

Summary
La comparaison et l'optimisation de deux méthodes d'enrichissement d'ADN organellaire végétal sont présentées: centrifugation différentielle traditionnelle et fractionnement de l'ADNc total basé sur l'état de méthylation. Nous évaluons la quantité et la qualité d'ADN qui en résultent, démontrons la performance dans le séquençage de prochaine génération de lecture courte et discutons le potentiel d'utilisation dans le séquençage à une seule molécule à longue lecture.
Abstract
Les génomes organellaires végétaux contiennent de gros éléments répétitifs qui peuvent faire l'objet d'un couplage ou d'une recombinaison pour former des structures complexes et / ou des fragments sub-génomiques. Les génomes organellés existent également dans des mélanges à l'intérieur d'une cellule ou d'un type de tissu (hétéroplasmique) donné, et une abondance de sous-types peut changer tout au long du développement ou sous stress (déplacement sous-stoechiométrique). Les technologies de séquençage de la prochaine génération (NGS) sont nécessaires pour obtenir une compréhension plus approfondie de la structure et de la fonction du génome organellaire. Les études de séquençage traditionnelles utilisent plusieurs méthodes pour obtenir l'ADN organellaire: (1) Si une grande quantité de tissu de départ est utilisée, elle est homogénéisée et soumise à une centrifugation différentielle et / ou à une purification par gradient. (2) Si une quantité plus petite de tissu est utilisée ( c'est-à-dire, si les graines, le matériau ou l'espace est limité), le même processus est effectué comme dans (1), suivi d'une amplification complète du génome pour obtenir un ADN suffisant. (3) L'analyse de bioinformatique peut être utilisée pour seqL'ADN génomique total et d'analyser les lectures organiques. Toutes ces méthodes ont des défis et des compromis inhérents. Dans (1), il peut être difficile d'obtenir une telle quantité importante de tissu de départ; Dans (2), l'amplification du génome entier pourrait introduire un biais de séquençage; Et dans (3), l'homologie entre génomes nucléaires et organellars pourrait entraver l'assemblage et l'analyse. Dans les plantes à grands génomes nucléaires, il est avantageux d'enrichir l'ADN organellaire pour réduire les coûts de séquençage et la complexité des séquences pour les analyses de bioinformatique. Ici, nous comparons une méthode de centrifugation différentielle traditionnelle avec une quatrième méthode, une approche CpG-méthyle modifiée, pour séparer l'ADN génomique total en fractions nucléaires et organellaires. Les deux méthodes produisent suffisamment d'ADN pour le NGS, l'ADN qui est très enrichi pour les séquences organellaires, mais à des rapports différents dans les mitochondries et les chloroplastes. Nous présentons l'optimisation de ces méthodes pour le tissu des feuilles de blé et discutons des avantages majeurs et dDes avantages de chaque approche dans le contexte de l'apport d'échantillon, la facilité du protocole et l'application en aval.
Introduction
Le séquençage du génome est un outil puissant pour disséquer la base génétique sous-jacente des traits importants de la plante. La plupart des études de séquençage génomique se concentrent sur le contenu du génome nucléaire, car la majorité des gènes sont situés dans le noyau. Cependant, les génomes organellars, y compris les mitochondries (à travers les eucaryotes) et les plastides (dans les plantes, la forme spécialisée, le chloroplaste, les travaux de la photosynthèse) apportent des informations génétiques importantes essentielles au développement de l'organisme, à la réponse au stress et à la condition physique globale 1 . Les génomes organellaires sont généralement inclus dans les extractions totales d'ADN destinées au séquençage du génome nucléaire, bien que des méthodes pour réduire les nombres d'organelles avant l'extraction de l'ADN soient également utilisées 2 . De nombreuses études ont utilisé les résultats de séquençage des extractions totales d'ADNg pour assembler les génomes organellaires 3 , 4 , 5 ,Xref "> 6 , 7. Cependant, lorsque la cible de l'étude est de se concentrer sur les génomes organellars, l'utilisation de l'ADNc total augmente les coûts de séquençage car de nombreuses lectures sont" perdues "pour les séquences d'ADN nucléaire, en particulier dans les plantes à grands génomes nucléaires De plus, en raison de la duplication et du transfert de séquences organellaires dans le génome nucléaire et entre les organites, résoudre la position de cartographie correcte des lectures de séquençage au génome approprié est un défi bioinformatique 2 , 8. La purification des génomes organellaires du génome nucléaire est une Stratégie pour réduire ces problèmes. D'autres stratégies de bioinformatique peuvent être utilisées pour séparer la carte à des régions d'homologie entre les mitochondries et les chloroplastes.
Alors que les génomes organellaires provenant de nombreuses espèces de plantes ont été séquencés, on sait peu à peu l'ampleur de la diversité du génome organellaireDisponible dans les populations sauvages ou dans les piscicultures cultivées. Les génomes organellés sont également connus pour être des molécules dynamiques qui subissent un réaménagement structurel important en raison de la recombinaison entre les séquences répétées 9 . En outre, plusieurs copies du génome organellaire sont contenues dans chaque organelle, et des organites multiples sont contenues dans chaque cellule. Toutes les copies de ces génomes ne sont pas identiques, ce qui est connu sous le nom d'hétéroplasme. Contrairement à l'image canonique des «cercles maîtres», il existe maintenant une preuve croissante d'une image plus complexe des structures du génome organellaire, y compris les cercles sub-génomiques, les chromosomes linéaires, les concatémas linéaires et les structures ramifiées 10 . L'assemblage des génomes organellaires végétaux est encore plus compliqué par leurs dimensions relativement importantes et leurs répétitions inversées et directes importantes.
Protocoles traditionnels pour l'isolement organellaire, la purification de l'ADN et la génomique subséquente Le séquençage est souvent encombrant et nécessite de gros volumes d'entrée de tissus, avec plusieurs grammes à plus de centaines de grammes de tissu de feuilles jeunes nécessaires comme point de départ 11 , 12 , 13 , 14 , 15 , 16 , 17 . Cela rend le séquençage du génome organellaire inaccessible lorsque le tissu est limité. Dans certaines situations, les quantités de semences sont limitées, par exemple lorsqu'elles doivent s'effectuer sur une base générationnelle ou sur des lignes stériles masculines qui doivent être maintenues par passage. Dans ces situations, l'ADN organellaire peut être purifié puis soumis à une amplification complète du génome. Cependant, l'amplification du génome complet peut introduire un biais significatif de séquençage, ce qui est un problème particulier lors de l'évaluation de la variation structurelle, des structures sous-génomiques et des niveaux d'hétéroplasmie> 18. Les progrès récents dans la préparation de la bibliothèque pour les technologies de séquençage à lecture courte ont permis de surmonter les obstacles à faible intrusion pour éviter l'amplification du génome entier. Par exemple, le kit de préparation de la bibliothèque Illumina Nextera XT permet d'utiliser aussi peu que 1 ng d'ADN comme entrée 19 . Cependant, les préparations de bibliothèques standard pour les applications de séquençage à lecture prolongée, telles que les technologies de séquençage PacBio ou Oxford Nanopore, nécessitent encore une quantité relativement élevée d'ADN d'entrée, ce qui peut constituer un défi pour le séquençage du génome organellaire. Récemment, de nouveaux protocoles de séquençage conçus par l'utilisateur et à lecture longue ont été développés pour réduire les quantités d'intrants et aider à faciliter le séquençage du génome dans les échantillons où l'obtention de microgrammes d'ADN est difficile 20 , 21 . Cependant, l'obtention de fractions organellaires pures et de masse moléculaire élevée pour alimenter ces préparations de bibliothèque reste un défi.
Nous avons cherchéO Comparer et optimiser les méthodes organiques d'enrichissement et d'isolement de l'ADN appropriées pour les NGS sans besoin d'amplification du génome entier. Plus précisément, notre objectif était de déterminer les meilleures pratiques pour enrichir l'ADN organellaire de masse moléculaire élevée à partir de matériaux de départ limités, comme un sous-échantillon de feuille. Ce travail présente une analyse comparative des méthodes à enrichir pour l'ADN organellaire: (1) un protocole de centrifugation différentielle traditionnel modifié par rapport à (2) un protocole de fractionnement de l'ADN basé sur l'utilisation d'une méthode de transfert de protéines de domaine CpG-liaison au méthyle d'ADN disponible dans le commerce 22 appliqué au tissu végétal 23 . Nous recommandons les meilleures pratiques pour l'isolement de l'ADN organellaire à partir de feuilles de blé, ce qui peut être facilement étendu à d'autres plantes et types de tissus.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Génération de matériaux végétaux pour l'isolement organellaire et l'extraction d'ADN
- Croissance standard des semis de blé
- Plantez des graines dans de la vermiculite dans de petites casseroles carrées avec 4 à 6 graines par coin. Transférer dans une serre ou une chambre de croissance avec un cycle léger de 16 h, 23 ºC jour / 18 ºC de nuit.
- Eau les plantes chaque jour. Fertiliser les plantes avec ¼ c. À thé d'engrais granulés 20-20-20 NPK après germination et 7 jours après germination.
- Étiologie alternative des semis de blé
- Suivez l'étape 1.1, mais placez les pots dans une chambre de croissance sombre, 23 ° C pendant 16 h / 18 ºC pendant 8 h. Alternativement, couvrez les plantes dans la serre ( p. Ex., Avec un récipient de stockage, mais une ventilation appropriée doit être maintenue).
- Croissance et collecte des tissus
- Cultivez les plantes pendant 12 à 14 jours. Pour la plupart des génotypes de bléS, 75 - 100 plants proposent environ 10 à 12 g de tissu, ce qui est suffisant pour deux extractions organellaires en utilisant la méthode de centrifugation différentielle (section 2); Une seule plante est nécessaire si l'on utilise l'approche par pulvérisation à base de méthylation de CpG d'ADN pour fractionner l'organelaire à partir d'ADN nucléaire (section 3).
- Si l'on utilise l'approche de centrifugation différentielle, collecter le tissu frais et procéder immédiatement au traitement des échantillons, comme décrit dans la section 2.
- Si l'on utilise l'approche CpG-methyl pulldown, récoltez des sections de 20 mg de tissu de feuilles jeunes dans des tubes de microcentrifugeuse (utilisez des tissus standard ou étiolés, voir les résultats représentatifs ). Gélose instantanée sur l'azote liquide et congélation à -80 ºC jusqu'à l'utilisation. Procédez au fractionnement progressif de l'ADN, comme décrit dans la section 3.
2. Méthode n ° 1: Extraction d'ADN utilisant une centrifugation différentielle (DC)
REMARQUE: le diffUn protocole de centrifugation erroné a été modifié à partir de deux publications qui optimisent les conditions pour isoler les deux organelles mais enrichissent les mitochondries 17 , 24 . Le protocole résultant nécessite moins de temps et utilise moins de produits chimiques toxiques que les méthodes précédentes. Plus précisément, nous avons apporté des modifications aux tampons et aux étapes de lavage, y compris l'addition de polyvinylpyrrolidone (PVP) au tampon d'extraction STE et l'élimination de l'étape finale de lavage dans le tampon NETF, qui contient du fluorure de sodium (NaF).
Attention: La préparation et l'utilisation du tampon STE doivent être effectuées sous une hotte chimique avec un équipement de protection individuelle approprié, car ce tampon contient du 2-mercaptoéthanol (BME).
- Les choses à faire avant de commencer
- Assurez-vous que tout l'équipement est extrêmement propre, et autoclavez tout équipement pouvant être autoclavé ( p. Ex., Bouteilles de meulage , centraux à grande vitesseTubes de fuge, etc. ).
REMARQUE: Des conseils de filtrage sont recommandés pour toutes les étapes nécessitant une pipetage afin d'éviter toute contamination croisée. - Voir la liste des équipements et des réactifs requis et préparer les tampons requis et les stocks de travail pour la méthode no 1 ( tableau 1 ). Refroidir les blocs de broyage cryogénique à -20 ºC et les rotors et tampons à 4 ºC, mettre la microcentrifugeuse à 4 ºC et allumer un bain-marie à 37 ºC.
- Assurez-vous que tout l'équipement est extrêmement propre, et autoclavez tout équipement pouvant être autoclavé ( p. Ex., Bouteilles de meulage , centraux à grande vitesseTubes de fuge, etc. ).
- Isolation des organites
- Récoltez 5 g de tissu frais et rincez-le dans de l'eau stérile et froide dans un bécher refroidi sur glace.
REMARQUE: conservez toujours les échantillons sur de la glace pendant toutes les opérations et les transports depuis et vers les centrifugeuses, les hottes, etc. Vous pouvez également travailler dans une chambre froide s'il y a accès à suffisamment d'espace et d'équipement pour exécuter le protocole. - À l'aide de ciseaux, couper des feuilles dans des morceaux de ~ 1 cm directement dans un tube de 50 ml contenant deux broyage en céramiqueDes cylindres.
REMARQUE: Nettoyer ou changer les ciseaux entre les échantillons afin d'éviter toute contamination croisée. - S'il n'y a pas d'homogénéisateur de tissu, utilisez un mortier et un pilon et suivez les étapes 2.2.4 à 2.2.9.
- Couper le tissu de feuilles dans un mortier pré-refroidi sur glace. Moudre les échantillons pendant 2 à 3 min dans 15 ml de STE (dans la hotte).
- Verser le tampon (laisser le tissu dans le mortier) à travers un entonnoir contenant une couche de tissu de filtration pré-humide et stérile (taille de pore de 22 à 25 μm, voir le protocole principal pour plus de détails) dans un autre tube de 50 ml . Ajouter un supplément de 10 ml de STE sur le mortier et le pilon et homogénéiser à nouveau.
- Verser le tissu homogénéisé et le tampon dans le même entonnoir. Rincez le mortier et le pilon avec 10 ml de STE et versez-le dans l'entonnoir. Pressez et essorez le tissu de filtration dans l'entonnoir pour récupérer autant de liquides que possible.
REMARQUE: Changer les gants entre les échantillons afin d'éviter toute contamination croisée. Continuez avec le proTocol à l'étape 2.2.10.
- Ajouter 20 ml de STE (dans la hotte) à chaque tube de 50 ml.
- Placez les échantillons dans des blocs de meulage cryogéniques pré-refroidis dans un appareil de meulage de tissu et broyez les échantillons pendant 2 x 30 s à 1750 tr / min. Faites pivoter les positions de l'échantillon et placez les échantillons sur de la glace pendant environ 1 minute entre les broyages.
REMARQUE: Un mortier et un pilon, un mélangeur ou un autre dispositif de broyage / homogénéisation de tissu peuvent être utilisés dans cette étape. Cependant, chaque méthode affectera la qualité de l'ADN résultante à différents degrés et, par conséquent, la longueur et la qualité de l'ADN devraient être évaluées avant de continuer avec les applications en aval. - Insérez un entonnoir dans un tube propre de 50 ml placé dans de la glace. Placez une couche de tissu de filtration dans l'entonnoir et pré-mouillez-le avec 5 ml de STE. Ne pas jeter l'écoulement.
- Verser le tissu homogénéisé dans l'entonnoir. Rincer le tube de broyage avec 15 ml de STE, récapituler et inverser le tube pour rincer les murs et le couvercle, et verser dans le champEl.
- Retirez délicatement les pierres de céramique, puis serrez et essorez le linge de filtration dans l'entonnoir.
REMARQUE: Changer les gants entre les échantillons afin d'éviter toute contamination croisée. - Enrouler les bouchons de tubes avec du parafilm pour éviter les déversements. Centrifuger à 2 000 xg pendant 10 min à 4 ºC.
- Aspirer soigneusement le surnageant à l'aide d'une pipette sérologique (éviter de perturber la pastille) et la placer dans un tube de centrifugation à grande vitesse de 50 mL (si les tubes n'ont pas de joints d'étanchéité étroits, enrouler les bouchons avec du parafil pour éviter les déversements). Jeter les pastilles.
- Équilibrer les tubes à 0,1 g en utilisant STE et centrifuger le surnageant résultant pendant 20 min à 18 000 xg et 4 ºC. Pour équilibrer les tubes, placez un petit bécher de glace sur la balance, tuez l'échelle et pesez les échantillons sur de la glace pour les garder au froid. Sinon, utilisez une balance et une hotte dans une chambre froide.
- Jeter le surnageant. Ajouter 1 ml de ST à la pastille et reprendre doucement nousAvec un pinceau doux. Ajouter 24 mL de ST (volume final de 25 mL) et mélanger / tourbillonner ( c.-à-d. Presser le pinceau sur le côté du tube pour enlever tout le liquide).
- Équilibrez les tubes à moins de 0,1 g en utilisant ST. Centrifuger pendant 20 min à 18 000 xg et 4 ºC. Pendant ce temps, préparez la solution DNaseI (voir le tableau 1 pour les recettes de stock et de solution de travail). Pour chaque échantillon, faire une aliquote de 200 μL dans un tube de 1,5 mL.
- Jeter le surnageant, nettoyer le tube et remettre en suspension la pastille (toujours dans un tube centrifuge à grande vitesse) dans 300 μL de ST à l'aide d'un pinceau doux. Placez le pinceau dans le tube de 1,5 mL préalablement préparé contenant 200 μL de solution DNaseI et faites tourbillonner le pinceau pour éliminer toute pastille résiduelle collée dans la brosse. Pipettez la solution DNaseI dans le tube centrifuge à grande vitesse et tournez doucement pour mélanger.
- Incuber à 37 ° C pendant 30 min dans un bain-marie (envelopper le parafilm autour de la partie supérieure du tube pour éviter la fuite de condensationG dans le capuchon). Mélanger délicatement en remuant 2 fois pendant l'incubation.
- Pipeter doucement le mélange de pastilles hors du tube à l'aide d'une pointe de pipette avec un grand orifice et le placer dans un tube à faible liant de 1,5 ml. Ajouter 500 μL d'EDTA 400 mM, pH 8,0, au tube de centrifugation à grande vitesse et à une pipette douce pour éliminer toute la pastille résiduelle du tube. Transférer l'EDTA sur le même tube à 1,5 litre sous forme de pellet et mélanger délicatement par inversion.
- Centrifuger à 18 000 xg pendant 20 min à 4 ºC. Jeter le surnageant, tacher le tube et utiliser à la fois pour isoler l'ADN. Si nécessaire, congeler des granulés à -20 ºC, mais cela peut entraîner une réduction du rendement, car DNase résiduelle peut dégrader l'échantillon d'ADN s'il n'est pas traité immédiatement.
- Récoltez 5 g de tissu frais et rincez-le dans de l'eau stérile et froide dans un bécher refroidi sur glace.
- Extraction d'ADN à partir d'organelles isolées à l'aide d'une approche commerciale à base de colonne
REMARQUE: consultez le manuel du kit pour le protocole complet 25 , et voir ci-dessous les modifications. PrIl est préférable de passer directement de l'isolement organellaire à l'extraction de l'ADN. La congélation et la décongélation répétées réduiront la taille des fragments d'ADN et conduiront à une dégradation de l'ADN par DNaseI résiduelle. Limiter le vortex ou le pipettage vigoureux, car cela peut cisailler l'ADN. L'utilisation de tubes de microcentrifugation à faible liaison est recommandée pour maximiser la récupération de l'ADN.- Procédure d'extraction d'ADN
REMARQUE: lisez le protocole commercial détaillé 25 avant de commencer à vous assurer que les tampons sont correctement fabriqués / stockés et que les procédures de spin-column sont comprises.- Ajouter 180 μL de tampon ATL directement dans le tube avec la pastille (décongelée si préalablement congelée et équilibrée à température ambiante sur la table).
- Procédez à l'étape 3 dans le protocole pour "Purifications d'ADN à partir des tissus" dans le manuel du kit, avec les modifications suivantes: une lyse de 30 minutes à l'étape 3, incluent la digestion facultative de la RNase A et élucient dans 3 x 200 μL d'AE ( Chacun dans un seParate tube puis combine les élutions).
- Enregistrez une aliquote (au moins 20 μL) pour qPCR (voir étape 4.1). Pour quantifier avant la concentration, économisez 1 μL supplémentaire pour la quantification à haute sensibilité.
- Si désiré, procéder à la concentration de l'échantillon.
- Procédure d'extraction d'ADN
- Concentration d'échantillon avec des unités de filtrage commerciales
REMARQUE: voir le protocole commercial 26 pour plus de détails. En fonction de l'utilisation en aval, il se peut qu'il ne soit pas nécessaire d'effectuer une concentration d'échantillon ( p. Ex., Pour la PCR en bout de point et les applications qPCR). Cependant, pour la construction de la bibliothèque NGS, il sera probablement nécessaire de concentrer l'ADN organellaire dilué obtenu après l'extraction de l'ADN.- Procédure de colonne de concentration
- Pré-peser soigneusement (voir le tableau 2 ) l'unité de filtre vide (sans tube) sur un morceau de papier de travail propre sur une balance analytique numérique. Notez le poids.
- PiPette les élutions combinées dans l'unité de filtrage et pesez-les soigneusement.
REMARQUE: Le manuel commercial 26 indique que le volume maximal de l'unité de filtrage est de 500 μL, mais jusqu'à 575 μL peuvent être ajoutés à l'unité à la fois sans débordement. - Placez soigneusement l'unité de filtre remplie dans un tube (fourni avec les colonnes). Centrifuger à 500 xg pour le temps désiré pour atteindre le volume de concentré requis. Pour un volume d'échantillon de ~ 575 μL, un spin de 20 minutes entraînera habituellement un volume de concentré de 15 à 30 μL.
- Retirez le filtre du tube et pesez à nouveau. Utilisez le tableau pour déterminer si le volume désiré du concentré a été atteint. Sinon, centrifuger à nouveau à 500 xg pendant plus de temps et pesez encore; Répétez jusqu'à ce que le volume désiré du concentré soit atteint.
- Placez un nouveau tube (fourni avec les colonnes) sur le dessus de l'unité de filtrage et inversez. Centrifuger pendant 3 min à 1000 xg pour transférer le coCentré sur le tube.
- Déterminez le volume récupéré. Ce sera généralement de ~ 3 à 5 μL de moins que le volume calculé, en raison de la rétention du filtre. Si trop concentré, diluer avec de l'eau stérile ou TE pour obtenir le volume désiré.
- Quantifier l'ADN en utilisant une quantification de haute sensibilité (selon les instructions du fabricant).
- Procédure de colonne de concentration
3. Méthode n ° 2: Approche de fractionnement de méthyle (MF) pour enrichir l'ADN organellaire de l'ADN génomique total
NOTE: Ce protocole a été modifié à partir d'un protocole d'extraction d'ADN Genomic Tip Kit développé par l'utilisateur pour les plantes et les champignons 27 et le protocole commercial Microbiome DNA Enrichment Kit 28 . En théorie, tout protocole d'isolement d'ADN qui donne un ADN de masse moléculaire élevée peut être utilisé pour le déroulage. Pour un séquençage à lecture courte, toute extraction produisant principalement des fragments de 15 kb est adéquate pour une utilisation dans le déroulage. Pour leNg-read sequence, des fragments plus importants peuvent être souhaitables. Par conséquent, nous avons optimisé ce protocole pour produire un ADN de poids moléculaire élevé.
- Isolation de l'ADN total
REMARQUE: voir la liste des équipements requis et des réactifs et préparer les tampons requis et les stocks de travail pour la méthode no 2 ( tableau 1 ). Ajouter des enzymes de lysing au stock de tampon de lyse pour obtenir la solution de traitement du tampon de lyse. Allumez le thermomixer et réglez-le à 37 ° C. Allumez le bain-marie à 50 ° C et placez le tampon QF dans le bain. Placer 70% d'EtOH dans le congélateur et mettre la microcentrifugeuse à 4 ° C.- Extraction totale d'ADN à l'aide de colonnes commerciales d'extraction d'ADN
REMARQUE: Avant de commencer, lisez le manuel commercial 29 pour des informations détaillées concernant l'utilisation des colonnes d'échange d'anions par gravité. Les colonnes peuvent être configurées à l'aide d'un rack spécialisé ou placées sur les tubes à l'aide des bagues en plastique fournies. Toutes les étapes, y compris le gDes astuces d'émotion, devraient être autorisées à se faire par écoulement par gravité, et le liquide résiduel ne devrait PAS être forcé.- Moudre 20 mg de tissu congelé dans de l'azote liquide dans un tube à 2 ml à faible liant en utilisant des pilons à broyer portatifs conçus pour des tubes de 2 ml.
- Ajouter 2 mL de solution de tampon de lyse (les tubes seront très complets).
- Incuber dans un thermomixer à 37 ° C pendant 1 h avec une agitation douce à 300 tr / min. Si un thermomixer n'est pas disponible, l'incubation sur un bloc de chaleur et le mélange par un léger flicking toutes les 15 minutes est une alternative appropriée.
- Ajouter 4 μL de RNase A (100 mg / mL, concentration finale de 200 μg / mL). Inverser pour mélanger et incuber dans un thermomixer pendant 30 min à 37 ° C, avec une agitation douce à 300 tr / min.
- Ajouter 80 μl de protéinase K (20 mg / ml, concentration finale de 0,8 mg / mL), inverser pour mélanger et incuber dans un thermomixer pendant 2 h à 50 ° C, avec une agitation douce à 300 tr / min.
- Centrifuger pendant 20 min à 4 ° C et 15.000 xg pour granuler les débris insolubles.
- Pendant que les échantillons sont centrifugés, équilibrer les colonnes avec 1 mL de Buffer QBT et laisser la colonne s'effondrer par écoulement par gravité.
- Utilisez une pointe de pipette à large diamètre pour appliquer rapidement l'échantillon (éviter la pastille) à la colonne équilibrée et lui permettre de circuler complètement dans la colonne. Si l'échantillon devient trouble, filtrer ou centrifuger à nouveau avant l'application sur la colonne (voir le manuel commercial pour les détails 29 ).
- Une fois que l'échantillon est entré dans la résine, lavez la colonne avec 4 x 1 mL de tampon QC.
- Suspendre la colonne sur un tube de microcentrifugeuse propre, à 2 ml, à faible liaison. Eluire l'ADN génomique avec 0,8 ml de Buffer QF préchauffé à 50 ° C.
- Précipiter l'ADN en ajoutant 0,56 ml (0,7 volume de tampon d'élution) d'isopropanol à température ambiante à l'ADN élué.
- Mélanger par inversion (10X) et centrifuger immédiatement pendant 20 min à 15 000 xg et 4 ° C. Se soucierEnlever complètement le surnageant sans perturber la pastille vitreuse, lâchement attachée.
- Laver la pastille d'ADN centrifugée avec 1 ml d'éthanol froid à 70%. Centrifugez pendant 10 min à 15 000 xg et 4 ° C.
- Retirez soigneusement le surnageant (soyez prudent avec cette étape également) sans perturber la pastille. Sécher à l'air pendant 5 à 10 min et ré-endiguer l'ADN dans 0,1 ml de tampon d'élution (EB). Dissoudre l'ADN pendant une nuit à température ambiante. Évitez les pipettes, ce qui peut cisailler l'ADN.
- Quantifier les échantillons en utilisant un dosage de quantification d'ADN à haute sensibilité (selon les instructions du fabricant).
- Extraction totale d'ADN à l'aide de colonnes commerciales d'extraction d'ADN
- Fractionnement à base de perles d'ADN méthylé et non méthylé
NOTE: Une publication récente a démontré l'utilisation d'un kit 28 disponible dans le commerce qui profite d'une approche déroulante en utilisant une protéine de domaine de liaison au méthyle spécifique de CpG fusionnée au fragment Fc IgG humain (protéine MBD2-Fc) à la fractionA mangé des génomes organellaires de plantes (non méthylés) à partir de génome nucléaire (fortement méthylé) 23 . L'efficacité de fractionnement dans les échantillons de blé n'a pas été préalablement testée à l'aide de ce kit MF commercial 28 .- Les choses à faire avant de commencer
- Préparez à nouveau 80% d'éthanol (au moins 800 μL par réaction). Réglez le tampon de liaison / lavage 5x pour décongeler sur la glace et préparez 5 mL de 1x tampon par échantillon (diluer le tampon 5X avec de l'eau stérile sans nuclease et garder la glace pendant le protocole).
- Préparer des billes magnétiques liées à la protéine MBD2-Fc
- Préparez le nombre requis de jeux de talons. Évaluez les réactions à utiliser entre 1 et 2 μg d'ADN d'entrée totale, nécessitant 160 à 320 μl de perles. Notez que les réactions énumérées ci-dessous sont pour 1 μg d'ADN d'entrée totale, donc elles nécessitent 160 μL de perles. Échelle les réactions selon les besoins.
- À l'aide de pointes à puits larges, faites pipeter doucement la protéine A magnétique BMélanger les boules de mouton vers le haut et vers le bas pour créer une suspension homogène. En variante, tourner doucement le tube de perles pendant 15 min à 4 ° C.
REMARQUE: Ne pas tourbillonner les perles. - Procédez selon les instructions selon les instructions du fabricant 28 .
- Capture d'ADN nucléaire méthylé
- Pour chaque échantillon individuel, ajouter 1 μg d'ADN d'entrée à un tube contenant 160 μL de billes magnétiques liées à MBD2-Fc.
- Ajouter 5 fois le tampon de liaison / lavage en fonction du volume de l'échantillon d'ADN pour une concentration finale de 1x (volume de 5x bind / tampon de lavage pour ajouter (μL) = volume d'ADN d'entrée (μL) / 4). Pipettez l'échantillon vers le haut et vers le bas quelques fois pour le mélanger en utilisant une pointe de pipette à large diamètre.
- Faire tourner les tubes à température ambiante pendant 15 min. Pipettez doucement les échantillons avec une pointe de pipette à large diamètre et faites passer les échantillons 2 à 3 fois tout au long de l'incubation pour éviter l'agglomération des bourrelets.
REMARQUE: le pipettage et le flickiNg est essentiel pour assurer un effondrement efficace de l'ADN méthylé.
- Collecte d'ADN organellaire enrichi et non méthylé
- Tourner brièvement le tube contenant l'ADN et le mélange de cordons magnétiques liés à MBD2-Fc. Placez le tube sur un support magnétique pendant au moins 5 minutes pour collecter les perles sur le côté du tube. La solution devrait apparaître claire.
- En utilisant des pointes à large éventail, retirez soigneusement le surnageant nettoyé sans perturber les perles. Transférer le surnageant (contient un ADN non méthylé, enrichi en organelle) à un tube de microcentrifugeuse propre, à faible liaison et à 2 mL. Rangez cet échantillon à -20 ou -80 ° C, ou passez directement à l'étape 3.2.6 pour la purification.
- Elute a capturé l'ADN nucléaire à partir des billes magnétiques liées à MBD2-Fc
- Si la fraction nucléaire est également souhaitée, suivre les instructions du fabricant 28 pour éluer l'ADN nucléaire à partir des billes magnétiques liées à MBD2-Fc; Purifier comme décrit à l'étape 3.2.7.
- Purification d'acide nucléique à base de perles
- Assurez-vous que les grains de purification sont à température ambiante et sont bien mélangés. Procédez avec le protocole selon les instructions du manuel MF kit 28 .
REMARQUE: l'échantillon peut maintenant être utilisé pour la construction de la bibliothèque NGS ou une autre analyse en aval.
- Assurez-vous que les grains de purification sont à température ambiante et sont bien mélangés. Procédez avec le protocole selon les instructions du manuel MF kit 28 .
4. Quantification d'échantillon et contrôle de qualité
- QPCR analyse pour évaluer l'enrichissement organellaire
REMARQUE: les paramètres de réaction et d'analyse du qPCR répertoriés ici ont été conçus pour être utilisés sur un Roche LightCycler 480 et peuvent être ajustés pour différents équipements et réactifs. Si qPCR n'est pas disponible, la PCR et la visualisation finale sur un gel d'agarose peuvent être utilisées comme mesure qualitative de la pureté de l'échantillon, en utilisant les mêmes amorces et les mêmes conditions décrites ici. Les tailles d'amplicon seront de ~ 150 pb pour tous les ensembles d'amorces. Voir le tableau 3 pour la séquence d'amorcesCes et les jumelages.- Configuration de la réaction qPCR
- Pour mettre en place une réaction individuelle de 20 μL de qPCR, faites pipeter soigneusement le suivant dans un puit unique d'une plaque qPCR à 96 puits: 10 μL de 2x SYBR Green I Master; 2 μL du mélange d'amorces avant et arrière de 10 μM (pour une concentration finale de 0,5 μM); 2 μL de gabarit (dans la plage de la courbe standard); Et 6 μl de H 2 O stérile et exempt de nucléases. Pour réduire les erreurs de pipetage, il est préférable de créer un mélange maître avec tous les composants de réaction sauf le modèle. Ajoutez le mélange maître à la plaque qPCR, puis ajoutez le modèle d'intérêt pour chaque puits. Trois répétitions techniques pour chaque échantillon devraient être effectuées pour minimiser les effets de l'erreur de pipettage.
NOTE: En fin de compte, le rapport des cycles de quantification nucléaire à organellaire est comparé entre les échantillons, de sorte que de légères différences de concentration sont acceptables. Cependant, les concentrations d'ADN devraient être à peu près dans la limite deCh autre. - Sceller la plaque avec un film d'étanchéité qPCR de haute qualité. Mélanger doucement les échantillons, en prenant soin d'éviter la création de bulles. Tourner brièvement la plaque pendant 2 min à 4 ° C pour collecter l'échantillon et éliminer les petites bulles.
- Chargez la plaque dans la machine. Exécutez le programme qPCR selon les directives énumérées ci-dessous.
- Pour mettre en place une réaction individuelle de 20 μL de qPCR, faites pipeter soigneusement le suivant dans un puit unique d'une plaque qPCR à 96 puits: 10 μL de 2x SYBR Green I Master; 2 μL du mélange d'amorces avant et arrière de 10 μM (pour une concentration finale de 0,5 μM); 2 μL de gabarit (dans la plage de la courbe standard); Et 6 μl de H 2 O stérile et exempt de nucléases. Pour réduire les erreurs de pipetage, il est préférable de créer un mélange maître avec tous les composants de réaction sauf le modèle. Ajoutez le mélange maître à la plaque qPCR, puis ajoutez le modèle d'intérêt pour chaque puits. Trois répétitions techniques pour chaque échantillon devraient être effectuées pour minimiser les effets de l'erreur de pipettage.
- QPCR paramètres de réaction
REMARQUE: Ce sont les paramètres par défaut, à l'exception du cycle de recuit de l'étape d'amplification. Réglez ce paramètre pour tenir compte des amorces spécifiques si celles utilisées diffèrent des amorces présentées dans ce protocole.- Pré-incuber à 95 ° C pendant 5 min, avec un taux de rampe de 4.4 ° C / s.
- Effectuer 45 cycles d'amplification de (1) 95 ° C pendant 10 s, avec un taux de rampe de 4.4 ° C / s; (2) 60 ° C pendant 20 s, avec un taux de rampe de 2,2 ° C / s; Et (3) 72 ° C pendant 10 s, avec un taux de rampe de 4.4 ° C / s (données acquises pendant (3)).
- Utilisez une optioCycle de courbure de fond de 95 ° C pendant 5 s, avec un taux de rampe de 4.4 ° C / s; 65 ° C pendant 1 min, avec un taux de rampe de courant de 2.2 ° C / s; Et 97 ° C, avec un mode d'acquisition continu.
- Utilisez un cycle de refroidissement de 40 ° C pendant 30 s, avec un taux de rampe de 1,5 ° C / s.
- Paramètres d'analyse
- Sélectionnez le modèle SYBR. Vérifiez les paramètres du programme dans le bouton Expérience. Une fois la plaque chargée, le dosage peut être démarré et les réglages peuvent être ajustés pendant l'exécution du test.
- Affectez des échantillons à l'aide de l'éditeur d'échantillons. Sélectionnez Abs Quant comme flux de travail et désignez les échantillons comme inconnus, des normes ou des contrôles négatifs. Désignez les répliques et remplissez les exemples de noms de la première de chaque réplique. Ajouter les concentrations et les unités aux normes.
- Configurer les sous-ensembles pour l'analyse; Ceux-ci sont attribués dans l'éditeur de sous-ensemble.
- Pour l'analyse, sélectionnez Abs Quant / 2nd Derivative Max dans la liste "Créer une nouvelle analyse".Importez la courbe standard sauvegardée de l'extérieur (le cas échéant), puis cliquez sur Calculer; Le rapport contiendra les informations sélectionnées.
- Pour effectuer une quantification absolue précise pour la détermination du nombre de copies ou de la concentration, utilisez une courbe standard représentative de l'échantillon testé ( par exemple, l' ADN organellaire isolé des méthodes ci-dessus). Étant donné que la quantité d'ADN mitochondrial nécessaire pour préparer une courbe standard est trop élevée pour être atteinte avec une quantité raisonnable de tissu, ne pas utiliser les calculs de numéros de copie fournis par le logiciel, mais plutôt examiner les valeurs du point de passage (Cp) pour déterminer l'enrichissement relatif D'organelaire par rapport à l'ADN nucléaire dans les échantillons. Comparez ces quantités relatives à celles de l'ADN génomique total (voir les résultats représentatifs ). Efficacité des amorces de test sur cinq dilutions 1:10 d'ADN génomique total à partir de semis de blé entièrement cultivés à la lumière de deux semaines (efficacités représentatives signalées dansE légende de la figure 2 ).
- Configuration de la réaction qPCR
- L'électrophorèse sur gel à champ pulsé (PFGE)
REMARQUE: Ce protocole est basé sur les directives du fabricant pour effectuer le PFGE pour résoudre l'ADN de masse moléculaire élevée. Voir la table des matériaux.- Préparation du gel et des échantillons
- Suivez les directives pour la préparation du gel et des échantillons et adaptez-les au système disponible.
- Exécuter les paramètres
- Suivez les instructions pour la mise en place du système d'électrophorèse et utilisez les paramètres suivants: temps de commutation initial de 2 s, temps de commutation final de 13 s, durée de fonctionnement de 15 h et 16 min, V / cm de 6 et angle inclus de 120 ° .
- Tacher et imaginer le gel
- Tachez le gel avec un colorant de choix ( par exemple, bromure d'éthidium ou une alternative appropriée) et une image avec un système de documentation de gel approprié.
- Préparation du gel et des échantillons
- Utilisez 1 ng d'ADN comme entrée pour le kit de préparation de la bibliothèque d'ADN, selon les instructions du fabricant.
- Barcode et regroupe les échantillons pour le séquençage en une seule course. Effectuer le séquençage selon les directives du fabricant.
REMARQUE: les paramètres de mise en commun et de séquençage peuvent être modifiés selon les espèces d'intérêt, le niveau de couverture souhaité et la plate-forme utilisée pour séquencer les bibliothèques. Par exemple, une voie HiSeq a beaucoup plus de sortie qu'une voie MiSeq, tant d'autres échantillons peuvent être multiplexés. Séquence d'un sous-ensemble plus petit d'échantillons pour déterminer si les niveaux de couverture des génomes organellaires sont adéquats pour l'analyse en aval.- Examinez la qualité de lecture en utilisant FastQC 31 pour déterminer l'étendue de la coupe et du filtrage requise pour les données.
- Découpez et filtrez les lectures brutes en utilisant Trimmomatic 32 ou un autre programme comparable. Utilisez les paramètres suivants: ILLUMINACLIP 2:30:10 (pour enlever les adaptateurs), LEADING 3, TRAILING 3, SLIDINGWINDOW 4:10 et MINLEN 100.
- Mettez les lectures filtrées filtrées par la qualité et adaptées (PE) à la mitochondrie Spring chinoise (NCBI Reference Sequence NC_007579.1 33 ), au chloroplaste (NCBI Reference Sequence NC_002762.1 34 ) et au nucléaire 35 génomes de référence utilisant Bowtie2 36 , Avec les paramètres suivants: -I 0 -X 800 - sensible.
- Convertissez les fichiers d'alignement sam au format bam (samtools) et trimez les fichiers bam. Utilisez les fichiers bam pour calculer la couverture de l'ensemble du génome et la couverture par base avec les toiles de lit. Visualisez les résultats avec la fonction R-plot.
- Les choses à faire avant de commencer
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Les protocoles présentés dans ce manuscrit décrivent deux méthodes distinctes pour enrichir l'ADN organellaire du tissu végétal. Les conditions présentées ici reflètent l'optimisation du tissu de blé. Une comparaison des étapes clés dans les protocoles, l'apport de tissus requis et la sortie d'ADN sont décrites à la figure 1 . Les étapes du protocole DC testé suivent des conditions similaires à celles décrites précédemment ( Figure 1A ). Les tissus récoltés doivent être traités fraîchement et soumis à une centrifugation différentielle et / ou à des gradients pour isoler les organites intactes. L'ADN nucléaire est éliminé avant que les organelles ne soient lysés, et finalement, l'ADN est extrait et utilisé pour les applications en aval. En revanche, dans le protocole MF, le tissu végétal peut être récolté et stocké avant utilisation et des organites intactes ne sont pas nécessaires. Au lieu de cela, l'ADN nucléaire et organellaire est fractionné à partir du gDNA total basé sur Le statut de méthylation de l'ADN. Les deux protocoles donnent des quantités approximativement égales d'ADN organellaire ( figure 1C ). En ce qui concerne la production totale d'ADN organellaire par rapport à l'entrée tissulaire, le protocole MF est avantageux lorsque le tissu est limité, car un petit échantillon d'une seule plante peut être utilisé et la plante peut être amenée à développer pour une analyse plus approfondie. Typiquement, dans les protocoles DC, tous les tissus aériens de nombreuses plants sont nécessaires et ces plantes sont éliminées. Cependant, la méthode DC peut être optimisée pour enrichir spécifiquement un type organelle par rapport à l'autre, ce qui n'est pas possible avec l'approche MF. Il convient de mentionner que le temps total pour chaque protocole est approximativement équivalent, bien qu'il y ait moins de temps pratique dans l'approche MF.
Les deux méthodes enrichissent l'ADN organellaire, mais avec des proportions différentes de séquences de mitochondries et de plastides:
"> Des quantités très faibles d'ADN organellaire purifié sont obtenues à partir de l'une ou l'autre méthode (de l'ordre de 50 à 100 ng, figure 1C ). Pour évaluer les niveaux d'enrichissement du génome organellaire et la contamination du génome nucléaire dans l'ADN isolé à la fois du DC et du MF Méthodes, un dosage de qPCR a été utilisé. Dans ce dosage, l'abondance relative de trois amplicons ( c. -à-d., Spécifique au nucléaire, ACTIN , mitochondrial-specific, NAD3 et Chloroplastique spécifique, PSBB ) ont été évalués dans l'ADN génomique total, et la fraction d'ADN organellaire a été obtenue à partir des deux méthodes ( figure 2 ). Les valeurs du cycle de quantification (C q ) ont été examinées pour chaque échantillon ( figure 2A ) et parce que le C q est défini comme le cycle de PCR auquel la fluorescence de l'amplification cible augmente au-dessus du niveau de fluorescence de fond, C q et l'abondance cible ont un relation inverse. DansL'échantillon DC, le C q de NAD3 et PSBB sont, respectivement, ~ 17 et ~ 15 cycles plus tôt que l' ACTIN (qui a un C q de ~ 36) (voir la figure 2B pour les valeurs de C q et les niveaux d'enrichissement). Cela équivaut à des enrichissements théoriques de 167,181 et 47 790 fois pour NAD3 et PSBB , respectivement, Par rapport à ACTIN dans l'échantillon DC ( Figure 2B , voir la légende de la Figure 2 pour le calcul). Dans l'échantillon d'ADN génomique total, les enrichissements en plis pour NAD3 et PSBB par rapport à l' ACTIN sont seulement 158 et 10 701, respectivement. Il n'est pas surprenant de trouver une plus grande abondance des amplicons organellaires par rapport à l'amplicon nucléaire dans l'ADN génomique total, étant donné que les génomes organellaires existent en plus de copies par cellule que le génome nucléaire 37 et que le nombre d'organites peLa cellule r peut différer selon le type de tissu ou le stade de développement 38 , 39 . Dans l'ensemble, les données indiquent que la méthode DC enrichit préférentiellement pour les mitochondries, ce qui est à prévoir, car les vitesses de centrifugation sont optimisées pour isoler sélectivement les mitochondries et réduire la «contamination» des nuclées et des chloroplastes.La fraction non méthylée de l'ADNc total MF présente également un enrichissement substantiel des deux amplicons organellaires et devrait conserver les quantités relatives natives de ces cibles. Les enrichissements en plis pour NAD3 et PSBB par rapport à l' ACTIN dans la fraction non méthylée sont respectivement 20,551 et 1,703,253 ( figure 2A et 2B ). Dans la fraction méthylée, les enrichissements en plis pour NAD3 et PSBB par rapport à ACTIN sont respectivement de 31 et 823 indiEn ce sens que la protéine MBD2-Fc est hautement efficace à la mise au rebut de l'ADN nucléaire méthylé. Comme l'amplicon de chloroplastes a une plus grande abondance que l'amplicon mitochondrial dans l'ADN génomique total (~ 6 C q plus tôt), la fraction méthylée (~ 5 C q plus tôt) et les échantillons de fraction non méthylée (~ 6 C q plus tôt), cela suggère que L'abondance native de ces amplicons n'est pas modifiée de manière substantielle par le dérangement MDB2. Nous nous concentrons ici sur la fraction non méthylée (organellaire) due à l'intérêt de séquencer ces génomes spécifiquement. Cependant, si le génome nucléaire est le principal intérêt, le MF et le séquençage ultérieur de la fraction méthylée produiraient une couverture de génome nucléaire beaucoup plus élevée que le séquençage total de l'ADN génomique, en raison de la réduction de la "contamination" de l'ADN organellaire.
Il convient de noter que si qPCR n'est pas disponible, la PCR en point final (en utilisant les mêmes amorces que pour qPCR) fournit la qualitaÉvaluation de la pureté organellaire. Dans ce cas, des échantillons d'ADN organellaire pur montrent une amplification pour les amplicons mitochondriaux et plastidiques, mais aucune amplification détectable de l'amplicon nucléaire sur le gel d'agarose, alors que l'ADN génomique total montre une amplification pour les trois ensembles d'amorces, comme l'ont démontré les études précédentes 11 , 12 .
L'ADN organellaire isolé des deux méthodes convient à NGS:
Les lectures de séquençage PE trimancées et nettoyées (voir l'étape 4.3) ont été cartographiées dans des génomes de référence organellaires de blé précédemment publiés, et la quantité de lectures utilisées pour la cartographie de chaque échantillon variait de ~ 800 000 à 1 100 000 lectures ( Figure 3I ). Les résultats de la cartographie de nouveaux résultats de séquençage d'Illumina au chloroplasme de blé disponible et aux génomes des mitochondries sont compatibles avec les res de qPCRAvec la méthode DC produisant de l'ADN qui est plus enrichi en ADN mitochondrial ( Figure 3A et 3B , ~ 80% et ~ 10% de la carte des lectures aux génomes mitochondriaux (mt) et chloroplastes (cp), respectivement) et la méthode MF Produisant de l'ADN qui reflète vraisemblablement l'abondance native des deux génomes organellaires ( figures 3A et 3B , ~ 20% et ~ 80% de la carte des lectures aux génomes mt et cp, respectivement). Dans les deux méthodes, la couverture théorique (voir la légende de la figure 3 pour le calcul) des deux génomes organellars de blé dépasse la couverture de 100X (et s'étend jusqu'à une couverture de ~ 2 000 X pour le génome du chloroplaste dans la fraction non méthylée de la méthode MF), même Lorsque 12 bibliothèques sont multiplexées ( Figure 3C et 3D ; les 6 bibliothèques incluses dans cette analyse ont été regroupées avec 6 bibliothèques supplémentaires pour une analyse distincte, pour un total de 12 bibliothèquesRegroupés dans une seule voie de séquençage). Une vue plus détaillée de la couverture a été atteinte en examinant la fraction du génome couverte à des profondeurs spécifiques, ainsi que les niveaux de couverture par base ( figure 3E- 3 ). Pour la méthode MF, la couverture moyenne par base était de ~ 300 - 450X pour le génome de mt et de 4 000 à 5 000 X pour le génome de cp. Pour la méthode DC, la couverture moyenne par base était de ~ 900 - 1 300 et ~ 500 à 700 X pour les génomes mt et cp, respectivement. Cependant, il y avait une petite fraction des génomes de mt et cp qui avaient une couverture extrêmement basse ou élevée, et cela a été observé dans l'ADN organellaire dérivé de l'une ou l'autre méthode ( Figure 3I ). Les régions de couverture supérieure à la moyenne correspondent vraisemblablement à des régions d'homologie entre les génomes organellaires, et les régions à faible couverture peuvent indiquer des SNP ou d'autres petites variantes entre les cultivars que nous avons séquencés et les références publiées. À l'appui de cette notion, ces pointesDe couverture élevée ont été les plus prononcés pour l'ADN mt dérivé de la méthode MF ( figures 3E et 3I ), probablement en raison de la couverture élevée du génome cp dans cette méthode. De façon inexplicable, la couverture du génome cp est plus inégale dans la méthode MF que la méthode DC ( Figure 3G et 3H ), ce qui pourrait être dû à de légers biais dans le basculement MBD2-Fc le long de l'ADN cp. D'autres expériences seront nécessaires pour déterminer pourquoi c'est le cas. Quoi qu'il en soit, les génomes mt et cp ont une couverture relativement uniforme avec les deux méthodes et pas de grandes zones de couverture manquante, ce qui peut être démontré par l'examen de la fraction de génomes séquencés à une profondeur donnée ( Figure 3E -3H ). En outre, les niveaux de couverture pour les deux génomes sont considérés comme suffisants pour l'analyse en aval, comme l'analyse variante. S'il est jugé nécessaire pour l'analyse de variantes rares, en réduisant le nombreR des échantillons groupés permettrait une plus grande couverture. Alternativement, un nombre beaucoup plus important d'échantillons peut être regroupé sur une voie HiSeq, tout en obtenant une profondeur de séquençage encore plus grande, bien que lors d'un sacrifice à la longueur de la séquence, car les bibliothèques HiSeq sont actuellement limitées à la longueur PE150 contrairement aux bibliothèques PE300 MiSeq.
Pour examiner les niveaux de contamination du génome nucléaire en utilisant une approche cartographique, les catégories de cartographie de lecture PE ont été examinées. Les lectures PE peuvent correspondre à un génome de référence dans une variété de configurations. Lorsque les 1 et 2 alignent la référence de manière tête à tête avec une certaine distance "attendue" entre les deux partenaires (en fonction de la taille d'insertion moyenne de la bibliothèque et généralement spécifiée comme paramètre d'entrée dans le logiciel de cartographie ), Ces lectures de PE sont censées cartographier "de manière concordante". En revanche, la cartographie «discordante» est la situation dans laquelle les mates sont représentés avec un écart inférieur ou supérieur à ce qui est attenduAu génome de référence ou à la carte dans des configurations alternatives (tête-à-queue ou queue-à-queue). Si un seul compagnon s'harmonise avec le génome de référence, on dit que la lecture de PE ne correspond pas de manière concordante ou discordante au génome de référence. Dans les trois catégories de lecture-mappage, les lectures PE peuvent s'aligner sur le génome de référence une ou plusieurs fois.
Pour l'ADN organellaire isolé DC et MF, lire le cartographie au génome mitochondrial était principalement dans la catégorie concordante concordante d'une seule fois ( Figure 4A ), alors que les lectures sont cartographiées dans le génome du chloroplaste dans des proportions relativement égales d'une manière concordante une fois et de manière concordante supérieure à Une fois ( figure 4B ), probablement en raison des grandes répétitions inversées présentes dans le génome du chloroplaste et aussi aux niveaux de couverture extrêmement élevés. Cependant, moins de lectures de PE ont été mappées au génome nucléaire et ont largement été cartographiées plus d'une fois dans unNi de manière concordante ni discordante ( c.-à-d. Qu'un seul compagnon est capable de cartographier). Ceux-ci sont plus susceptibles de cartographier «hors cible» aux séquences du génome nucléaire, qui sont homologues aux génomes organellaires ou aux régions mal assemblées. Seule une quantité mineure de lectures (<5%) est cartographiée dans le génome nucléaire de manière concordante, ce qui indique de faibles niveaux de contamination par le génome nucléaire dans l'ADN organellaire isolé de la méthode DC ou MF ( Figure 4C ), comme en témoigne également les résultats qPCR ( Figure 2A ). La fraction nucléaire après le basculement de MBD2-Fc à partir de tissus non étiolés de printemps chinois a également été séquencée pour déterminer l'efficacité de la pulldown à l'élimination de l'ADN non méthylé. Moins de 1% des lectures dans la bibliothèque dérivée de la fraction nucléaire mappée aux génomes de référence organellars, alors que ~ 45% de toutes les lectures sont cartographiées dans le génome nucléaire ( Figure 4 ). Cependant, la plupart des lectures sont cartographiées de manière discordante, wCe qui reflète vraisemblablement les niveaux élevés de désassemblage et de fragmentation dans le génome de référence nucléaire du blé. Quoi qu'il en soit, les résultats suggèrent que le basculement MBD2-Fc est très efficace pour éliminer l'ADN organellaire non méthylé à partir d'ADN nucléaire méthylé. Il convient de noter que, parce que l'ADN enrichi en organelle résultant de ces méthodes contient un mélange de séquences de mitochondries et de chloroplastes, et parce que les similitudes de séquence résultant du transfert de gène ancien entre ces organelles restent dans leurs génomes, l'affectation appropriée des lectures à la spécificité Les génomes doivent être résolus de manière bioinformatique.
L'étiolisation des tissus foliaires ne modifie pas de manière appréciable les abondances d'Organelle:
Traditionnellement, les tissus étiolés sont préférés pour l'isolement de l'ADN mitochondrial des plantes afin de diminuer les niveaux de phénoliques et d'amidons, ce qui peut entraver l'extractioN ou applications en aval 13 . Pour déterminer si les niveaux d'enrichissement du génome organellaire pourraient être modifiés ou améliorés par des conditions de croissance, les tissus étiolés et non étiolés ont été soumis au protocole MF et au séquençage. Fait intéressant, l'étiolisation n'a pas sensiblement changé le pourcentage de lectures qui ont été mappées aux génomes de référence organellaires ( figures 3A et 3B ) ou la couverture par base ( figure 3I ) par rapport aux conditions non étiolées. Nous avons également isolé l'ADN organellaire en utilisant une centrifugation différentielle, avec des tissus étiolés et non étiolés, et une faible différence d'enrichissement a été observée entre les différents tissus en utilisant qPCR (données non présentées). Cela suggère que des tissus non étiolés plus physiologiquement pertinents peuvent être utilisés pour des études de séquençage organellaire, sans changement appréciable d'enrichissement.
Le contrôle de qualité suggère queL'ADN de MF est le plus approprié pour le séquençage à lecture prolongée:
Comme le séquençage à lecture prolongée devient plus accessible aux chercheurs, l'isolement de l'ADN de masse moléculaire élevée devient de plus en plus important. Pour évaluer l'ADN organellaire isolé avec l'une ou l'autre méthode d'intactité et de qualité, PFGE a été utilisé. L'ADN génomique total migre généralement comme un frottis diffus dans PFGE, et le poids moléculaire est déterminé par le protocole et comment l'ADN a été stocké et traité après l'extraction. L'ADN génomique total isolé avec des conseils génomiques devrait dépasser 50 kb, ce qui a été vérifié à l'aide de PFGE ( Figure 5 , voie 2). L'ADN génomique total à partir des conseils génomiques est utilisé comme entrée dans la Trousse d'enrichissement Microbiome pour fractionner l'ADN nucléaire à partir d'ADN organellaire. La fraction nucléaire obtenue après le fractionnement diminue en taille, mais reste centrée autour de 50 kb ( figure 5 , voie 4). Ce n'est pas suLe soulèvement, étant donné que la manipulation relativement plus difficile de la fraction nucléaire comme élution à partir de billes liées à MBD2-Fc nécessite une digestion par la chaleur et la protéinase K. En raison de la masse limitée, la fraction organellaire n'a pas été exécutée sur PFGE, mais une analyse ultérieure avec le TapeStation a indiqué une ADN> 50 kb (données non représentées). L'ADN organellaire obtenu avec la centrifugation différentielle a une masse moyenne de ~ 20 kb, probablement causée par le protocole d'isolement organellaire étendu et l'extraction et la concentration ultérieures d'ADN à base de colonne. L'isolement organellaire à base de gradient et les méthodes d'extraction d'ADN alternatives peuvent maintenir une plus grande taille de fragment d'ADN. Quoi qu'il en soit, l'ADN de la taille obtenue dans ce protocole peut être utilisé pour générer des lectures séquentielles de 10 ou 15 kb si des soins sont pris pendant la préparation de la bibliothèque.
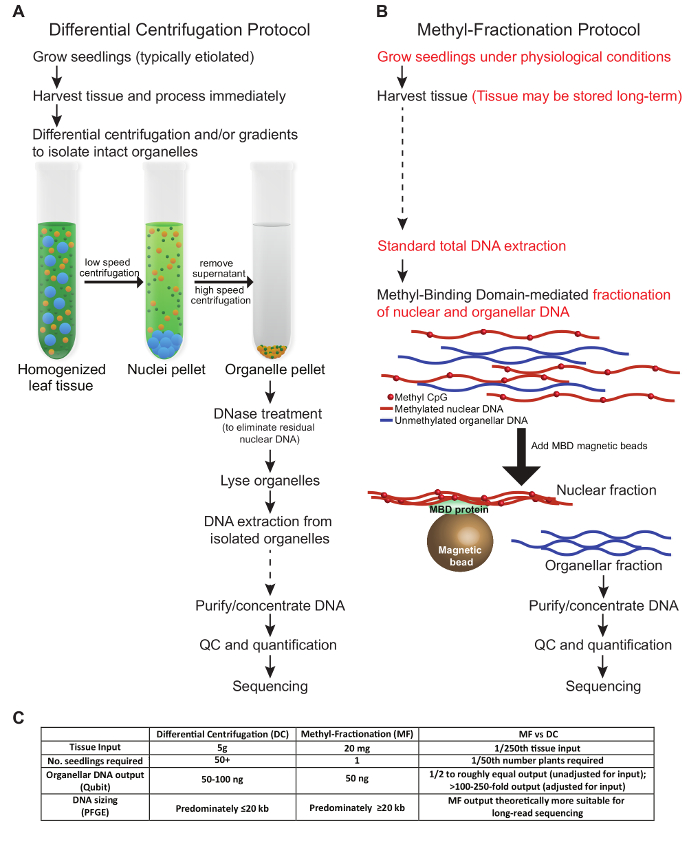
Figure 1: une vue comparative de deux methoDs Enrichir pour l'ADN végétal végétal. Un protocole DC traditionnel ( A ) est contrasté avec le protocole MF ( B ). Il est recommandé d'éviter de congeler et de décongeler les échantillons; Cependant, les étapes à laquelle les échantillons peuvent être stockés à long terme sont indiquées par des flèches pointillées ( A et B ). Les principales différences entre les protocoles sont mises en surbrillance en rouge ( B ). ( C ) Le tableau compare les méthodes en termes d'entrée de tissu, le nombre de plantes requises, la production d'ADN et la taille d'ADN résultante. Cliquez ici pour voir une version plus grande de ce chiffre.

Figure 2: Évaluation de la contamination de l'ADN nucléaire dans l'ADN organellaire isolée en utilisant deux méthodes. (
( B ) Le tableau montre les valeurs C q , qui sont affichées sur le graphique en ( A ), et l'enrichissement en plis des amplicons organellaires par rapport à l' ACTIN . * Fold enrichissement = 2 (Cq ACTIN - Cq Target) . La formule suppose une efficacité parfaite de 2 pour chaque jeu d'amorces, puisque le déviat mineurL'ion de chaque ensemble d'amorces de 2 est négligeable et aurait peu d'effet sur le calcul et la tendance générale ( ACTIN = 1.961, NAD3 = 1.95 et PSBB = 1.989). Les efficacités des amorces ont été évaluées en faisant une courbe standard avec une série de cinq dilutions 1:10 d'ADN génomique total. Cliquez ici pour voir une version plus grande de ce chiffre.

Figure 3: Cartographie de lecture et couverture théorique des génomes chloroplastes et mitochondriaux. Pourcentage de lectures cartographiées dans les génomes de référence mitochondriaux ( A ) ou chloroplastes ( B ) chinois. Couverture théorique correspondante du genoïde de référence mitochondrial ( C ) ou chloroplaste ( D ) de printemps chinoisMes, en supposant des tailles de génome de 450 et 135 kb, respectivement, calculées en utilisant les nombres de lecture total et le pourcentage de lectures de cartographie dans les différents génomes. Répartition génomique de la couverture de l'ADN organellaire à partir de la méthode MF ( E et G ) ou de la méthode DC ( F et H ). Les données dans les panneaux E - H proviennent de l'échantillon etiolé du printemps chinois, mais tous les autres échantillons ont montré une tendance similaire. ( I ) Couverture moyenne, inférieure et plus élevée par base pour tous les échantillons dans les panneaux A - D . Les étiquettes d'échantillons incluant "E" désignent des échantillons etiolés, et "NE" désigne des échantillons non étiolés. DC indique l'ADN isolé avec la méthode de centrifugation différentielle et Unmethylated indique l'ADN qui est dans la fraction non méthylée après pulldown avec MBD2-Fc (protocole MF). Les échantillons étiquetés «Chris» désignent le blé Triticum aestivum'Chris'. CS désigne des échantillons de blé Triticum aestivum 'Spring chinois. Note: En raison de l'homologie de séquence entre le chloroplaste, les mitochondries et les génomes nucléaires résultant d'un transfert génique ancien entre les génomes organellaires ainsi que entre les génomes organellaires et nucléaires, un petit pourcentage de lectures brutes peut être cartographié en génomes multiples. En outre, les lectures qui ne correspondent pas au génome de référence organellaire ne sont pas représentées sur cette figure. Par conséquent, les pourcentages affichés ici ( A et B ) ne totalisent pas 100%. Cliquez ici pour voir une version plus grande de ce chiffre.

Figure 4: Mapping de lecture PE au génome nucléaire de blé. Pourcentage de catégories de PE Lire les types de cartographie dans les génomes de référence mitochondriaux (A) , chloroplastes (B) ou nucléaires (C) chinois. - E désigne des échantillons étiolés et - NE désigne des échantillons non étiolés. DC indique l'ADN isolé avec la méthode de centrifugation différentielle, Unmethylated indique l'ADN qui est dans la fraction non méthylée après pulldown avec MBD2-Fc dans le protocole MF, et Methylated désigne la fraction nucléaire après le basculement MBD2-Fc. Les échantillons étiquetés «Chris» désignent le blé Triticum aestivum «Chris». CS désigne des échantillons de blé Triticum aestivum 'Spring chinois'. Les lectures non mappées ne sont pas affichées. Cliquez ici pour voir une version plus grande de ce chiffre.
Oad / 55528 / 55528fig5.jpg "/>
Figure 5: Examen de la qualité de l'ADN à l'aide de PFGE. L'ADN génomique total de blé (piste 2), l'ADN organellaire de blé obtenu à partir de la centrifugation différentielle (voie 3) et la fraction nucléaire après MF avec l'approche déroulante MBD2-Fc (voie 4) ont été soumis à PFGE sur un gel d'agarose à 1% avec un Échelle étendue de 1 kb utilisée comme marqueur (pistes 1 et 5). Cliquez ici pour voir une version plus grande de ce chiffre.
| Nom du tampon | Recette | Remarques | méthode |
| STE Buffer | Saccharose 400 mM, Tris 50 mM pH 7,8, EDTA 20 mM pH 8,0, polyvinylpyrrolidone à 0,6% (p / v) (PVP), albumine de sérum bovin (BSA) 0,2% (p / v), 0.1% (v / v) de β-mercaptoéthanol (BME) | Le mélange tampon contenant uniquement du saccharose, du Tris et de l'EDTA peut être préparé jusqu'à un mois à l'avance et maintenu à 4 ° C. PVP, BSA et BME devraient être ajoutés frais à une fraction aliquote de la quantité requise de tampon juste avant utilisation. | Méthode n ° 1 |
| ST Buffer | Saccharose 400 mM, Tris 50 mM pH 7,8, polyvinylpyrrolidone à 0,6% (p / v) (PVP), albumine de sérum bovin (BSA) 0,1% (p / v) | Le mélange tampon contenant uniquement du saccharose et du Tris peut être préparé jusqu'à un mois à l'avance et maintenu à 4 ° C. Notez que le tampon ST ne contient pas d'EDTA ou de BME et contient une concentration inférieure de BSA. | Méthode n ° 1 |
| Stock DNase | 2 mg / ml d'ADNase dans du NaCl 0,15 M jusqu'à une concentration en stock de 2 mg / ml | Conserver des aliquotes de 200 ul à -20 ° C. Préparer une solution de traitement DNase (200 μl de solution DNase par échantillon) voirTableau 1 ci-dessous. Voir le protocole complet ci-dessous pour tous les détails de la digestion par DNase. La solution de traitement DNase devrait être préparée à l'état frais. Pour arrêter la réaction de DNase, une solution d'EDTA de pH 400 mM est nécessaire (la concentration finale nécessaire pour arrêter la réaction est de 0,2 M EDTA, voir le protocole complet pour plus de détails). | Méthode n ° 1 |
| Solution de travail DNase | 0,25 mg / ml de DNase et 20 mM de MgCl2 dans le tampon ST | Préparez frais, 200 ul par échantillon. Les concentrations indiquées sont pour le volume final de réaction, de sorte mélanger: 62,5 ul de 2 mg / ml de DNase ( par rapport au volume réactionnel final de 500 pl), 4 pl 1 M MgCl 2 (sur la base de 200 ul de volume de solution de DNase), et 133,5 pi de tampon ST pour Un volume final de 200 μl. | Méthode n ° 1 |
| Tampon de lyse | EDTA 20 mM pH 8,0; Tris 10 mM pH 7,9; Guanidine-HCl 500 mM; NaCl 200 mM; 1% Triton X-100; 0,5 mg / ml d'enzymes de lyse deTrichoderma harzianum | Mélanger tous les ingrédients, à l'exception de lyser les enzymes et les conserver à température ambiante. Les enzymes de lysing doivent être ajoutées à une petite aliquote pour une utilisation immédiate. | Méthode n ° 2 |
Tableau 1: Recettes des tampons maison et des stocks de travail.
| Feuille de travail de concentration | |||||||
| NOM DE L'ÉCHANTILLON | Poids du dispositif vide (g) | Poids du dispositif rempli (g) | Volume rempli (ul, rempli moins les poids vides) | Poids après la 1ère rotation (20 min *, g) | Volume après 1ère rotation (ul, rempliMoins les poids vides) | Poids après la deuxième rotation (X min *, g) | Volume après la deuxième rotation (ul, rempli moins les poids vides) |
| Notez que le volume récupéré réel sera d'environ un peu moins que le volume calculé. | |||||||
Tableau 2: Feuille de travail de concentration.
| prénom | Spécificité génomique | Source de séquence de gènes | Séquence (5 '- 3') |
| Ta_ACTIN - F | Nucléaire | Échiquier de Gramene IWGSC_CSS_1AS_scaff_3272162: 10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | Nucléaire | Comme ci-dessus | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | Mitochondrial | Adhésion NCBI EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | Mitochondrial | Comme ci-dessus | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | Chloroplaste | Accession au NCBI KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | Chloroplaste | Comme ci-dessus | GCTGCCTGTTTCCTTGTAGTT |
Tableau 3: Liste des amorces qPCR.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
À ce jour, la plupart des études de séquençage organellaire se concentrent sur les méthodes DC traditionnelles pour enrichir l'ADN spécifique. Des méthodes pour isoler des organites de diverses plantes ont été décrites, y compris la mousse 40 ; Monocoques telles que le blé 15 et l'avoine 11 ; Et des dicotylédones comme l'arabidopsis 11 , le tournesol 17 et le colza 14 . La plupart des protocoles se concentrent sur les tissus foliaires 13 , 14 , 15 , 16 , 17 , certains ayant été adaptés pour divers types de tissus, y compris les graines 11 . L'isolement des organites à partir des protoplastes a également été démontré 41 . Cependant, cela ne convient pas à tous les systèmes, ni n'est possible lorsque le tissu d'intérêt est limité. Beaucoup de ces orgaLes méthodes d'isolement nellar ont été conçues pour récupérer des organites intactes pour des expériences spécifiques, telles que des études physiologiques. Ces protocoles sont lourds et exigent généralement l'utilisation de gradients de densité, tels que les gradients de saccharose ou de Percoll, qui sont très efficaces pour isoler des fractions organellaires spécifiques, mais nécessitent une grande entrée de tissu ( c'est- à- dire plus de 5 g et plus de kilogrammes, selon Le type de tissu). Cependant, la méthode DC peut être optimisée pour enrichir des fractions cellulaires spécifiques, telles que les mitochondries ou les chloroplastes, en modifiant les vitesses de rotation et les gradients de densité. En revanche, l'approche MF nécessite beaucoup moins de matière de départ (20 mg), mais les ADN mitochondriaux et plastiques seront présents par leur abondance relative dans le tissu utilisé pour l'extraction de l'ADN. Néanmoins, le protocole MF offre une approche alternative pour isoler l'ADN organellaire mixte et est particulièrement avantageux pour commencer avec de petites quantités de tissu.
T O évaluer la pureté de l'échantillon après l'isolement des organites, la plupart des études à ce jour utilisent uniquement la PCR en phase finale et l'électrophorèse sur gel 11 , 12 . Cela donne une mesure qualitative équitable de la pureté de l'échantillon. Cependant, de faibles niveaux d'amplification peuvent ne pas être visualisés sur un gel d'agarose. Peu de rapports incluent des mesures plus quantitatives du contrôle de la qualité, telles q qCR 14 . Pour une évaluation quantitative de la pureté de l'échantillon d'ADN isolée des deux méthodes, nous avons utilisé qPCR et séquençage pour déterminer la quantité d'ADN nucléaire dans l'échantillon, ainsi que les proportions relatives de l'ADN mitochondrial versus chloroplaste. Les deux méthodes évaluées ici sont efficaces pour éliminer l'ADN nucléaire. Les deux méthodes produisent un mélange d'ADN mitochondrial et de chloroplastes, bien que dans des proportions différentes.
Les plantes en croissance dans l'obscurité (étiolisation) sont rapportées pour faciliter l'isolement organellaire en raison d'une réduction des composés phénoliquesPourtant, dans cette comparaison, nous n'avons pas trouvé un avantage appréciable pour travailler avec des tissus étiolés sur des échantillons légers. Bien que la proportion de chloroplastes spécialisés soit probablement plus élevée lors de la croissance légère, le nombre total de plastides, comme Reflété dans la proportion de lectures de cartographie dans le génome du chloroplaste, est inchangé dans des conditions de lumière différentes. Par conséquent, pour les analyses fonctionnelles en aval, telles que l'évaluation de l'hétéroplasme dans différents tissus ou sous différents facteurs de stress ou pour des analyses d'expression, nous recommandons d'effectuer un séquençage génomique sur Plantes cultivées dans des conditions physiologiquement pertinentes.
Pour une application avec des technologies de séquençage à lecture courte, les deux techniques comparées ici fournissent une quantité et une qualité suffisantes d'ADN. Cependant, pour obtenir de longues lectures de> 20 kb pour les applications de séquençage à une seule molécule, une quantité plus importante d'ADN de qualité supérieure est nécessaire. Par exemple, idéalement,> 1 μg d'orga pureL'ADN de blé nellé avec un poids moléculaire> 20 kb est nécessaire pour les protocoles internes et à faible entrée pour les préparations de bibliothèques d'insertion de 20 kb 42 . Les nouveaux protocoles développés par l'utilisateur et à faible entrée peuvent réduire les besoins en ADN ( c.- à-d. À 50 ng ou même moins 20 ), mais le défi demeure d'avoir une ADN de haute qualité et de haut poids moléculaire dans les préparations de la bibliothèque. Il est essentiel qu'une majorité de l'ADN soit> 20 kb, car des fragments plus petits seront préférentiellement insérés dans le SMRTbell et élimineront la distribution de taille de la bibliothèque 43 . Nous avons essayé un certain nombre de protocoles d'extraction d'ADN maison et un certain nombre de protocoles commerciaux pour l'extraction de l'ADN (non représenté). Pour le tissu de feuilles de blé, le meilleur équilibre entre la quantité et la qualité de l'ADN, en particulier la longueur, a été obtenu en utilisant un kit commercial 27 , 29 . Selon les espèces végétales et les tissus d'intérêt, alternatiLes protocoles d'extraction peuvent être également adaptés ou plus fructueux. Néanmoins, nous concluons que l'extraction totale d'ADN génomique à haut poids moléculaire> 50 kb de taille, suivie d'un fractionnement avec l'approche pulvérisée MBD2-Fc 28 , permet d'obtenir un séquençage à lecture prolongée à partir d'un matériau de départ limité. Les travaux futurs devraient tester les limites du matériau de départ requis après le fractionnement pour la préparation de la bibliothèque longue durée et le séquençage ultérieur à lecture prolongée. De manière critique, cette approche pourrait fournir une méthode robuste pour isoler l'ADN d'un sous-échantillon d'une seule feuille qui convient à un séquençage à lecture prolongée, sans amplification complète du génome. Nous prévoyons que cette approche sera facilement adaptable aux types de tissus supplémentaires et largement applicable à d'autres espèces végétales. Il sera particulièrement utile dans les situations où les quantités de tissus sont limitantes, comme le séquençage à des générations individuelles dans un schéma de croisement ou dans des types de tissus plus rares.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs déclarent qu'ils n'ont pas d'intérêts concurrents.
La mention de noms commerciaux ou de produits commerciaux dans cette publication vise uniquement à fournir des informations spécifiques et n'implique pas une recommandation ou une approbation par le ministère de l'Agriculture des États-Unis. USDA est un fournisseur et un employeur offrant l'égalité des chances.
Acknowledgments
Nous souhaitons remercier le ministère du Service d'agriculture et de recherche agricole de l'Amérique du Nord et de la National Science Foundation (IOS 1025881 et IOS 1361554). Nous remercions R. Caspers pour la maintenance des serres et les soins des plantes. Nous remercions également le Centre de génomique de l'Université du Minnesota, où les préparations et le séquençage de la bibliothèque Illumina ont été réalisés. Nous sommes également reconnaissants pour les commentaires des éditeurs de journaux et de quatre examinateurs anonymes qui ont renforcé notre manuscrit. Nous remercions également l'OCDE pour une bourse à SK pour intégrer ces protocoles pour des projets collaboratifs avec des collègues au Japon.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
