

Summary
2つの植物細胞小器官DNA濃縮方法の比較および最適化が提示される:従来の分画遠心分離およびメチル化状態に基づく全gDNAの分画。得られたDNAの量と品質を評価し、短期間の次世代シーケンシングでの性能を実証し、長時間読み取り単一分子シークエンシングで使用する可能性について検討します。
Abstract
植物オルガネラゲノムは、複雑な構造および/またはサブゲノム断片を形成するために対合または組換えを受ける可能性がある、大きな反復要素を含む。オルガネラゲノムはまた、所与の細胞または組織型(ヘテロプラスミー)内での混合物として存在し、多量のサブタイプは、発達中またはストレス下で変化し得る(化学量論的部分シフト)。オルガネラゲノムの構造と機能をより深く理解するためには、次世代シーケンシング(NGS)技術が必要です。伝統的な配列決定研究は、細胞小器官DNAを得るためにいくつかの方法を使用する:(1)大量の出発組織を使用する場合、それをホモジナイズし、分画遠心分離および/または勾配精製に供する。 (種子、材料、又は空間が限られている場合、すなわち、)(2)組織の少量が使用される場合、同じプロセスが(1)十分なDNAを得るために、全ゲノム増幅に続いてのように行われます。 (3)バイオインフォマティクス分析は、seq総ゲノムDNAを調べ、オルガネラの読みを解析する。これらの方法にはすべて固有の課題とトレードオフがあります。 (1)では、そのような多量の出発組織を得ることは困難であり得る。 (2)において、全ゲノム増幅は配列決定バイアスを導入し得る; (3)において、核ゲノムとオルガネラゲノムとの間の相同性が、アセンブリおよび分析を妨害する可能性がある。大きな核ゲノムを有する植物では、オルガネラDNAを濃縮して、バイオインフォマティクス分析の配列決定コストおよび配列複雑性を低減することが有利である。ここでは、伝統的な微分遠心分離法を第4の方法、適合したCpG-メチルプルダウン法と比較して、全ゲノムDNAを核および細胞小器官画分に分離する。いずれの方法も、ミトコンドリアおよび葉緑体において異なる比率であるにもかかわらず、細胞小器官配列に対して高度に濃縮されたNGS、すなわちDNAに対して十分なDNAを生じる。我々は、小麦の葉組織のためのこれらの方法の最適化を提示し、主要な利点およびdサンプル入力、プロトコルの容易性、および下流のアプリケーションの文脈における各アプローチの長所である。
Introduction
ゲノム配列決定は、重要な植物形質の根底にある遺伝的根拠を解明するための強力なツールです。ほとんどのゲノムシーケンシング研究は、遺伝子の大部分が核内に位置するため、核ゲノムの内容に焦点を当てています。しかし、(真核生物にわたる)ミトコンドリアと色素体(植物中で、特殊な形式、葉緑体、光合成で動作)を含む細胞小器官のゲノムは、生物の開発、ストレス応答、および全体的なフィットネス1に不可欠な重要な遺伝情報を貢献します。オルガネラゲノムは、通常、核ゲノム配列決定を目的とした全DNA抽出に含まれますが、DNA抽出に先立つオルガネラ数を減らす方法も使用されています2 。多くの研究は、オルガネラゲノムを組み立てるために、総gDNAの抽出からの配列決定結果を使用していた3、4、5、外部参照"> 6、7。しかし、多くの人がしている読み取るための研究の目標は、合計のgDNAは、シーケンシングコストを増大させ使用して、細胞小器官のゲノムに焦点を当てているとき、『特に大きな核ゲノムを有する植物で、核DNA配列に』失われましたまた、による核ゲノム中へのオルガネラ配列の複製、転写および細胞小器官の間に、配列決定の正しいマッピング位置を解決することは、適切なゲノムに読み出しバイオインフォマティクス2,8に挑戦されている。核ゲノムからオルガネラゲノムの精製が1でありますミトコンドリアと葉緑体との間の相同性領域にマッピングされる読み取りを分離するために、さらなるバイオインフォマティクス戦略を使用することができる。
多くの植物種からのオルガネラゲノムが配列決定されているが、オルガネラゲノム多様性の幅についてはほとんど知られていない野生の集団または栽培された繁殖プールで利用可能である。オルガネラゲノムはまた、反復配列間の組換えのために重要な構造的再編成を受ける動的分子であることが知られている9 。さらに、オルガネラゲノムの複数のコピーが各オルガネラ内に含まれ、複数のオルガネラが各細胞内に含まれる。これらのゲノムの全てのコピーが同一であるとは限らず、ヘテロプラスミーとして知られている。 「マスターサークル」の正統図とは対照的に、サブゲノム円、線状染色体、線状コンカテマー、分枝構造など、細胞小器官ゲノム構造のより複雑な画像の証拠が増えています10 。植物オルガネラゲノムの集合は、それらの比較的大きなサイズおよび実質的な逆方向反復および直接反復によってさらに複雑になる。
オルガネラ単離、DNA精製およびその後のゲノムに関する従来のプロトコール E配列は、多くの場合、煩雑であり、いくつかのグラム、組織入力の大容量を必要とするように上方に始点11、12、13、14、15、16、17、必要に応じて、若い葉の組織のグラム数百。これは、組織が限られている場合、細胞小器官のゲノム配列決定が不可能になります。いくつかの状況では、交配によって維持されなければならない雄性不稔性系統または世代別に配列決定する必要がある場合など、種子の量は限られる。このような状況では、細胞小器官のDNAを精製してから全ゲノム増幅を行うことができます。しかしながら、全ゲノム増幅は、構造変化、サブゲノム構造、およびヘテロプラスミーレベルを評価する際に特に問題となる、有意な配列決定バイアスを導入することができる> 18。短リードシーケンシング技術のライブラリ作成における最近の進歩は、全ゲノム増幅を回避するために低入力障壁を克服してきた。例えば、Illumina Nextera XTライブラリー調製キットでは、1 ngのDNAを入力19として使用できます。しかしながら、PacBioまたはOxford Nanopore配列決定技術のような長時間配列決定アプリケーションのための標準ライブラリー調製は、オルガネラゲノム配列決定のための課題を提起することができる比較的多量の入力DNAを依然として必要とする。最近、新しいユーザー作られた、長読みシーケンシングプロトコルは、入力量を低減し、DNAのマイクログラム-量を得ることが21、20困難なサンプルではゲノム配列決定を促進するために開発されてきました。しかしながら、これらのライブラリー調製物に供給するための高分子量の純粋なオルガネラ画分を得ることは、依然として課題である。
私たちは全ゲノム増幅を必要とせずに、NGSに適したオルガネラDNA濃縮および単離法を比較し最適化する。具体的には、葉のサブサンプルなどの限られた出発物質から高分子量細胞小器官DNAを濃縮するためのベストプラクティスを決定することでした。この研究は、(1)市販のDNA CpG-メチル結合ドメインタンパク質プルダウンアプローチの使用に基づく(2)DNA分画プロトコルに対する、改変された伝統的な微分遠心分離プロトコルである細胞小器官DNAを濃縮する方法の比較分析を提示する22は、組織23を植える適用しました。我々は、小麦葉組織からのオルガネラDNAの単離のためのベストプラクティスを推奨しており、これは他の植物および組織タイプに容易に拡張することができる。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
臓器単離およびDNA抽出のための植物材料の生成
- 小麦の苗の標準的な成長
- バーミキュライトの植物の種子を角のついた4〜6個の種子を持つ小さな四角い鉢に入れる。 16時間の光サイクル、23℃/ 18℃の夜間温室または成長チャンバーに移す。
- 毎日植物に水を注ぎます。発芽時および発芽後7日目に、顆粒20-20-20 NPK肥料の¼ティースプーンで植物を肥料化する。
- 小麦の苗の代替的栽培
- ステップ1.1に従いますが、ポットを暗室で23℃、16時間/ 18℃で8時間置きます。あるいは、温室内の植物を覆う( 例えば、貯蔵容器を用いるが、適切な換気が維持されなければならない)。
- 成長および組織収集
- 12〜14日間植物を栽培してください。ほとんどの小麦遺伝子型苗の75〜100苗は約10〜12gの組織を産生し、これは分画遠心分離法(セクション2)を用いた2回の細胞小器官の抽出に十分である。核DNAから細胞小器官を分画するためにDNA CpG-メチル化に基づくプルダウン法を利用する場合には、1つの植物のみが必要である(第3節)。
- ディファレンシャル遠心法を利用する場合は、組織を新鮮なものにして、セクション2で説明したように、サンプルの処理に直ちに進んでください。
- CpGメチルプルダウンアプローチを利用する場合、マイクロ遠心チューブに若い葉組織の収穫20mgのセクション( 代表的な結果を参照して、いずれかの標準の成長または黄化組織を使用)。液体窒素でスナップフリーズし、使用するまで-80℃で凍らせます。セクション3で説明したように、DNAのプルダウン分画に進みます。
2.方法#1:示差遠心分離(DC)を用いたDNA抽出
注:differential遠心分離プロトコルは、両方の細胞小器官を単離が、ミトコンドリア17、24を濃縮するために条件を最適化された2巻の刊行物から改変しました。得られたプロトコールは、従来の方法よりも時間がかかりにくく、毒性の少ない化学物質を使用する。具体的には、STE抽出緩衝液へのポリビニルピロリドン(PVP)の添加およびフッ化ナトリウム(NaF)を含有するNETF緩衝液中の最終洗浄工程の除去を含む、緩衝液および洗浄工程を改変した。
注意:STE緩衝液の調製および使用は、このバッファーに2-メルカプトエタノール(BME)が含まれているため、適切な個人用保護具を備えた化学薬品フードの下で行う必要があります。
- 開始前のこと
- すべての装置がきれいになっていることを確認して、オートクレーブが可能な装置をオートクレーブしてください( 例:粉砕シリンダー、高速セントリフュージチューブなど )。
注:クロスコンタミネーションを避けるために、ピペッティングが必要なすべての手順に、フィルターチップを使用することをお勧めします。 - 必要な機器と試薬のリストを参照し、方法#1( 表1 )のために必要な緩衝液と作業ストックを準備する。極低温粉砕ブロックを-20℃に冷却し、ローターとバッファーを4℃に冷却し、微量遠心分離機を4℃に設定し、37℃の水槽に入れます。
- すべての装置がきれいになっていることを確認して、オートクレーブが可能な装置をオートクレーブしてください( 例:粉砕シリンダー、高速セントリフュージチューブなど )。
- オルガネラの分離
- 新鮮な組織を5g採取し、冷たいビーカーの氷冷した滅菌水ですすいでください。
注記:遠心分離機、ヒュームフードなどのすべての操作および輸送中は、サンプルを氷上に置いておきます。プロトコールを実行するのに十分なスペースと設備があれば、冷蔵室で作業してください。 - はさみを使用して、葉の組織を2cmのセラミック研削を含む50mLのチューブに直接〜1cm片に切断するシリンダー。
注:クロスコンタミネーションを避けるために、サンプル間のハサミを清掃または交換してください。 - 組織ホモジナイザーがない場合は、乳鉢と乳棒を使用して、ステップ2.2.4-22.9を置き換える。
- 氷上のあらかじめ冷却した乳鉢に葉組織を切断する。 15mLのSTE(ヒュームフード内)で2〜3分間サンプルを粉砕する。
- バッファーをあらかじめ濡らした滅菌濾過クロス(〜22μm〜25μmの孔径;詳細は主なプロトコールを参照)の1層を含む漏斗を通して緩衝液(モルタル中に組織を残す)から別の50mLチューブ。さらに10 mLのSTEを乳鉢に加え、乳棒で掻き混ぜ、再度ホモジナイズする。
- ホモジナイズした組織とバッファーを同じ漏斗に注ぎます。乳鉢をすすぎ、10mLのSTEで乳化し、漏斗に注ぐ。できるだけ多くの液体を回収するために、濾布を絞って漏斗に絞ってください。
注:クロスコンタミネーションを避けるために、サンプル間の手袋を交換してください。プロと続けるステップ2.2.10でトコール。
- 各50 mLチューブに20 mLのSTE(ヒュームフード内)を添加する。
- 試料を、組織粉砕装置中の予め冷却した極低温粉砕ブロックに入れ、1,750rpmで2×30秒間試料を粉砕する。サンプルの位置を回転させ、粉砕の間に約1分間氷上にサンプルを置く。
注:乳鉢と乳棒、ブレンダー、または他の組織粉砕/均質化装置をこの工程で使用することができる。しかし、各方法は結果として得られるDNA品質に異なる程度で影響を与えるため、DNAの長さおよび品質を評価してから、下流のアプリケーションを続行する必要があります。 - 氷中に置かれた清潔な50mLチューブに漏斗を入れる。ろ液の一層を漏斗に入れ、5mLのSTEであらかじめぬらします。フロースルーを廃棄しないでください。
- ホモジナイズした組織を漏斗に注ぎます。 15 mLのSTEで粉砕チューブをすすいだ後、チューブを逆さにして反転させて壁と蓋をすすぎ、ファーンに注ぎますエル。
- 慎重に陶器の石を取り除き、濾過布を絞って漏斗に絞ってください。
注:クロスコンタミネーションを避けるために、サンプル間の手袋を交換してください。 - こぼれないようにパラフィルムでチューブキャップを包みます。 4℃で10分間2,000 xgで遠心します。
- 慎重に血清ピペットを使用して上清を吸引し(ペレットを乱さないように)、50 mLの高速遠心分離チューブに入れます(密封ガスケットがない場合はパラフィンでチューブキャップを包んでください)。ペレットを捨てる。
- STEを使用してチューブを0.1g以内に収め、得られた上清を18,000 xgおよび4℃で20分間遠心します。チューブのバランスを取るために、氷の小さなビーカーを天びんに置き、風袋を秤量し、サンプルを氷上で秤量して冷たく保ちます。あるいは、冷たい部屋で天びんとヒュームフードを使用してください。
- 上清を捨てる。ペレットに1 mLのSTを加え、穏やかに再懸濁するやわらかな絵筆を描く。 24mLのST(25mLの最終容量)を加え、混合/渦巻き( すなわち、チューブの側面のペイントブラシを押してすべての液体を除去する)。
- STを使用してチューブを0.1g以内に収めます。 18,000 xgおよび4℃で20分間遠心分離する。一方、DNaseI溶液を調製してください(ストック溶液および作業溶液レシピについては表1を参照)。各サンプルについて、1.5mLチューブで200μLのアリコートを1つ作成します。
- 上清を捨て、チューブをブロットし、軟ペイントブラシを使用して300μLのST中でペレット(高速遠心チューブに依然として)を再懸濁する。 200μLのDNaseI溶液を含む先に調製した1.5mLチューブにペイントブラシを置き、ペイントブラシを渦巻かせてブラシに付着した残留ペレットを除去する。 DNaseI溶液をピペットで高速遠心チューブに戻し、穏やかに混合して混和させます。
- 37℃で30分間、水浴中でインキュベートする(チューブ上部のパラフィルムを包んで、凝縮漏出を防ぐgをキャップに入れる)。インキュベーションの間に2回旋回させることにより静かに混合する。
- ピペットチップを用いてペレット混合物をチューブからゆっくりとピペットで広げ、1.5mLの低結合チューブに入れる。 500μLの400mM EDTA、pH 8.0を高速遠心管に加え、ゆっくりとピペットで残ったペレットをすべてチューブから取り出します。ペレット混合物と同じ1.5mLの低結合チューブにEDTAを移し、穏やかに転倒混和する。
- 4℃で18,000 xgで20分間遠心する。上清を捨て、チューブをブロットし、DNA単離のためにすぐに使用する。必要に応じて、-20℃でペレットを凍結しますが、残存するDNaseIがすぐに処理されないとサンプルDNAを分解させる可能性があるため、収率が低下する可能性があります。
- 新鮮な組織を5g採取し、冷たいビーカーの氷冷した滅菌水ですすいでください。
- 市販のカラムを用いた単離されたオルガネラからのDNA抽出
注記:完全プロトコールについては、キットハンドブックを参照してください( 25 )。 Prオルガネラ単離からDNA抽出へ直接移行することが好ましい。反復凍結および融解は、DNA断片サイズを減少させ、残留DNaseIによるDNA分解をもたらす。ボルテックスや激しいピペッティングはDNAを剪断することができますので、制限してください。 DNA回収を最大にするために、低結合マイクロ遠心チューブの使用を推奨します。- DNA抽出手順
注:バッファが適切に作成/保存され、スピンカラム手順が理解されていることを確認する前に、詳細な商用プロトコール25をお読みください。- ペレットを用いて180μLのBuffer ATLをチューブに直接添加する(予め凍結してベンチトップ上で室温に平衡化していれば解凍する)。
- 手順3で30分の溶解、オプションのRNase A消化を含むキットハンドブックの「DNA Purifications from Tissues」の手順3に進み、3×200μLのAEに溶出する。それぞれのパラメートチューブを開き、溶出液を合わせる)。
- qPCRのためにアリコート(少なくとも20μL)を保存する(ステップ4.1参照)。濃縮前に定量するために、高感度定量のために1μLをさらに保存します。
- 必要に応じて、サンプルの濃縮を進めてください。
- DNA抽出手順
- 市販のフィルターユニットでのサンプル濃度
注:詳細については、商用プロトコル26を参照してください。下流の用途に応じて、( 例えば、エンドポイントPCRおよびqPCRアプリケーションのための)サンプル濃縮を行う必要はないかもしれない。しかし、NGSライブラリー構築のためには、DNA抽出後に得られる希薄オルガネラDNAを濃縮する必要があると思われる。- 濃縮カラム手順
- デジタル分析天びん上の清潔な秤量紙に空のフィルターユニット(チューブなし)を注意深く事前に計量してください( 表2参照)。体重を記録する。
- Pi合わせた溶出液をフィルターユニットに注入し、再び注意深く秤量する。
注:商用マニュアル26によると、フィルタユニットの最大容量は500μLですが、オーバーフローなしで最大575μLをユニットに一度に追加できます。 - 充填されたフィルターユニットをチューブ(カラムに付属)に慎重に置きます。 500xgで所望の時間遠心分離して、必要な濃縮物の容量を達成する。 〜575μLのサンプル量では、20分のスピンは通常15〜30μLの濃縮液量になります。
- フィルターユニットをチューブから取り外し、再度秤量します。この表を使用して、所望の濃縮物量が達成されたかどうかを決定する。そうでない場合は、500 xgでもう一度短時間遠心し、再び体重を測定します。所望の濃縮物の容量に達するまで繰り返す。
- 新しいチューブ(カラムに付属)をフィルターユニットの上に置き、反転させます。 1000 xgで3分間遠心分離して、チューブに移す。
- 回復されたボリュームを決定します。これは通常、フィルターの保持のために、計算された容量より3〜5μL少なくなります。過剰に濃縮された場合は、滅菌水またはTEで希釈して所望の容量を達成する。
- 高感度定量を使用してDNAを定量する(製造業者の指示に従って)。
- 濃縮カラム手順
方法#2:全ゲノムDNAからのオルガネラDNAを濃縮するメチル分画(MF)アプローチ
注:このプロトコールは、植物および真菌27および市販のMicrobiome DNA Enrichment Kitプロトコール28について、ユーザーが開発したゲノムチップキットDNA抽出プロトコルから変更されました。理論的には、高分子量のDNAを生じる任意のDNA単離プロトコールをプルダウンに使用することができる。短リードシーケンシングの場合、プルダウンでの使用には、主に15 kb以上の断片を生じる抽出が適切です。 For lo読取りシーケンシングでは、より大きなフラグメントが望ましい場合があります。したがって、我々は高分子量のDNAを得るためにこのプロトコルを最適化した。
- 全DNAの単離
注記:必要な機器と試薬のリストを参照し、方法#2( 表1 )のために必要なバッファーと作業ストックを準備します。溶解緩衝液ストックに溶解酵素を加えて、溶解緩衝液を作用溶液にする。サーモミキサーの電源を入れ、37℃に設定します。水浴を50℃にし、QF緩衝液を浴に入れる。冷凍庫に70%EtOHを置き、微量遠心分離機を4℃に設定する。- 市販のDNA抽出カラムを用いた全DNA抽出
注:開始する前に、重力流動陰イオン交換カラムの使用に関する詳細については、コマーシャルハンドブック29を参照してください。特別なラックを使用してカラムを設置するか、付属のプラスチックリングを使用してチューブの上に置くことができます。 gを含むすべてのステップエノミックチップは、重力流によって進行させるべきであり、残留液体は強制的に通過させてはならない。- 2mLチューブ用に設計された手持ち式粉砕乳棒を使用して、2mLの低結合チューブ中の液体窒素中の凍結組織20mgを粉砕する。
- 2mLの溶解緩衝液を加える(チューブは非常にいっぱいになる)。
- 300 rpmで穏やかに攪拌しながら、37℃のサーモミキサーで1時間インキュベートする。サーモミキサーが利用できない場合は、ヒートブロック上でインキュベートし、15分ごとに穏やかなフリックで混合することが適しています。
- RNアーゼA(100mg / mL、最終濃度200μg/ mL)4μLを添加する。 37℃で30分間、サーモミキサー中で混合し、インキュベートし、300rpmで穏やかに攪拌する。
- 80μLのプロテイナーゼK(20mg / mL、最終濃度0.8mg / mL)を添加し、混合物に転化させ、300rpmで穏やかに攪拌しながら、50℃で2時間サーモミキサー中でインキュベートする。
- 4℃、1分間で20分間遠心分離する。不溶性破片をペレット化するために5,000×g。
- サンプルを遠心分離しながら、カラムを1 mLのBuffer QBTで平衡化し、カラムを重力流によって空にします。
- ワイドボアピペットチップを使用して、平衡したカラムにサンプルをすばやく塗布し(ペレットを避ける)、完全にカラムに流してください。サンプルは、カラムに適用する前に、再び曇り、フィルターや遠心分離機になった場合(詳細は29商業用ハンドブックを参照してください)。
- サンプルが完全に樹脂に入ったら、4×1mLのBuffer QCでカラムを洗浄する。
- 清潔な2 mLの低結合マイクロ遠心チューブにカラムを懸濁する。 50℃であらかじめ温めた0.8 mLのBuffer QFでゲノムDNAを溶出する。
- 溶出したDNAに0.56mL(0.7容量の溶出バッファー)の室温イソプロパノールを加えてDNAを沈殿させます。
- 逆転(10X)して混合し、すぐに15,000×gおよび4℃で20分間遠心分離する。お手入れゆるく付着したペレットを妨害することなく上清を完全に除去する。
- 冷たい70%エタノール1mLで遠心分離したDNAペレットを洗浄する。 15,000 xgおよび4℃で10分間遠心分離する。
- ペレットを乱すことなく上清を注意深く除去する(このステップでも注意する)。 5〜10分間空気乾燥させ、0.1mLの溶出緩衝液(EB)にDNAを再懸濁する。 DNAを室温で一晩溶解させる。 DNAを剪断するピペッティングは避けてください。
- 高感度のDNA定量アッセイを使用してサンプルを定量する(製造者の指示に従う)。
- 市販のDNA抽出カラムを用いた全DNA抽出
- メチル化および非メチル化DNAのビーズに基づく分画
注:最近の刊行物は、ヒトIgG Fc断片(MBD2-Fcタンパク質)に融合されたCpG特異的メチル結合ドメインタンパク質を利用して、分画するプルダウンアプローチを利用する市販のキット28の使用を示した核ゲノム(高度にメチル化された)含量から植物オルガネラゲノム(非メチル化)を食べた23 。コムギ試料中の分画効率は、この市販のMFキット28を用いて以前に試験されなかった。- 開始前のこと
- 80%エタノール(反応あたり少なくとも800μL)を新鮮に調製する。氷上で解凍するために5x結合/洗浄緩衝液をセットし、サンプルあたり1x緩衝液5mLを調製する(無菌のヌクレアーゼフリー水で5倍に希釈し、プロトコール中に氷上に保つ)。
- MBD2-Fcタンパク質結合磁気ビーズを調製する
- 必要数のビーズセットを準備します。 1〜2μgの全入力DNAを使用するように反応をスケールすると、160〜320μLのビーズが必要です。下に列挙した反応は1μgの全入力DNAに対応するため、160μLのビーズが必要であることに注意してください。必要に応じて反応を拡大します。
- ワイドボアのチップを使用して、Protein A Magnetic B均一な懸濁液を作るためにスラリーを上下させる。代わりに、4℃で15分間ビーズのチューブを静かに回転させます。
注:ビーズをボルテックスしないでください。 - メーカーの説明書28あたりの方向に進みます。
- メチル化された核DNAを捕獲する
- 個々のサンプルについて、1μgの入力DNAを160μLのMBD2-Fc結合磁気ビーズを含むチューブに加える。
- 最終濃度が1x(添加する5倍の結合/洗浄緩衝液の容量(μL)=入力DNAの容量(μL)/ 4)になるようにDNA入力試料の容量を考慮して、必要に応じて5x結合/洗浄緩衝液を加える。サンプルを数回上下にピペットし、ワイドボアピペットチップを使用して混合します。
- チューブを室温で15分間回転させる。サンプルをゆっくりとワイドボアピペットチップでピペットにかけ、ビーズ凝集を防ぐためにインキュベーション中2〜3回サンプルをフリックします。
注:ピペッティングとフリッキーメチル化DNAの効率的なプルダウンを確実にするために重要である。
- 豊富な非メチル化オルガネラDNAを集める
- DNAおよびMBD2-Fc結合磁気ビーズ混合物を含むチューブを短時間スピンさせる。チューブを磁気ラックに少なくとも5分間置いてビーズをチューブの側面に集める。その解決策は明確に見えるはずです。
- ワイドボアのチップを使用して、ビーズを乱すことなく清澄な上清を注意深く取り除きます。上清(メチル化されていないオルガネラが豊富なDNAが含まれています)を清潔で低結合の2mL微量遠心チューブに移します。このサンプルを-20℃または-80℃で保存するか、精製のためにステップ3.2.6に直接進んでください。
- MBD2-Fc結合磁気ビーズからの核DNAの捕獲
- 核分画もまた望むならば、MBD2-Fc結合磁性ビーズから核DNAを溶出させるために製造業者の説明書28に従う;ステップ3.2.7で説明したように精製する。
- ビーズベースの核酸精製
- 精製ビーズが室温で、完全に混合されていることを確認してください。 MFキットのマニュアル28の手順に従ってプロトコールを進めてください。
注:このサンプルは、NGSライブラリー構築または別の下流解析に使用できるようになりました。
- 精製ビーズが室温で、完全に混合されていることを確認してください。 MFキットのマニュアル28の手順に従ってプロトコールを進めてください。
4.サンプル定量および品質管理
- オルガネラ濃縮を評価するためのqPCRアッセイ
注記:ここに記載されたqPCR反応およびアッセイパラメーターは、Roche LightCycler 480での使用のために設計されており、さまざまな機器および試薬に合わせて調整する必要があります。 qPCRが利用できない場合、エンドポイントPCRおよびアガロースゲル上での可視化を、本明細書に記載されたのと同じプライマーおよび条件を使用して、サンプル純度の定性的尺度として使用することができる。アンプリコンのサイズはすべてのプライマーセットに対して〜150 bpになります。プライマーシーケンシングについては表3を参照セックスとペアリング。- qPCR反応セットアップ
- 20μLのqPCR反応を個別に設定するには、96ウェルqPCRプレートの1つのウェルに次のものを慎重にピペットで入れます:10μLの2x SYBR Green I Master; 2μLの10μMフォワードおよびリバースプライマーミックス(0.5μMの最終濃度用)。鋳型2μL(標準曲線の範囲内)。 6μLの滅菌ヌクレアーゼフリーH 2 Oを含む。ピペッティング誤差を減らすために、テンプレートを除くすべての反応成分を含むマスターミックスを作製することが好ましい。 qPCRプレートにマスターミックスを添加し、目的のテンプレートを各ウェルに添加する。ピペッティングエラーの影響を最小限に抑えるために、各サンプルについて3回の技術的複製を行う必要があります。
注:最終的には、核と細胞小器官の定量サイクルの比がサンプル間で比較されるため、濃度のわずかな差は許容されます。しかし、DNA濃度はおよそeach。 - 高品質のqPCRシーリング・フィルムでシートをシールします。サンプルを静かにボルテックスし、気泡の発生を避けるよう注意してください。簡単に4℃で2分間プレートを回転させてサンプルを集め、小さな泡を除去する。
- プレートを機械にセットします。下記のガイドラインに従ってqPCRプログラムを実行してください。
- 20μLのqPCR反応を個別に設定するには、96ウェルqPCRプレートの1つのウェルに次のものを慎重にピペットで入れます:10μLの2x SYBR Green I Master; 2μLの10μMフォワードおよびリバースプライマーミックス(0.5μMの最終濃度用)。鋳型2μL(標準曲線の範囲内)。 6μLの滅菌ヌクレアーゼフリーH 2 Oを含む。ピペッティング誤差を減らすために、テンプレートを除くすべての反応成分を含むマスターミックスを作製することが好ましい。 qPCRプレートにマスターミックスを添加し、目的のテンプレートを各ウェルに添加する。ピペッティングエラーの影響を最小限に抑えるために、各サンプルについて3回の技術的複製を行う必要があります。
- qPCR反応パラメーター
注:これらは、増幅ステージのアニールサイクルを除いて、デフォルトのパラメータです。使用するプライマーが本プロトコールで提示されているプライマーと異なる場合は、この設定を調整して特定のプライマーに対応させます。- 95℃で5分間、4.4℃/ sの上昇率でプレインキュベートする。
- 4.4℃/ sの上昇率で、(1)95℃で10秒間45回の増幅サイクルを実施する。 (2)20℃で60℃、2.2℃/ sのランプ速度; (3)72℃10秒間、4.4℃/ sの上昇率((3)で得られたデータ)。
- オプティオを使用する4.4℃/ sのランプ速度で5秒間95℃の融解曲線サイクル; 65℃で1分間、2.2℃/ sのランプ速度;および97℃、連続取得モード。
- 40°Cの冷却サイクルを30秒間、1.5°C / sのランプ速度で使用してください。
- アッセイパラメータ
- SYBRテンプレートを選択します。実験ボタンでプログラムのパラメータを確認します。プレートがロードされると、アッセイを開始することができ、アッセイが実行されている間、設定を調整することができる。
- サンプルエディタを使用してサンプルを割り当てます。 Abs Quantをワークフローとして選択し、サンプルを未知、標準、または陰性対照として指定します。レプリケートを指定し、各レプリケートの最初のサンプル名を記入します。濃度と単位を基準に加える。
- 分析のためのサブセットを設定する。これらはサブセットエディタで割り当てられます。
- 分析するには、「新規分析の作成」リストからAbs Quant / 2nd Derivative Maxを選択します。外部保存された標準曲線をインポートし(該当する場合)、次に計算を押します。レポートには選択した情報が含まれます。
- コピー数または濃度の決定のための正確な絶対定量を行うためには、試験されているサンプル( 例えば、上記の方法から単離されたオルガネラDNA)を代表する標準曲線を使用する。標準曲線を作成するために必要なミトコンドリアDNAの量は、妥当な量の組織で達成するには高すぎるため、ソフトウェアによって提供されるコピー数計算を利用せず、クロスポイント(Cp)値を調べて相対濃縮を決定するサンプル中の核DNAと比較してオルガネラの存在を示している。これらの相対量を全ゲノムDNAの相対量と比較する( 代表的結果参照)。完全に軽く増殖した2週齢コムギ苗からの全ゲノムDNAの5倍の1:10希釈物に対するプライマーの効率を試験する(代表的な効率は、図2の伝説)。
- qPCR反応セットアップ
- パルスフィールドゲル電気泳動(PFGE)
注:このプロトコールは、高分子量DNAを分解するためにPFGEを実施する製造業者のガイドラインに基づいています。 材料表を参照してください。- ゲルとサンプルの準備
- ゲルとサンプルの準備に関するガイドラインに従って、利用可能なシステムに合わせてください。
- パラメータを実行する
- 電気泳動システムを設定するためのガイドラインに従い、初期スイッチ時間2秒、最終スイッチ時間13秒、ランタイム15時間16分、V / cm 6、および120°の夾角。
- ゲルの汚れと画像
- 選択した色素( 例えば、エチジウムブロマイドまたは適切な代替物)でゲルを染色し、適切なゲルドキュメンテーションシステムで画像化する。
- ゲルとサンプルの準備
- 製造元の指示に従って、DNA Library Prep Kitの入力として1 ngのDNAを使用してください。
- 1回の操作でシーケンシング用のバーコードとサンプルをプールします。製造元のガイドラインに従ってシーケンシングを行います。
注:プーリングおよびシーケンシングパラメータは、目的の種、所望のカバレッジレベル、およびライブラリーの配列に使用されるプラットフォームに応じて変更することができます。例えば、HiSeqレーンはMiSeqレーンより実質的に多くの出力を有するので、より多くのサンプルを多重化することができる。オルガネラゲノムのカバレッジレベルが下流分析に適しているかどうかを判断するために、サンプルのより小さなサブセットをシーケンスします。- FastQC 31を使用して読み取り品質を調べ、データに必要なトリミングとフィルタリングの程度を判断します。
- Trimmomatic 32または他の同等のプログラムを使用して未加工の読み込みをトリムおよびフィルタリングします。次の設定を使用してください:ILLUMINACLIP 2:30:10(アダプターを取り外すため)、3回、3回、SLIDINGWINDOW4:10、およびMINLEN 100。
- 、品質濾過およびアダプタートリミングペアエンド(PE)は、中国のスプリングミトコンドリア(NCBI参照配列NC_007579.1 33)、葉緑体(NCBI参照配列NC_002762.1 34)、及びBowtie2 36を用いて、核35の参照ゲノムに読み出す地図-I 0 -X 800 - sensitiveと設定します。
- samアラインメントファイルをbam形式(samtools)に変換し、bamファイルをソートします。 bamファイルを使用して、ベッドツールでゲノム全体のカバレッジとベースごとのカバレッジを計算します。 R-プロット関数を使用して結果を視覚化します。
- 開始前のこと
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
この原稿で提示されたプロトコルは、植物組織からオルガネラDNAを富化するための2つの異なる方法を記載している。ここに示した条件は、小麦組織の最適化を反映しています。プロトコルの重要なステップ、必要な組織入力、およびDNA出力の比較を図1に示します。我々がテストしたDCプロトコールのステップは、以前に記載されたものと同様の条件に従う( 図1A )。収穫された組織は、新たに処理され、インタクトな細胞小器官を単離するために示差遠心分離および/または勾配に供されなければならない。オルガネラが溶解される前に核DNAが除去され、最後にDNAが抽出され、下流の用途に使用される。対照的に、MFプロトコルでは、使用前に植物組織を採取して保存することができ、完全な細胞小器官は不要である。その代わりに、核および細胞小器官のDNAは、 DNAのメチル化状態。両方のプロトコールは、ほぼ同量の細胞小器官DNAを産生する( 図1C )。組織入力に関連する全細胞小器官DNA出力に関しては、MFプロトコルは組織が限られている場合に有利であり、単一の植物からの小さな試料を使用することができ、植物をさらなる分析のために増殖させることができる。典型的には、DCプロトコルでは、多くの実生のすべての空中組織が必要であり、これらの植物は廃棄される。しかしながら、DC法は、一方のオルガネラ型を他方のオルガネラ型よりも特に豊富にするように最適化することができ、これはMF法では不可能である。 MFのアプローチでは、実習時間が少なくても、各プロトコルの合計時間はほぼ同等であることに言及することは重要です。
両方の方法は、ミトコンドリアおよびプラスチド配列の異なる割合を有するにもかかわらず、オルガネラDNAを濃縮する:
オルガネラゲノムの濃縮およびDCとMFの両方から単離されたDNAにおける核ゲノムの汚染のレベルを評価するために(約50〜100ng; 図1C )このアッセイでは、3つのアンプリコン( すなわち、核特異的、 ACTIN ;ミトコンドリア特異的、 NAD3 ;およびNAD3 )の相対的な存在量が測定された。 葉緑体特異的、 PSBB )を全ゲノムDNAで評価し、両方の方法から細胞小器官DNA画分を得た( 図2 )。定量化サイクル(C q )値を各試料( 図2A )について調べ、C qは標的増幅からの蛍光がバックグラウンド蛍光レベルを上回って増加するPCRサイクルとして定義されるので、C qおよび標的存在量は逆関係。にDCサンプル、 NAD3およびPSBBの C qは、それぞれACTIN ( 〜36のC qを有する)よりも約 17および15サイクル早い(C q値および富化レベルについては図2Bを参照)。これは、 NAD3およびPSBBのそれぞれ理論的に167,181倍および47,790倍濃縮に相当し、 DCサンプル中のACTINと比較した( 図2B 、計算のための図2の凡例参照)。総ゲノムDNAサンプルにおいて、 ACTINに対するNAD3およびPSBBの 倍率は、それぞれわずか158および10,701である。オルガネラゲノムが核ゲノム37よりも細胞当たりより大きなコピー数で存在すること、および細胞小器官の数が存在することを考えると、全ゲノムDNA中の核アンプリコンに比べてオルガネラアンプリコンの存在度が高いことを見出すことは驚くことではないR細胞は、組織タイプまたは発生段階38、39に応じて異なっていてもよいです。全体として、このデータは、遠心分離速度がミトコンドリアを選択的に単離し、核および葉緑体の「汚染」を低減するために最適化されるので、DC法がミトコンドリアを優先的に濃縮することを示している。MF総gDNAの非メチル化画分も、オルガネラアンプリコンの両方の実質的な濃縮を示し、これらの標的の天然の相対量を保持すると予想される。非メチル化画分中のACTINに対するNAD3およびPSBBの倍率は、それぞれ20,551および1,703,253である( 図2Aおよび2B )。メチル化画分において、 ACTINに対するNAD3およびPSBBの倍濃縮物は、それぞれ31および823であり、 indiMBD2-Fcタンパク質がメチル化核DNAのプルダウンで非常に効率的であると述べている。葉緑体アンプリコンは、全ゲノムDNA(約6Cq早期)、メチル化画分(~ 5Cq早期)、および非メチル化画分(~ 6Cq早期)サンプルにおいてミトコンドリアアンプリコンよりも豊富に存在するので、これは、これらのアンプリコンの天然の存在量は、MDB2プルダウンによって実質的に変化しない。我々は、これらのゲノムを特異的に配列決定することに興味があるため、ここでは非メチル化(オルガネラ)画分に焦点を当てる。しかし、核ゲノムが主要な関心事である場合、MFおよびその後のメチル化画分のシーケンシングは、オルガネラDNAの「汚染」の減少のために、全ゲノムDNA配列決定よりもはるかに高い核ゲノムカバレッジをもたらす。
qPCRが利用可能でない場合、エンドポイントPCR(qPCRと同じプライマーを使用する)が品質を提供することは注目に値するオルガネラの純度の評価。この場合、純粋なオルガネラDNAサンプルは、ミトコンドリアおよびプラスチドアンプリコンの増幅を示すが、アガロースゲル上の核アンプリコンの増幅は検出されないが、全ゲノムDNAは、以前の研究11 、 12 。
両方の方法から単離されたオルガネラDNAはNGSに適している:
トリミングされ洗浄されたPEシーケンシングリード(ステップ4.3参照)は、以前に発表された小麦オルガネラリファレンスゲノムにマッピングされ、各サンプルをマッピングするために使用されたリードの量は、〜800,000から1,100,000までの範囲であった( 図3I )。マッピングデノボイルミナシーケンスからの結果は、利用可能な小麦の葉緑体とミトコンドリアゲノムに読み込み、qPCRの解像度と一致しています(ミトコンドリア(mt)および葉緑体(cp)ゲノムにマップされた図3Aおよび3B 、〜80%および〜10%の読み取り値)およびMF法を用いて、おそらく2つのオルガネラゲノムの本来の豊富さを反映するDNAを産生する( 図3Aおよび3B 、それぞれ、mtおよびcpゲノムへのリードの〜20%および〜80%がマッピングされる)。どちらの方法においても、両方の小麦細胞小器官ゲノムの理論上の適用範囲(計算のための図3の凡例を参照)は、100Xの適用範囲を超えており(また、MF法の非メチル化画分の葉緑体ゲノムについては2,000Xまでの範囲) 12個のライブラリーが多重化されている場合( 図3Cおよび3D ;この分析に含まれる6個のライブラリーは別個の分析のために6個のライブラリーとともにプールされ、合計12個のライブラリー単一のシークエンシングレーンにプールされている)。カバレッジのより詳細な見通しは、特定の深度でカバーされたゲノムの割合、ならびに塩基ごとのカバレッジレベル( 図 3E〜3I )を調べることによって達成された。 MF法では、ベースあたりの平均カバレッジは、mtゲノムで約300〜450X、cpゲノムでは4,000〜5,000Xでした。 DC法では、mtおよびcpゲノムの平均塩基対当たりの被覆率は、それぞれ約900-1,300および約500-700Xであった。しかし、mtおよびcpゲノムの両方には、非常に低いまたは高いカバレッジを有する少数の画分しか存在せず、これはどちらの方法から得られた細胞小器官DNAにおいても見られた( 図3I )。オルガネラゲノム間の相同性領域に相当する可能性があり、低いカバレッジ領域は、配列決定された品種と公開された参考文献との間のSNPまたは他の小さな変異体を示し得る。この概念を支持するために、これらのスパイクは、おそらくこの方法においてcpゲノムの高いカバレッジのために、MF法( 図3Eおよび3I )由来のmtDNAについて最も顕著であった。説明不可能なことに、cpゲノムのカバレッジは、DC法( 図3Gおよび3H )よりもMF法で不均一であり、これはcp DNAに沿ったMBD2-Fcプルダウンのわずかな偏りに起因する可能性がある。なぜこれが当てはまるのかを判断するには、さらなる実験が必要になります。それにもかかわらず、mtおよびcpゲノムはどちらの方法でも比較的均一なカバレッジを有し、カバレッジの欠如はほとんどなかった。これは、所定の深度で配列決定されたゲノムの割合を調べることによって証明することができる( 図 3E〜3H )。さらに、両方のゲノムのカバレッジのレベルは、変異分析のような下流分析に十分であると考えられる。まれな変種の分析に必要と思われる場合は、r個のプールされたサンプルは、より大きなカバレッジを達成するだろう。 HiSeqライブラリーは現在、PE300 MiSeqライブラリーとは対照的にPE150の長さに制限されているため、より多くのサンプルをHiSeqレーンにプールすることができますが、シーケンシングの長さを犠牲にしています。
マッピングアプローチを用いて核ゲノム汚染のレベルを調べるために、PE読み取りマッピングカテゴリーを調べた。 PE読み取りは、様々な構成で参照ゲノムにマッピングすることができる。リード1とリード2がヘッドツーヘッド方式でリファレンスと整列し、2つのメイトの間に特定の "予想される"距離がある場合(ライブラリの平均挿入サイズに基づいており、通常はマッピングソフトウェアの入力パラメータとして指定されます)、これらのPE読み取りは「一致して」マッピングされていると言われています。対照的に、「不一致」のマッピングは、仲間が予想よりも小さいかより大きなものをマップする状況です参照ゲノムまたはマップに交互配置(頭から尾まで、または尾から尾まで)で交配することができます。 1つのメイトだけが参照ゲノムに整列するならば、そのPE読取りは、参照ゲノムに一致しても不一致でもないと言われる。 3つのすべての読み取りマッピングカテゴリにおいて、PE読み取りは、参照ゲノムに1回または複数回整列させることができる。
DCおよびMF単離された細胞小器官DNAの両方について、ミトコンドリアゲノムへの読み取りマッピングは、主に、整列した一致した1つの時間カテゴリー( 図4A )であったが、葉緑体ゲノムには、1倍とほぼ同率( 図4B )、葉緑体ゲノムに存在する大きな逆方向反復および非常に高いカバレッジレベルに起因する可能性がある。しかし、核ゲノムにマップされたPE読み取りは少なく、調和も不一致もしていない( すなわち、 1人の仲間だけがマップすることができる)。これらは、オルガネラゲノムまたはミスアセンブルされた領域に相同である、核ゲノム中の配列に「オフターゲット」をマッピングする可能性が最も高い。わずかな量はまた、定量PCR結果( 図で反射されたように(<5%)は、DC又はMF法( 図4C)から単離されたオルガネラDNAで核ゲノム汚染の低いレベルを示す、調和し核ゲノムにマッピングされた読み出し図2A )。 Chinese Spring非致死組織由来のMBD2-Fcプルダウン後の核画分もまた配列決定され、プルダウンが非メチル化DNAの除去においていかに効率的であるかを決定した。細胞小器官由来のゲノムにマッピングされた核フラクション由来のライブラリーの読み込みは1%未満ですが、すべての読み込みの約45%が核ゲノムにマップされています ( 図4 )。しかし、ほとんどの読み込みは不一致な方法でマップされます。w小麦核参照ゲノムにおける高レベルの誤組合および断片化を反映している可能性が高い。それにもかかわらず、結果は、MBD2-Fcプルダウンが、メチル化核DNAからの非メチル化細胞小器官DNAの除去において非常に効率的であることを示唆している。これらの方法から得られた細胞小器官に富むDNAはミトコンドリアと葉緑体配列の混合物を含んでおり、これらのオルガネラ間の古代の遺伝子伝達に起因する配列類似性がそのゲノムに残っているため、ゲノムは生物情報学的に解決されなければならない。
葉組織の切除は、オルガネラの豊富さを見事に変えない:
伝統的に、エトホルテーションされた組織は、フェノール性物質およびデンプンのレベルを低下させるために植物ミトコンドリアDNA単離に好ましいが、これは抽出物を妨害し得るnまたは下流のアプリケーション13 。オルガネラゲノムの濃縮レベルが成長条件によって変化し得るか、または改善され得るかどうかを決定するために、腐敗組織および非腐敗組織の両方をMFプロトコルおよび配列決定に供した。興味深いことに、寛解は、非致死条件と比較して、オルガネラ基準ゲノム( 図3Aおよび3B )または塩基ごとのカバレッジ( 図3I )にマッピングされた読み取りの割合を感知可能に変化させなかった。我々はまた、疎外された組織および非腸化された組織の両方を用いて、分画遠心分離を用いてオルガネラDNAを単離し、qPCRを用いて異なる組織間での濃縮の差はほとんど見られなかった(データ示さず)。これは、オルガネラの配列決定研究に生理的に関連性の高い非致死的な組織を用いることができることを示唆している。
品質管理はそれを提案するMF DNAは長時間読み取り配列に最適です:
長いリードシークエンシングが研究者にとってよりアクセスしやすくなるにつれて、高分子量DNAの単離がますます重要になってきています。いずれかの方法で単離された細胞小器官DNAをインタクト性および品質について評価するために、PFGEを用いた。総ゲノムDNAは、典型的にはPFGEのびまん性塗抹標本として移動し、分子量はプロトコルおよびDNAがどのように保存後保存後に処理されたかによって決定される。ゲノム先端で単離された全ゲノムDNAは50kbを超えるべきであり、これはPFGEを用いて検証された( 図5 、レーン2)。ゲノム先端からの全ゲノムDNAは、細胞小器官DNAから核を分取するために、Microbiome Enrichment Kitへの入力として使用されます。分画後に得られた核分画はサイズが減少するが、約50kbを中心に残っている( 図5 、レーン4)。これはsuではありませんMBD2-Fc結合ビーズからの溶出としての核画分の比較的粗い取り扱いが、熱およびプロテイナーゼK消化を必要とすることを考えると、驚くべきことである。限られた質量のため、オルガネラ画分はPFGE上では検出されなかったが、TapeStationでのその後の分析はDNA> 50kbを示した(データは示していない)。分画遠心分離で得られたオルガネラDNAは、平均オルガネラ単離プロトコールおよびその後のカラムに基づくDNAの抽出および濃縮に起因すると考えられる〜20kbの平均質量を有する。グラジエントベースの細胞小器官単離および代替のDNA抽出法は、より大きなDNA断片サイズを維持し得る。いずれにしても、このプロトコールで得られたサイズのDNAを用いて、ライブラリー調製中に注意を払うと10または15kbのシークエンシングリードを生成することができる。
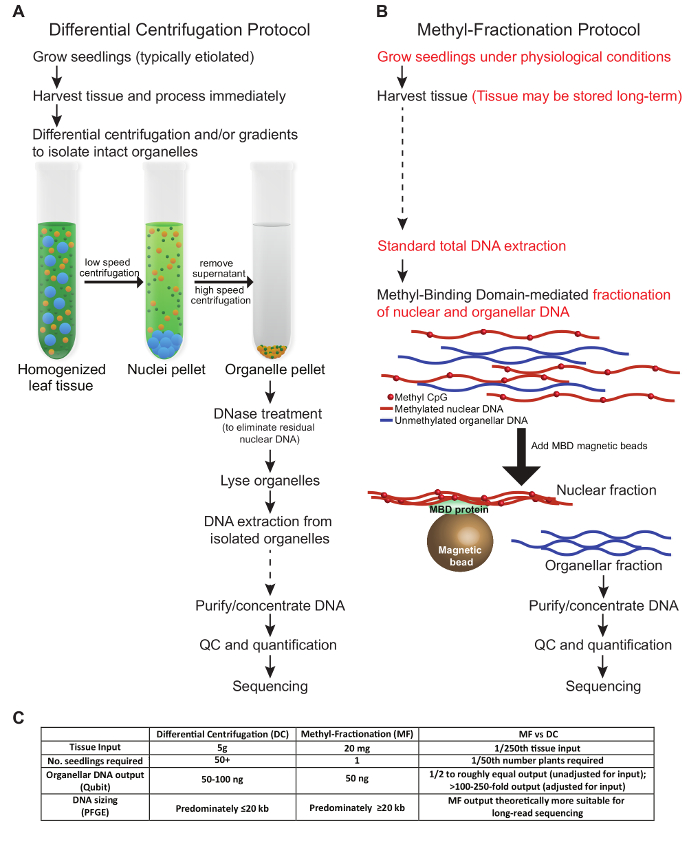
図1:2つのメゾの比較図植物細胞小器官DNAのために濃縮する。従来のDCプロトコル( A )はMFプロトコル( B )と対比されています。サンプルの凍結および融解を避けることを推奨します。しかしながら、サンプルを長期保存することができるステップは、破線の矢印( AおよびB )で示されている。プロトコル間の主な違いは赤で強調表示されています( B )。 ( C )この表は、組織入力、必要な植物の数、DNA出力、および結果として生じるDNAサイズの点で方法を比較する。 この図の拡大版を見るには、ここをクリックしてください。

図2:2つの方法を用いて分離したオルガネラDNAにおける核DNA汚染の評価。 (
( B )表は、( A )のグラフに示されているC q値、およびACTINに対するオルガネラアンプリコンの倍濃縮を示す 。 * Fold enrichment = 2 (Cq ACTIN - Cq Target) 。この式は、各プライマーセットについて2の完全効率を仮定している。なぜなら、2からの各プライマーセットのイオンはごくわずかであり、計算および全体的傾向( ACTIN = 1.961、 NAD3 = 1.95、およびPSBB = 1.989)にほとんど影響しない。プライマー効率は、全ゲノムDNAの一連の5つの1:10希釈液で標準曲線を作成することによって評価した。 この図の拡大版を見るには、ここをクリックしてください。

図3:葉緑体およびミトコンドリアゲノムの読み取りマッピングおよび理論的範囲。ミトコンドリア( A )または葉緑体( B )中国春の参照ゲノムにマップされた読み込みの割合。中国春ミトコンドリア( C )または葉緑体( D )参照遺伝子の対応する理論上の適用範囲mes、ゲノムサイズがそれぞれ450kbおよび135kbであると仮定して、総リード数および異なるゲノムにマッピングされたリードのパーセンテージを用いて計算した。 MF法( EおよびG )またはDC法( FおよびH )からのオルガネラDNAのためのカバレッジのゲノムワイド分布。データパネルでEは - Hは、中国スプリング黄化試料からのものであるが、他のすべての試料は同様の傾向を示しました。パネルA内のすべてのサンプルについて(I)の平均、最小、および最高毎ベースカバレッジ- D。 「E」を含む試料ラベルは漂白試料を示し、「NE」は非漂白試料を示す。 DCは分画遠心分離法で単離したDNAを示し、非メチル化はMBD2-Fc(MFプロトコル)でプルダウンした後の非メチル化画分に存在するDNAを示す。 "Chris"と名付けられたサンプルは小麦Triticum aestivum「クリス」 CSはコムギTriticum aestivum 'Chinese Springのサンプルを指定します。注:オルガネラゲノムとオルガネラゲノムとの間、およびオルガネラゲノムと核ゲノムとの間の古代の遺伝子移入に起因する葉緑体、ミトコンドリアおよび核ゲノム間の配列相同性のために、未処理の読み込みのわずかな割合が複数のゲノムにマッピングされ得る。さらに、どちらの細胞小器官参照ゲノムにもマップされていない読み取りは、この図には示されていません。したがって、ここに表示されるパーセンテージ( AおよびB )は合計100%ではありません。 この図の拡大版を見るには、ここをクリックしてください。

図4:小麦核ゲノムへのPE読み取りマッピング。 PEのカテゴリーのパーセンテージミトコンドリア(A) 、葉緑体(B) 、または核(C)中国春の参照ゲノムにマッピングタイプを読み込みます。 - Eは漂白サンプルを示し、 - NEは非漂白サンプルを示す。 DCは、分画遠心分離法で単離されたDNAを示し、非メチル化は、MFプロトコルでMBD2-Fcでプルダウン後の非メチル化画分にあるDNAを示し、Methylatedは、MBD2-Fcプルダウン後の核画分を示す。 「クリス」と表示されたサンプルは、コムギTriticum aestivum 'Chris'を示しています。 CSはコムギTriticum aestivum 'Chinese Spring'のサンプルを指定します。マッピングされていない読み取りは表示されません。 この図の拡大版を見るには、ここをクリックしてください。
oad / 55528 / 55528fig5.jpg "/>
図5:PFGEを用いたDNA品質の検討分画遠心分離(レーン3)から得られたコムギ総ゲノムDNA(レーン2)、小麦細胞小器官DNA(レーン3)、およびMBD2-Fcプルダウンアプローチを用いたMF後の核画分(レーン4)を、1%アガロースゲルでPFGEマーカーとして使用される1kbの拡大ラダー(レーン1およびレーン5)。 この図の拡大版を見るには、ここをクリックしてください。
| バッファ名 | レシピ | ノート | 方法 |
| STEバッファ | 0.6%(w / v)ポリビニルピロリドン(PVP)、0.2%(w / v)ウシ血清アルブミン(BSA)、0%.1%(v / v)β-メルカプトエタノール(BME) | スクロース、トリス、EDTAのみを含むバッファーミックスは1ヶ月前に調製し、4℃で保存することができます。 PVP、BSA、およびBMEは、使用直前に必要な量の緩衝液のアリコートに新たに添加する必要があります。 | 方法1 |
| STバッファ | 0.6%(w / v)ポリビニルピロリドン(PVP)、0.1%(w / v)ウシ血清アルブミン(BSA) | スクロースとトリスのみを含むバッファーミックスは、1ヶ月前までに調製し、4℃で保存することができます。 STバッファーはEDTAまたはBMEを含まず、BSAの濃度がより低いことに注意してください。 | 方法1 |
| DNaseストック | 0.15MのNaCl中の2mg / mlのDNaseを用いて、2mg / mlのストック濃度 | -20℃で200μlのアリコートを保存する。 DNaseワーキング溶液(1サンプルあたり200μlのDNase溶液)を調製するには、以下の表1。 DNase消化の詳細については、以下の全プロトコールを参照してください。 DNase作業溶液は新鮮な状態で調製する必要があります。 DNase反応を停止するには、400mM EDTA pH 8.0溶液が必要です(反応を停止させるのに必要な最終濃度は0.2M EDTAです、詳細はフルプロトコールを参照)。 | 方法1 |
| DNaseワーキングソリューション | ST緩衝液中の0.25mg / ml DNaseおよび20mM MgCl 2 | サンプルあたり200μlを新鮮に調製する。示された濃度は最終反応液量ですので、ミックス:62.5μl2 mg / ml DNase(最終500μl反応液量に基づく)、4μl1M MgCl 2 (200μlDNase溶液量に基づく)、および133.5μlのSTバッファー200μlの最終容量。 | 方法1 |
| 溶解緩衝液 | 20mM EDTA pH8.0; 10mMトリスpH7.9; 500mMグアニジン-HCl; 200mM NaCl; 1%Triton X-100; 0.5mg / mlの溶解酵素トリコデルマ・ハルジアナム | 溶解酵素を除くすべての成分を混合し、室温で保存する。溶解酵素は、直ちに使用するために少量ずつ新しいものに添加する必要があります。 | 方法2 |
表1:自家製のバッファーと作業用ストックのレシピ。
| 濃度ワークシート | |||||||
| サンプル名 | 空のデバイス重量(g) | 充填されたデバイスの重量(g) | 充填量(ul、充填量 - 空重量) | 1回目のスピン後の重量(20分*、g) | 第1スピン後の音量(ul、filled空の重量を引いた値) | 2回目のスピン後の重量(X min *、g) | 2回目のスピンの後の体積(ul、空白から空の重量を差し引いたもの) |
| 実際の回復されたボリュームは、計算されたボリュームよりも数ul少ないことに注意してください。 | |||||||
表2:濃度ワークシート。
| 名 | ゲノムの特異性 | 遺伝子配列ソース | シーケンス(5 '〜3') |
| Ta_ACTIN - F | 核 | グラミン足場IWGSC_CSS_1AS_scaff_3272162:10,633-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | 核 | 同上 | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | ミトコンドリア | NCBI加盟EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | ミトコンドリア | 同上 | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | 葉緑体 | NCBI受託番号KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | 葉緑体 | 同上 | GCTGCCTGTTTCCTTGTAGTT |
表3:qPCRプライマーのリスト。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
今日まで、ほとんどのオルガネラ配列研究は、特定のDNAを豊富にする伝統的なDC法を中心に研究しています。モス40を含む多様な植物からオルガネラを単離する方法が記載されている。小麦15および小麦11などの単子葉植物;アラビドプシス11 、ヒマワリ17 、ナタネ14などの双子葉植物が挙げられる。ほとんどのプロトコルは、いくつかの種子11を含む組織型の種々のために適合されたもので、葉組織13、14、15、16、17に焦点を当てます。プロトプラストからのオルガネラの単離もまた実証されている41 。しかしながら、これはすべてのシステムに受け入れられず、関心組織が限られている場合には実現可能でもない。これらのオルガの多く生理学的研究のような特定の実験のために無傷の細胞小器官を回収するためのネラール単離法を設計した。これらのプロトコールは面倒であり、典型的には、特定の細胞小器官画分を単離するのに非常に効率的であるが、大きな組織入力を必要とする( すなわち、 5g以上のキログラムを必要とする、スクロースまたはパーコール勾配のような密度勾配の使用を必要とする。組織タイプ)。しかし、DC法は、スピン速度および密度勾配を変化させることによって、ミトコンドリアまたは葉緑体などの特定の細胞分画を富化するように最適化することができる。対照的に、MFアプローチは出発材料(20mg)をはるかに必要とするが、ミトコンドリアおよびプラスチドのDNAは、DNA抽出に使用される組織中のそれらの相対存在量につき存在するであろう。それにもかかわらず、MFプロトコルは、混合オルガネラDNAを単離するための代替的なアプローチを提供し、少量の組織で開始する場合に特に有益である。
T Oオルガネラ単離後のサンプルの純度を評価するため、これまでのほとんどの研究は、エンドポイントPCRおよびゲル電気泳動11,12を使用します。これは、サンプルの純度の公正な定性的尺度を与える。しかし、低レベルの増幅は、アガロースゲル上で視覚化されないことがある。いくつかの報告にはqPCR 14などの品質管理のより定量的な尺度が含まれています。両方の方法から単離されたDNAサンプル純度の定量的評価のために、qPCRおよびシークエンシングを利用して、サンプル中の核DNAの量ならびにミトコンドリアDNAと葉緑体DNAの相対的比率を決定した。ここで評価した両方の方法は、核DNAを除去するのに有効です。両方の方法は、ミトコンドリアDNAと葉緑体DNAの混合物を異なる比率で産生するが、
暗闇の中で成長する植物(テルモレーション)は、フェノール系物質の減少によるオルガネラの単離を促進すると報告されているしかし、この比較では、光を発したサンプルよりも腐敗した組織での作業には大きな利点は見いだせなかったが、特殊な葉緑体の割合は、光が生育すると高くなると思われるが、葉緑体ゲノムにマッピングされている読みの割合に反映されているが、異なる光条件では変化しないため、異なる組織または異なるストレッサー下でのヘテロプラスミーの評価や発現解析などの下流機能解析では、生理学的に関連する条件下で成長した植物。
短リードシーケンシング技術を用いたアプリケーションでは、ここで比較した両方の技術が十分なDNA量と品質をもたらします。しかし、1分子シークエンシングアプリケーションで20kbを超える長い読み取りを達成するためには、より高品質のDNAがより多く必要です。例えば、理想的には、>1μgの純粋なorga分子量が20 kb以上のネラ小麦DNAは、20 kb挿入ライブラリー調製物のための社内の低入力プロトコールに必要です42 。新規ユーザーが開発した、低入力プロトコルは、DNAの要件( すなわち、50 ngのにまたは20でもそれ以下)を減らすことができるが、課題は、高品質、ライブラリの準備に入る高分子量のDNAを持っていません。より大きいDNAが> 20kbであることが不可欠である。なぜなら、より小さい断片が優先的にSMRTbellに挿入され、ライブラリー43のサイズ分布を捨てるからである。私たちは、自家製のDNA抽出プロトコールと、DNA抽出のためのいくつかの商業プロトコール(図示せず)を試みた。小麦葉組織について、DNAの量と質との間の最適なバランス、特に長さは、市販のキット27、29を用いて得ました。関心のある植物種および組織に応じて、オルタナティve抽出プロトコルは、同様に適しているか、またはより有益であり得る。それにもかかわらず、MBD2-Fcプルダウンアプローチ28による分画に続いて、> 50kbの高分子量ゲノムDNAの全抽出が、限定された出発物質からの長時間の配列決定に適していると結論する。今後の研究では、ロングインサートライブラリーの調製とそれに続くロングリードシーケンシングの分画に必要な出発物質の限界を試験する必要があります。重要なことに、このアプローチは、全ゲノム増幅を伴わずに、長い読み取り配列に適した単一葉のサブサンプルからDNAを単離する堅牢な方法を提供することができる。我々は、このアプローチが追加の組織タイプに容易に適応可能であり、他の植物種に広く適用可能であると予想している。それは、交差スキームまたはより希少な組織タイプにおける個々の世代での配列決定のような、組織量が制限されている状況において特に有用である。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者らは、競合する利益がないと宣言している。
この刊行物に記載されている商号または商品の説明は、特定の情報を提供する目的のみであり、米国農務省の推奨または保証を意味するものではありません。 USDAは平等な機会提供者と雇用者です。
Acknowledgments
私たちは、米国農務省農業研究庁と国立科学財団(IOS 1025881およびIOS 1361554)からの資金提供を認めたいと思います。私たちはR. Caspersに温室保全とプラントケアに感謝します。イルミナライブラリーの調製と配列決定が行われたミネソタ大学ゲノミクスセンターにも感謝します。私たちはまた、ジャーナル編集者と4人の匿名の査読者からのコメントに感謝して、私たちの原稿をさらに強化しました。我々はまた、OECDに、これらのプロトコルを日本の同僚との共同プロジェクトに統合するためのSKへのフェローシップに感謝する。
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
