,其中进行了Illumina图书馆的制备和测序。我们也感谢期刊编辑和四位匿名审稿人的意见,进一步加强了我们的稿件。我们也感谢经合组织与SK的合作,将这些协议项目与日本的同事整合起来。
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
