

Summary
2 가지 plant organellar DNA 농축 방법의 비교 및 최적화가 제시됩니다 : 전통적인 차동 원심 분리 및 메틸화 상태에 기초한 총 gDNA 분획. 우리는 결과로 나오는 DNA 양과 품질을 평가하고, 짧은 읽기 차세대 시퀀싱에서의 성능을 시연하며, 장시간 읽기 단일 분자 시퀀싱에서의 사용 가능성에 대해 논의합니다.
Abstract
식물 세포 소기관 게놈은 복잡한 구조 및 / 또는 서브 게놈 조각을 형성하기 위해 페어링 또는 재조합을 거칠 수있는 크고 반복적 인 요소를 포함합니다. Organellar 게놈은 또한 주어진 세포 또는 조직 유형 (heteroplasmy) 내의 혼합물에 존재하며, 아형의 다량은 발육 기간 동안 또는 스트레스하에있을 때 (부분 화학량 론적 이동) 변화 할 수있다. 세포 기관 게놈 구조와 기능에 대한 더 깊은 이해를 위해서는 차세대 시퀀싱 (NGS) 기술이 필요합니다. 전통적인 sequencing 연구는 organellar DNA를 얻기 위해 몇 가지 방법을 사용합니다 : (1) 대량의 시작 조직이 사용되면 균질화되고 차등 원심 분리 및 / 또는 기울기 정화가 이루어집니다. (종자, 재료 또는 공간이 한정되어있는 경우, 즉) (2) 조직의 작은 양이 사용된다면, 동일한 프로세스는 (1) 충분한 DNA를 얻기 위해 전체 게놈 증폭 다음과 같이 수행된다. (3) 생물 정보학 분석은 seq전체 게놈 DNA를 분석하고 세포 기관 판독을 분석합니다. 이러한 모든 방법에는 고유 한 문제점과 단점이 있습니다. (1)에서 이와 같이 많은 양의 시작 조직을 얻는 것이 어려울 수 있습니다. (2)에서, 전체 게놈 증폭은 시퀀싱 바이어스를 도입 할 수있다; (3)에서 핵과 세포 내 게놈 간의 상동 성은 집합과 분석을 방해 할 수있다. 거대한 핵 게놈을 가진 식물에서, organellar DNA를 풍부하게하여 생물 정보학 분석을위한 서열화 비용 및 서열 복잡성을 줄이는 것이 유리하다. 여기에서 전통적인 차동 원심 분리 방법과 네 번째 방법 인 CpG- 메틸 풀다운 방식을 비교하여 총 게놈 DNA를 핵 및 세포 내 분획으로 분리합니다. 두 가지 방법 모두 미토콘드리아와 엽록체의 비율이 다르긴하지만 세포 내 소기관 서열에 대해 고도로 농축 된 DNA 인 NGS에 충분한 양의 DNA를 생산합니다. 우리는 밀 잎 조직을위한 이러한 방법의 최적화를 제시하고 주요 이점과 d샘플 입력, 프로토콜 용이성 및 다운 스트림 응용 프로그램의 맥락에서 각 접근법의 단점.
Introduction
게놈 시퀀싱은 중요한 식물 형질의 기본 유전 적 기초를 분석하는 강력한 도구입니다. 대부분의 게놈 시퀀싱 연구는 대부분의 유전자가 핵에 위치하므로 핵 게놈 내용에 초점을 맞 춥니 다. 그러나, (진핵 생물에서) 미토콘드리아와 색소체 포함 organellar 게놈 (식물, 전문화 된 형태로, 엽록체, 광합성에서 작동)는 생명체의 개발, 스트레스 반응 및 전반적인 체력 1에 필수적인 중요한 유전 정보를 기여한다. Organellar 게놈은 일반적으로 핵 게놈 시퀀싱을위한 총 DNA 추출에 포함되지만 DNA 추출 이전에 세포 소기관 수를 줄이는 방법도 사용됩니다 2 . 많은 연구에서 전체 gDNA 추출물의 sequencing 결과를 사용하여 organellar 게놈 3 , 4 , 5 ,xref "> 6 , 7. 그러나 연구 대상이 세포 소기관 게놈에 초점을 맞추고있을 때 많은 gDNA가 핵산 염기 서열에"손실 "되어 있기 때문에 전체 핵산을 사용하면 시퀀싱 비용이 증가합니다. 적당한 게놈, bioinformatically 8 도전 2에 관한 것이다. 또한, 염기 서열의 정확한지도 위치를 해결 중복 핵 게놈 내로 소기관 사이 organellar 서열의 전사에 의한 읽기. 핵 게놈 organellar 게놈의 정제 하나이다 전략을 사용하여 미토콘드리아와 엽록체 사이의 상 동성 영역으로 매핑되는 판독을 분리 할 수 있습니다.
많은 식물 종의 세포 소기관 게놈이 서열화되어 있지만, 소기관 게놈 다양성의 폭은 거의 알려져 있지 않습니다야생 개체군 또는 경작 된 육종 풀에서 이용 가능하다. Organellar 게놈은 또한 반복 서열 9 사이의 재조합으로 인해 중요한 구조적 재 배열을 거친 동적 분자로 알려져있다. 또한, 세포 소기관 게놈의 여러 사본이 각 세포 소기관에 포함되어 있으며, 여러 세포 소기관이 각 세포 내에 포함되어 있습니다. 이 게놈의 모든 사본이 동일하다는 것은 이질균이라고 알려져 있습니다. "마스터 서클"의 정경과는 달리 하위 게놈 서클, 선형 염색체, 선형 concatamers 및 가지 형 구조 10을 포함하여 organellar 게놈 구조의 더 복잡한 그림에 대한 증거가 증가하고 있습니다. 식물 세포 소기관 게놈의 조립은 상대적으로 큰 크기와 상당한 거꾸로 된 직접 반복에 의해 더욱 복잡해진다.
세포 기관 분리, DNA 정제 및 후속 유전체에 대한 기존의 프로토콜 전자 시퀀싱은 종종 성가 시며 많은 양의 조직을 필요로하며 수백 g의 어린 잎 조직을 출발점 11 , 12 , 13 , 14 , 15 , 16 , 17로 필요로 합니다. 이렇게하면 조직이 제한되어있을 때 세포 내 게놈 시퀀싱을 사용할 수 없게됩니다. 어떤 상황에서는, 세대 간 (through generational basis) 또는 교차점을 통해 유지되어야하는 수컷 멸균 계통에서 필요할 때와 같이, 종자의 양은 제한적이다. 이러한 상황에서 세포 내 소기관 DNA를 정제 한 다음 전체 게놈 증폭을 수행 할 수 있습니다. 그러나 전체 게놈 증폭은 구조적 변이, 하위 게놈 구조 및 이종성 수준을 평가할 때 특히 중요한 문제인 시퀀싱 바이어스를 도입 할 수 있습니다> 18. 짧은 읽기 시퀀싱 기술에 대한 라이브러리 준비의 최근 발전은 전체 게놈 증폭을 피하기 위해 낮은 입력 장벽을 극복했습니다. 예를 들어, Illumina Nextera XT 라이브러리 준비 키트는 1 ng의 적은 DNA를 입력으로 사용할 수 있습니다 19 . 그러나 PacBio 또는 Oxford Nanopore 시퀀싱 기술과 같은 장시간 시퀀싱 응용 프로그램을위한 표준 라이브러리 준비에는 여전히 많은 양의 입력 DNA가 필요하며 이는 세포 내 게놈 시퀀싱에 대한 도전이 될 수 있습니다. 최근에, 입력량을 줄이고 마이크로 그램 - 양의 DNA를 얻기가 어려운 시료에서 게놈 시퀀싱을 용이하게하기 위해 사용자가 작성한 긴 판독 시퀀싱 프로토콜이 개발되었습니다 20 , 21 . 그러나, 고 분자량의 순수한 세포 내 소 분획을 이들 라이브러리 제제에 공급하는 것은 여전히 어려운 과제이다.
우리는o 전체 게놈 증폭없이 NGS에 적합한 세포 내 DNA 농축 및 분리 방법을 비교 및 최적화합니다. 구체적으로, 우리의 목표는 잎의 하위 샘플과 같이 제한된 출발 물질로부터 고 분자량 세포 기관 DNA를 풍부하게하는 우수 사례를 결정하는 것이 었습니다. 이 연구는 organellar DNA를 풍부하게하는 방법의 비교 분석을 제시한다 : (1) 상업적으로 이용 가능한 DNA CpG- 메틸 결합 도메인 단백질 풀다운 접근의 사용에 기초한 DNA 분획 프로토콜 (2) 수정 된 전통적인 분별 원심 분리 프로토콜 22 식물 조직에 적용 23 . 우리는 다른 식물 및 조직 유형으로 쉽게 확장 될 수있는 밀엽 조직으로부터 세포 기관 DNA를 분리하는 최상의 방법을 권장합니다.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. 세포질 분리 및 DNA 추출을위한 식물 재료의 생성
- 밀 묘목의 표준 성장
- 구석 당 4 ~ 6 개 종자가있는 작고 정방형의 냄비에 질석에 식물 종자를 심습니다. 온실 또는 성장 챔버로 16 시간주기, 23 ℃ / 18ºC 밤으로 옮긴다.
- 매일 식물에 물을주십시오. 발아시 및 발아 후 7 일째에 ½ 티스푼의 과립 20-20-20 NPK 비료로 식물을 비옥하게하십시오.
- 밀 묘종의 대안 적 순환
- 1.1 단계를 따르되 냄비를 진한 성장 챔버, 23 ° C에서 16 시간 / 18 ° C 동안 8 시간 동안 둔다. 또는 온실에서 식물을 덮으십시오 ( 예 : 저장 용기로, 그러나 적절한 환기가 유지되어야 함).
- 성장 및 조직 수집
- 12 ~ 14 일 동안 식물을 재배하십시오. 대부분의 밀 유전자형s, 75-100 묘목은 약 10-12 g의 조직을 생성하는데, 이는 차동 원심 분리 방법 (섹션 2)을 사용하는 두 개의 세포 기관 추출에 충분하다. 핵 DNA에서 세포 소기관을 분획하기 위해 DNA CpG- 메틸화 기반 풀다운 접근법을 이용하는 경우 단 하나의 식물 만이 필요하다 (3 절).
- 차별적 인 원심 분리 방법을 사용하는 경우, 조직을 신선하게 수집하고 섹션 2에서 설명한대로 즉시 샘플 처리로 진행하십시오.
- CpG- 메틸 풀다운 방법을 사용하는 경우, 어린 잎 조직 20mg을 마이크로 원심 분리기 튜브에 채취하십시오 (표준 성장 또는 반발 조직을 사용하십시오, 대표 결과 참조). 액체 질소에서 급속 동결하고 사용할 때까지 -80 ºC에서 동결하십시오. 3 절에서 설명한대로 DNA의 풀다운 분획으로 진행하십시오.
2. 방법 # 1 : 차동 원심 분리 (DC)를 이용한 DNA 추출
참고 : differential 원심 분리 프로토콜은 모두 세포 소기관을 분리하지만, 미토콘드리아 (17), (24)에 대해 풍부하게하는 조건을 최적화이 출판물에서 수정되었습니다. 결과 프로토콜은 시간 집약성이 적고 이전 방법보다 적은 독성 화학 물질을 사용합니다. 구체적으로 우리는 STE 추출 버퍼에 폴리 비닐 피 롤리 돈 (PVP)을 첨가하고 불화 나트륨 (NaF)이 들어있는 NETF 완충액에서 최종 세척 단계를 제거하는 것을 포함하여 완충액과 세척 단계를 수정했습니다.
주의 사항 : STE 완충액의 준비와 사용은 2- 메르 캅토 에탄올 (BME)을 함유하고 있으므로 적절한 개인 보호 장비가있는 화학 흄 후드에서 수행해야합니다.
- 시작하기 전에 할 일
- 모든 장비가 매우 깨끗한 지 확인하고, 오토 클레이브 될 수있는 모든 장비를 오토 클레이브하십시오 ( 예 : 분쇄 실린더, 고속 센 트리퓨전 튜브 등 ).
참고 : 교차 오염을 방지하기 위해 피펫 팅이 필요한 모든 단계에 필터 팁을 사용하는 것이 좋습니다. - 필요한 장비 및 시약 목록을보고 방법 # 1 ( 표 1 )에 필요한 완충액 및 작업 스톡을 준비하십시오. 극저온 분쇄 블록을 -20 ℃로 냉각시키고 로터와 완충액을 4 ℃로 냉각시키고, 미소 원심 분리기를 4 ℃로 설정 한 다음 37 ℃의 수조를 켜십시오.
- 모든 장비가 매우 깨끗한 지 확인하고, 오토 클레이브 될 수있는 모든 장비를 오토 클레이브하십시오 ( 예 : 분쇄 실린더, 고속 센 트리퓨전 튜브 등 ).
- 소기관의 격리
- 5g의 신선한 조직을 수확하고 얼음 위에서 식힌 비이커에 넣고 차가운 멸균 수에서 씻어냅니다.
참고 : 원심 분리기, 흄 후드 등의 모든 작업 및 운반 중에는 항상 시료를 얼음 위에 보관하십시오. 프로토콜을 수행하기에 충분한 공간과 장비를 사용할 수없는 경우에는 차가운 방에서 작업하십시오. - 가위를 사용하여 잎 조직을 ~ 1cm 조각으로 잘라내어 2 개의 세라믹 연삭이 들어있는 50 mL 튜브에 직접 담습니다.실린더.
참고 : 교차 오염을 방지하기 위해 샘플 사이의 가위를 청소하거나 교환하십시오. - 조직 호 모지 나이저가 없다면 박격포와 유봉을 사용하여 단계 2.2.4-22.9를 대체하십시오.
- 얼음에 미리 냉장 된 박격포에 잎 조직을 자르십시오. 15 mL의 STE (흄 후드 내)에서 2 ~ 3 분 동안 시료를 갈아서냅니다.
- 미리 젖은 멸균 여과포 (~ 22 - 25 μm의 공극 크기, 자세한 내용은 주 프로토콜 참조)의 한 층이 들어있는 깔때기를 통해 버퍼를 떼어 내고 (모르타르에 티슈를 남긴다) 다른 50 mL 튜브 . 박격포에 STE 10 mL를 추가로 첨가하고 다시 균질화한다.
- 균질화 된 조직과 완충액을 같은 깔대기에 붓는다. 박격포를 헹구고 10 mL의 STE로 갈아서 깔때기에 붓는다. 가능한 한 많은 액체를 회수하기 위해 여과 천을 깔때기에 끼 우고 조입니다.
참고 : 교차 오염을 방지하기 위해 샘플 사이의 장갑을 교환하십시오. 프로와 함께 계속단계 2.2.10에서 토 콜.
- 각 50 mL 관에 20 mL의 STE (흄 후드에)를 넣으십시오.
- 시료를 연삭 장치의 미리 냉각 된 극저온 연삭 블록에 넣고 2 x 30 초 동안 1,750 rpm으로 연삭합니다. 샘플 위치를 회전시키고 그라인드 사이에서 ~ 1 분 동안 얼음 위에 샘플을 놓습니다.
참고 :이 단계에서는 박격포와 유봉, 블렌더 또는 기타 조직 분쇄 / 균질화 장치를 사용할 수 있습니다. 그러나 각 방법은 생성 된 DNA 품질에 영향을 미치므로 DNA 길이와 품질을 평가해야 다운 스트림 응용 프로그램을 계속 진행할 수 있습니다. - 얼음에 담긴 깨끗한 50-mL 튜브에 깔대기를 넣으십시오. 여과 천을 한 겹으로 깔고 5 mL의 STE로 미리 적시십시오. 유출 물을 버리지 마십시오.
- 균질화 된 조직을 깔때기에 붓습니다. 분쇄 튜브를 STE 15 mL로 헹구고, 다시 채우고 튜브를 뒤집어 벽과 뚜껑을 헹구십시오.엘자.
- 조심스럽게 세라믹 스톤을 제거한 다음 여과 천을 깔때기로 짜서 짜내십시오.
참고 : 교차 오염을 방지하기 위해 샘플 사이의 장갑을 교환하십시오. - 유출을 방지하기 위해 튜브 캡을 파라 필름으로 감싸십시오. 4 ℃에서 10 분간 2,000 xg로 원심 분리하십시오.
- 조심스럽게 혈청 피펫을 사용하여 상등액을 흡인하고 (50 mL 고속 원심 분리 튜브에 넣으십시오 (튜브가 단단한 씰링 개스킷을 가지고 있지 않은 경우 유출을 피하기 위해 parafilm으로 튜브 캡을 감싸십시오). 알약을 버리십시오.
- STE를 사용하여 0.1g 이내로 튜브를 조화시키고 18,000 xg 및 4ºC에서 20 분 동안 생성 된 상등액을 원심 분리하십시오. 튜브의 균형을 맞추기 위해 저울 위에 작은 얼음 비커를 올려 놓고 저울을 올려 놓고 샘플을 얼음 위에 올려 놓아 차갑게 유지하십시오. 또는 차가운 방에서 저울과 흄 후드를 사용하십시오.
- 뜨는 물을 버린다. 펠릿에 1 mL의 ST를 넣고 부드럽게 다시 정지시킨다.부드러운 그림 붓. ST (최종 부피 25 mL) 24 mL와 혼합 / 소용돌이 ( 즉, 모든 액체를 제거하기 위해 튜브 측면의 붓을 누르십시오).
- ST를 사용하여 0.1g 이내로 튜브의 균형을 맞 춥니 다. 18,000 xg 및 4ºC에서 20 분간 원심 분리하십시오. 한편, DNaseI 솔루션을 준비하십시오 (재고 및 작동 솔루션 레시피는 표 1 참조). 각 샘플에 대해 1.5 mL 튜브에서 200 μL 분취 량으로 만듭니다.
- 상층 액을 버리고 튜브를 털어 내고 부드러운 페인트 브러시를 사용하여 300μL ST에서 펠렛 (고속 원심 분리 관에 남아 있음)을 다시 현탁하십시오. 이전에 준비한 DNaseI 용액 200 μL가 들어있는 1.5 mL 튜브에 그림 붓을 넣고 그림 붓을 소용돌이 쳐 브러시에 붙어있는 잔여 펠렛을 제거하십시오. DNaseI 용액을 다시 고속 원심 분리 관에 넣고 가볍게 혼합하여 혼합합니다.
- 37 ° C의 수온에서 30 분간 품어 낸다. (응축 누출을 방지하기 위해 튜브의 꼭대기를 감싼다.g). 부화 도중 2 번 소용돌이 치듯 부드럽게 섞는다.
- 부드럽게 넓은 오리피스와 피펫 팁을 사용하여 튜브 밖으로 펠렛 혼합물을 피펫과 1.5 ML 낮은 바인딩 튜브에 놓으십시오. 고속 centrifuge 튜브에 400 MM EDTA, 산도 8.0의 500 μL를 추가하고 부드럽게 피펫 튜브의 모든 잔여 펠렛을 얻을 피펫. 펠렛 혼합물과 같은 1.5 ML, 낮은 바인딩 튜브에 EDTA를 전송하고 부드럽게 반전에 의해 섞는다.
- 4 ° C에서 20 분 동안 18,000 xg에서 원심 분리하십시오. 상등액을 버리고 튜브를 털고 DNA 분리를 위해 즉시 사용하십시오. 필요한 경우, -20 ℃에서 펠렛을 동결 시키십시오. 그러나 잔류 DNaseI가 즉시 처리되지 않으면 샘플 DNA를 분해 할 수 있으므로 수율 감소가 발생할 수 있습니다.
- 5g의 신선한 조직을 수확하고 얼음 위에서 식힌 비이커에 넣고 차가운 멸균 수에서 씻어냅니다.
- 상용 컬럼 기반 접근법을 사용하여 격리 된 기관으로부터의 DNA 추출
참고 : 전체 프로토콜 25 는 키트 안내서를 참조하고 수정 사항은 아래를 참조하십시오. Pr세포 기관 분리로부터 DNA 추출까지 직접 진행하는 것이 바람직하다. 반복 된 동결 및 해동은 잔류 DNaseI에 의한 DNA 단편 크기를 줄이고 DNA 분해를 유발합니다. vortexing 또는 활발한 pipetting을 제한하십시오, 이것은 DNA를 전단시킬 수 있습니다. DNA 회수율을 최대화하려면 저 결합 마이크로 원심 분리 관을 사용하는 것이 좋습니다.- DNA 추출 과정
참고 : 완충제가 적절히 제조 / 저장되고 스핀 - 컬럼 절차가 이해되도록하기 전에 상세한 상용 프로토콜 25 를 읽으십시오.- 펠렛과 튜브에 직접 버퍼 ATL의 180 μL를 (이전에 동결하고 벤치 탑에 상온으로 평형 경우 해동)에 추가합니다.
- 키트 핸드북의 "DNA Purifications from Tissues"프로토콜의 3 단계를 진행합니다. 다음 수정 사항이 적용됩니다. 3 단계에서 30 분 분해하고, RNase A 분해 옵션을 포함하고, 3 x 200 μL의 AE에서 용리하십시오 각자parate tube)에 넣고 용출액을 합친다.
- qPCR에 대한 분량 (최소 20 μL)을 저장하십시오 (4.1 단계 참조). 집중시키기 전에 정량하기 위해 고감도 정량을 위해 1μL를 추가로 저장하십시오.
- 원하는 경우, 시료 농축을 계속하십시오.
- DNA 추출 과정
- 상업용 필터 유닛으로 샘플 농도
참고 : 자세한 내용은 상용 프로토콜 26 을 참조하십시오. 다운 스트림 용도에 따라, 시료 농축 ( 예 : 종점 PCR 및 qPCR 적용)을 수행 할 필요가 없습니다. 그러나 NGS 라이브러리 구축을 위해서는 DNA 추출 후 희석 된 세포 내 DNA를 농축하는 것이 필요합니다.- 농도 칼럼 절차
- 디지털 분석 저울에 깨끗한 무게의 종이에 빈 필터 장치 (튜브가없는)를 조심스럽게 미리 칭량하십시오 ( 표 2 참조). 무게를 기록하십시오.
- 파이혼합 된 용리액을 필터 장치에 넣고 신중하게 다시 무게를 재십시오.
참고 : 상업용 설명서 26에 따르면 필터 장치의 최대 부피는 500 μL이지만 최대 575 μL를 오버플로없이 즉시 장치에 추가 할 수 있습니다. - 채워진 필터 장치를 조심스럽게 튜브 (컬럼과 함께 제공됨)에 넣으십시오. 필요한 집중 량을 달성하기 위해 원하는 시간 동안 500 xg에서 원심 분리하십시오. ~ 575 μL의 샘플 볼륨의 경우, 20 분 스핀은 보통 15 - 30 μL의 농축 볼륨을 초래합니다.
- 튜브에서 필터 장치를 꺼내고 다시 무게를 잰다. 표를 사용하여 원하는 농축 량이 달성되었는지 확인하십시오. 그렇지 않은 경우, 짧은 시간 동안 500 xg에서 다시 원심 분리하고 다시 무게를 측정하십시오. 원하는 농축 물의 양에 도달 할 때까지 반복하십시오.
- 새 튜브 (컬럼과 함께 제공)를 필터 장치 상단에 놓고 뒤집습니다. 1000 xg에서 3 분간 원심 분리기를 옮기고튜브로 이동합니다.
- 복구 된 볼륨을 결정하십시오. 이것은 일반적으로 필터 유지로 인해 계산 된 부피보다 3 ~ 5 μL 적습니다. 과도하게 농축 된 경우 멸균 수 또는 TE로 희석하여 원하는 용량을 얻으십시오.
- 고감도 정량을 사용하여 DNA를 정량화하십시오 (제조업체 지침에 따라).
- 농도 칼럼 절차
3. 방법 # 2 : 총 게놈 DNA에서 세포질 DNA를 풍부하게하는 메틸 분획 화 (MF) 접근법
참고 :이 프로토콜은 식물과 곰팡이 27 및 상업 Microbiome DNA 보충 키트 프로토콜 28에 대한 사용자가 개발 한 게놈 팁 키트 DNA 추출 프로토콜에서 수정되었습니다. 이론 상으로는 고 분자량 DNA를 생성하는 DNA 분리 프로토콜이 풀다운에 사용될 수 있습니다. 짧은 읽기 시퀀싱의 경우, 주로 15 kb 이상인 조각이 풀다운에 사용하기에 적합합니다. lo를 위해판독 시퀀싱을 사용하면 더 큰 조각이 바람직 할 수 있습니다. 따라서, 우리는 고 분자량 DNA를 산출하기 위해이 프로토콜을 최적화했습니다.
- 전체 DNA의 분리
참고 : 필요한 장비 및 시약 목록을보고 방법 # 2 ( 표 1 )에 필요한 버퍼 및 작업 스톡을 준비하십시오. 용해 버퍼 효소를 용해 버퍼 스톡에 첨가하여 용해 버퍼 작동 용액을 만든다. Thermomixer를 켜고 37 ° C로 설정하십시오. 50 ° C로 수 욕조를 켜고 욕조에 QF 버퍼를 놓으십시오. 냉동기에 70 % EtOH를 넣고 microcentrifuge를 4 ° C로 설정합니다.- 상업용 DNA 추출 컬럼을 이용한 총 DNA 추출
참고 : 시작하기 전에 중력 흐름 음이온 교환 컬럼의 사용에 대한 자세한 정보는 상업용 핸드북 29 를 읽으십시오. 컬럼은 전용 랙을 사용하여 설치하거나 제공된 플라스틱 링을 사용하여 튜브 위에 놓을 수 있습니다. g를 포함한 모든 단계에녹 팁은 중력 흐름에 의해 진행되어야하며 잔류 액체는 강제로 배출되어서는 안됩니다.- 2 mL 튜브 용으로 설계된 휴대용 연마 분을 사용하여 2 mL 저 결합 튜브에서 액체 질소로 냉동 된 조직 20 mg을 갈아줍니다.
- 용해 완충액 작업 용액 2 mL를 넣으십시오 (튜브가 너무 꽉 차 있습니다).
- 300 rpm으로 부드럽게 교반하면서 37 ° C에서 1 시간 동안 thermomixer에서 품어 낸다. Thermomixer를 사용할 수없는 경우 열 차단기에서 배양하고 매 15 분마다 부드럽게 섞어서 혼합하는 것이 좋습니다.
- RNase A (100 mg / mL, 최종 농도 200 μg / mL) 4 μL를 넣으십시오. 반전하여 37 ℃에서 30 분 동안 thermomixer에서 배양하고 300 rpm에서 완만하게 교반한다.
- 80 μl의 proteinase K (20 mg / mL, 최종 농도는 0.8 mg / mL)를 첨가하여 혼합하고, thermomixer에서 2 시간 동안 50 ° C에서 300 rpm으로 부드럽게 교반합니다.
- 4 ° C에서 1 분 20 분 동안 원심 분리기불용성 잔해를 펠렛 화하기 위해 5,000 xg.
- 시료를 원심 분리하는 동안 1 mL의 Buffer QBT로 컬럼을 평형시키고 컬럼을 중력 흐름으로 비울 수있게하십시오.
- 와이드 보어 피펫 팁을 사용하여 시료를 평형 칼럼에 즉시 뿌리고 (펠렛을 피함) 컬럼을 통해 완전히 흐르게하십시오. 샘플을 컬럼에 적용하기 전에 다시 흐림, 필터 나 원심 분리기를지면 (29 상세 상업적 핸드북 참조).
- 일단 시료가 수지에 완전히 들어간 다음 4 x 1 mL의 Buffer QC로 컬럼을 씻으십시오.
- 깨끗한 2 ML, 낮은 바인딩 microcentrifuge 튜브 위에 열을 일시 중단합니다. 50 ° C에서 미리 예열 된 완충액 QF 0.8 mL로 게놈 DNA를 용출시킨다.
- 용리 된 DNA에 상온의 이소프로판올 0.56 mL (용리 완충액 0.7 부피)를 첨가하여 DNA를 침전시킨다.
- 반전 (10X)하여 혼합하고 즉시 15,000 xg 및 4 ° C에서 20 분간 원심 분리하십시오. 케어유리질이 느슨하게 붙어있는 펠렛을 방해하지 않고 상청액을 완전히 제거한다.
- 차가운 70 % 에탄올 1 ML과 centrifuged DNA 펠렛을 씻으십시오. 15,000 xg 및 4 ° C에서 10 분간 원심 분리하십시오.
- 조심스럽게 펠렛을 방해하지 않고 뜨는 (이 단계에서도 신중해야 함)을 제거하십시오. 5 ~ 10 분 동안 공기 건조하고 0.1 ML 용출 버퍼 (EB)에 DNA를 resuspend. DNA를 실온에서 밤새 녹인다. DNA를 전단시킬 수있는 피펫 팅을 피하십시오.
- 고감도 DNA 정량 분석을 사용하여 샘플을 정량화하십시오 (제조업체 지침에 따라).
- 상업용 DNA 추출 컬럼을 이용한 총 DNA 추출
- 비드 기반 메틸화 및 비 메틸화 DNA 분획
참고 : 최근의 간행물은 인간 IgG Fc 단편 (MBD2-Fc 단백질)에 융합 된 CpG- 특이적인 메틸 결합 도메인 단백질을 이용하여 풀다운 방식을 이용하여 상업적으로 이용 가능한 키트 28핵 게놈 (고도로 메틸화 된)으로부터 식물 세포 소기 게놈 (비 메틸화)을 먹었습니다 23 . 밀 시료의 분획 효율은이 상업용 MF 키트 28을 사용하여 이전에 테스트하지 않았습니다.- 시작하기 전에 할 일
- 갓 80 % 에탄올 (반응 당 최소 800 μL)을 준비하십시오. 얼음에 해동하고 샘플 당 1x 버퍼 5 ML을 준비 5x 바인딩 / 세척 버퍼를 설정 (멸균, nuclease 무료 물로 5X 버퍼를 희석하고 프로토콜 동안 얼음에 계속).
- MBD2-Fc 단백질 바인딩 된 자기 구슬을 준비
- 필요한 개수의 비드 세트를 준비하십시오. 1 ~ 2 μg의 총 입력 DNA를 사용하기 위해 반응을 조정하십시오. 160 - 320 μL의 구슬이 필요합니다. 아래에 나열된 반응은 총 입력 DNA 1 μg에 해당하므로 160 μL의 구슬이 필요합니다. 필요에 따라 반응 규모를 조정하십시오.
- 와이드 보어 팁을 사용하여 Protein A Magnetic B를 부드럽게 피펫 팅하십시오슬러리를 상하로 흔들어 균질 한 현탁액을 만듭니다. 대안으로 4 ℃에서 15 분 동안 구슬 튜브를 부드럽게 회전시킵니다.
참고 : 구슬을 와동하지 마십시오. - 제조업체 지침에 따른 지시 사항을 따르십시오 28 .
- 메틸화 핵 DNA 캡처
- 각각의 개별 시료에 1 μg의 입력 DNA를 160 μL의 MBD2-Fc 결합 자성 비드가 들어있는 튜브에 첨가합니다.
- 최종 농도 1x (5x 바인드 / 씻어 버퍼의 용량 (μL) = 입력 DNA (μL) / 4의 부피)에 대한 DNA 입력 샘플의 부피를 고려하여 적절하게 5x 결합 / 세척 버퍼를 추가하십시오. 샘플을 위아래로 몇 번 피펫 과이드 보어 피펫 팁을 사용하여 섞는다.
- 튜브를 실온에서 15 분간 회전시킨다. 시료를 넓은 구멍의 피펫 팁으로 부드럽게 피펫을 부어서 부풀기를 방지하기 위해 잠복기 전체에 2 ~ 3 회 fl습니다.
참고 : 피펫 팅 및 플리 키ng는 메틸화 된 DNA의 효율적인 풀다운을 보장하는 데 중요합니다.
- 농축 된 비 메틸화 세포 기관 DNA 수집
- DNA와 MBD2 - Fc - 바인딩 마그네틱 비드 혼합물을 포함하는 튜브를 간단히 스핀. 적어도 5 분 동안 자성 선반에 튜브를 놓고 튜브의 측면에 구슬을 수집하십시오. 해결책은 명확하게 나타납니다.
- 광구 팁을 사용하여 조심스럽게 구슬을 방해하지 않고 맑은 상등액을 제거하십시오. 상청액 (메틸화되지 않은 세포 기관질이 풍부한 DNA 포함)을 깨끗한 저 결합 2 mL 미세 원심 분리 튜브에 옮긴다. 이 샘플을 -20 또는 -80 ° C에서 보관하거나 정제를 위해 3.2.6 단계로 직접 진행하십시오.
- MBD2-Fc- 결합 자성 비드로부터 포획 된 핵 DNA를 용출시켰다
- 핵 분획도 원할 경우 제조사의 지침 28 에 따라 MBD2-Fc 결합 자성 비드에서 핵산 DNA를 용리한다. 단계 3.2.7에서 설명한대로 정제하십시오.
- 구슬 기반 핵산 정제
- 정화 비드가 실온에 있고 완전히 혼합되어 있는지 확인하십시오. MF 키트 설명서 28 의 지침에 따라 프로토콜을 계속 진행하십시오.
참고 : 이제 샘플을 NGS 라이브러리 구성 또는 다른 다운 스트림 분석에 사용할 수 있습니다.
- 정화 비드가 실온에 있고 완전히 혼합되어 있는지 확인하십시오. MF 키트 설명서 28 의 지침에 따라 프로토콜을 계속 진행하십시오.
4. 샘플 정량 및 품질 관리
- 세포 기관 농축을 평가하기위한 qPCR 분석
참고 : 여기에 나열된 qPCR 반응 및 분석 파라미터는 Roche LightCycler 480에서 사용하도록 설계되었으며 다른 장비 및 시약에 맞게 조정해야 할 수 있습니다. qPCR을 사용할 수없는 경우 여기에 설명 된 것과 동일한 프라이머 및 조건을 사용하여 종점 PCR 및 아가 로스 겔에서의 시각화를 시료 순도의 정량적 측정으로 사용할 수 있습니다. Amplicon 크기는 모든 프라이머 세트에 대해 ~ 150 bp입니다. 프라이머 시퀸은 표 3 을 참조하십시오.ces 및 pairings.- qPCR 반응 설정
- 20 μL qPCR 반응을 개별적으로 설정하려면 다음을 96- 웰 qPCR 플레이트의 단일 웰에 조심스럽게 피펫 팅하십시오. 2 x SYBR Green I Master 10 μL; 2 μL 10 μM 정방향 및 역방향 프라이머 믹스 (0.5 μM의 최종 농도); 템플릿 2 μL (표준 곡선의 범위 내); 멸균 된 nuclease-free H 2 O 6 μL. pipetting 오류를 줄이려면 템플릿을 제외한 모든 반응 구성 요소와 마스터 믹스를 만드는 것이 좋습니다. qPCR 플레이트에 마스터 믹스를 추가하고 관심있는 템플릿을 각 웰에 첨가하십시오. 피펫 팅 오류의 영향을 최소화하기 위해 각 시료에 대해 3 번의 기술 복제를 수행해야합니다.
참고 : 궁극적으로 핵과 세포 간 정량 사이클의 비율을 표본간에 비교하므로 농도의 약간의 차이가 허용됩니다. 그러나 DNA 농도는 대략적으로 범위 내에 있어야합니다기타 채널. - 고품질의 qPCR 씰링 필름으로 플레이트를 밀봉하십시오. 부드럽게 거품을 생성하지 않도록주의하면서 샘플을 와동시킵니다. 샘플을 수집하고 작은 거품을 제거하기 위해 4 ° C에서 2 분 동안 플레이트를 짧게 회전시킵니다.
- 플레이트를 기계에로드하십시오. 아래 나열된 지침에 따라 qPCR 프로그램을 실행하십시오.
- 20 μL qPCR 반응을 개별적으로 설정하려면 다음을 96- 웰 qPCR 플레이트의 단일 웰에 조심스럽게 피펫 팅하십시오. 2 x SYBR Green I Master 10 μL; 2 μL 10 μM 정방향 및 역방향 프라이머 믹스 (0.5 μM의 최종 농도); 템플릿 2 μL (표준 곡선의 범위 내); 멸균 된 nuclease-free H 2 O 6 μL. pipetting 오류를 줄이려면 템플릿을 제외한 모든 반응 구성 요소와 마스터 믹스를 만드는 것이 좋습니다. qPCR 플레이트에 마스터 믹스를 추가하고 관심있는 템플릿을 각 웰에 첨가하십시오. 피펫 팅 오류의 영향을 최소화하기 위해 각 시료에 대해 3 번의 기술 복제를 수행해야합니다.
- qPCR 반응 파라미터
참고 : 이것은 증폭 단계의 어닐링 사이클을 제외하고는 기본 매개 변수입니다. 사용 된 프라이머가이 프로토콜에 제시된 프라이머와 다른 경우이 프라이머를 수용하도록이 설정을 조정하십시오.- 4.4 ℃ / s의 상승 속도로 5 분 동안 95 ° C에서 사전 배양하십시오.
- 4.4 ℃ / s의 상승 속도로 (1) 95 ℃에서 10 초 동안 45 번의 증폭 사이클을 수행하십시오. (2) 20 ° C에서 60 ° C, 2.2 ° C / s의 상승 속도; (3) 72 ℃에서 10 초 동안, 4.4 ℃ / s의 상승 속도 ((3)에서 얻어진 데이터).
- optio 사용4.4 ℃ / s의 램프 속도로 5 초 동안 95 ° C의 용융 곡선 사이클; 65 ° C에서 1 분간, 램프 속도는 2.2 ° C / s. 및 97 ° C, 연속 수집 모드.
- 30 ° C에서 40 ° C의 냉각주기를 1.5 ° C / s의 램프 속도로 사용하십시오.
- 분석 매개 변수
- SYBR 템플리트를 선택하십시오. 실험 버튼에서 프로그램 매개 변수를 확인하십시오. 일단 플레이트가로드되면 분석이 시작될 수 있으며 분석이 실행되는 동안 설정이 조정될 수 있습니다.
- 샘플 편집기를 사용하여 샘플을 지정하십시오. Abs Quant를 워크 플로로 선택하고 샘플을 알 수 없음, 표준 또는 음성 컨트롤로 지정합니다. 복제본을 지정하고 각 복제본의 첫 번째 샘플 이름을 채 웁니다. 농도와 단위를 기준에 추가하십시오.
- 분석을위한 부분 집합을 설정하십시오. 이것들은 부분 집합 편집기에서 지정됩니다.
- 분석을 위해 "새 분석 작성"목록에서 Abs Quant / 2nd Derivative Max를 선택하십시오.외부 저장 표준 곡선 (해당되는 경우)을 가져온 다음 계산을 누르십시오. 보고서에는 선택한 정보가 포함됩니다.
- 카피 수 또는 농도를 결정하기 위해 정확한 절대 정량을 수행하려면, 시험되는 샘플을 대표하는 표준 곡선 ( 예 : 위의 방법으로 분리 된 organellar DNA)을 사용하십시오. 표준 곡선을 준비하기 위해 필요한 미토콘드리아 DNA의 양은 너무 많아 적당량의 조직을 얻을 수 없으므로 소프트웨어가 제공하는 사본 번호 계산을 사용하지 말고 교차점 (Cp) 값을 조사하여 상대적 농축을 결정합니다 샘플에서 핵 세포 DNA와 비교 한 세포 소기관의 이 상대적 양을 전체 게놈 DNA의 양과 비교하십시오 ( 대표 결과 참조). 완전히 빛이 자란 2 주된 밀 묘목의 총 게놈 DNA 5 개 1 : 10 희석액에 대한 프라이머 효율 테스트 (대표적인 효능은그림 2의 전설).
- qPCR 반응 설정
- 펄스 필드 겔 전기 영동 (PFGE)
참고 :이 프로토콜은 고 분자량 DNA를 분해하기 위해 PFGE를 수행하는 제조업체의 지침을 기반으로합니다. Materials Table을 참조하십시오 .- 겔과 샘플 준비
- 겔 및 샘플 준비에 대한 지침을 따르고 사용 가능한 시스템에 적용하십시오.
- 매개 변수를 실행하십시오.
- 전기 영동 시스템 설정에 대한 지침을 따르고 다음과 같은 매개 변수를 사용하십시오 : 초기 전환 시간 2 초, 최종 전환 시간 13 초, 실행 시간 15 시간 16 분, V / cm 6 및 포함 각도 120 ° .
- 젤의 얼룩과 이미지
- 젤을 염료 ( 예 : ethidium bromide 또는 적절한 대안)로 염색하고 적합한 젤 문서화 시스템을 사용하여 이미지를 만듭니다.
- 겔과 샘플 준비
- DNA 라이브러리 준비 키트의 입력으로 1 ng의 DNA를 제조업체의 지침에 따라 사용하십시오.
- 한 번에 시퀀싱을위한 샘플을 바코드 및 풀링합니다. 제조업체 지침에 따라 시퀀싱을 수행하십시오.
참고 : 풀링 및 시퀀싱 매개 변수는 관심 종, 원하는 커버리지 수준 및 라이브러리를 시퀀싱하는 데 사용되는 플랫폼에 따라 변경 될 수 있습니다. 예를 들어 HiSeq 레인은 MiSeq 레인보다 실질적으로 더 많은 출력을 가지므로 더 많은 샘플이 다중화 될 수 있습니다. 세포 소기관 게놈의 적용 범위 수준이 하류 분석에 적합한 지 여부를 결정하기 위해 샘플의 더 작은 하위 집합을 시퀀싱합니다.- FastQC 31 을 사용하여 판독 품질을 검사하여 데이터에 필요한 트리밍 및 필터링 범위를 결정합니다.
- Trimmomatic 32 또는 다른 유사한 프로그램을 사용하여 원시 읽기를 정리하고 필터링하십시오. 다음 설정을 사용하십시오 : ILLUMINACLIP 2시 30 분 10 초 (어댑터 제거), 3 단계, 3 단계, SLIDINGWINDOW 4시 10 분, 그리고 MINLEN 100.
- 품질 필터링 및 어댑터 트림 페어링 엔드 (PE)는 중국어 스프링 미토콘드리아 (NCBI 레퍼런스 시퀀스 NC_007579.1 33), 엽록체 (NCBI 레퍼런스 시퀀스 NC_002762.1 34)을 사용하여 핵 Bowtie2 36 35 개 참조 게놈 읽 약도 -I 0 -X 800 - 민감합니다.
- sam 정렬 파일을 bam 형식 (samtools)으로 변환하고 bam 파일을 정렬합니다. bam 파일을 사용하여 bedtools로 게놈 전체 적용 범위 및베이스 별 적용 범위를 계산합니다. R- 플롯 기능으로 결과를 시각화하십시오.
- 시작하기 전에 할 일
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
이 원고에 제시된 프로토콜은 식물 조직에서 세포 기관 DNA를 풍부하게하는 두 가지 방법을 설명합니다. 여기에 제시된 조건은 밀의 조직 최적화를 반영합니다. 프로토콜의 주요 단계, 필요한 조직 입력 및 DNA 출력의 비교는 그림 1에 설명되어 있습니다. 우리가 테스트 한 DC 프로토콜의 단계는 이전에 설명한 것과 비슷한 조건을 따른다 ( 그림 1A ). 수확 된 조직은 신선하게 처리해야하며 차별화 된 원심 분리 및 / 또는 그라디언트를 적용하여 손상되지 않은 세포 기관을 분리해야합니다. 소기관이 용해되기 전에 핵 DNA가 제거되고 마지막으로 DNA가 추출되어 하류 적용에 사용됩니다. 대조적으로, MF 프로토콜에서, 식물 조직은 사용 전에 수확되고 저장 될 수 있으며, 손상되지 않은 세포 소기관은 필요하지 않습니다. 그 대신, 핵 및 세포 기관 DNA는 전체 gDNA에서 분획됩니다. DNA의 메틸화 상태. 두 프로토콜 모두 거의 같은 양의 세포 기관 DNA를 생성합니다 ( 그림 1C ). 조직 입력에 대한 총 세포 내 DNA 출력의 측면에서, MF 프로토콜은 단일 식물의 작은 샘플을 사용할 수 있고 조직을 추가 분석을 위해 성장시킬 수 있으므로 조직이 제한되어있을 때 유리합니다. 일반적으로 DC 프로토콜에서는 많은 묘목의 모든 공중 조직이 필요하며 이러한 식물은 폐기됩니다. 그러나, DC 방법은 MF 접근법으로는 불가능한 하나의 세포 소기관 유형에 대해 특히 농축되도록 최적화 될 수있다. MF 접근법에 직접 참여하는 시간은 적지 만 각 프로토콜의 총 시간은 대략 같습니다.
두 방법 모두 미토콘드리아와 플라 스티드 서열의 비율이 다른 경우에도 오르 셀라 DNA를 풍부하게합니다 :
"> 정제 organellar DNA의 매우 낮은 양 (~ 50의 순서 - 100 NG;도 1c)에있어서 어느 하나로부터 얻어진다. organellar 게놈 농축 핵 게놈 오염의 수준을 평가하는 DNA는 DC 및 MF 모두 분리에 방법을 사용하여 qPCR 분석법을 사용 하였다.이 분석에서, 3 개의 앰플 리콘 ( 즉, 핵 특이 적, ACTIN , 미토콘드리아 특이 적, NAD3 및 엽록체 특이 적, PSBB )을 전체 게놈 DNA에서 평가하고, 세포 내 DNA 분획을 두 방법 모두로부터 얻었다 ( 도 2 ). 각 샘플 ( 그림 2A )에 대해 정량 사이클 (C q ) 값을 조사했으며, C q 는 표적 증폭으로부터의 형광이 배경 형광 수준 이상으로 증가하는 PCR 사이클로 정의되었으므로 C q 와 표적 풍부는 역관계. 에서직류 샘플 NAD3 및 PSBB의 C의 Q는 각각 ~ 17 ~ 15 회 이전 (의 C의 Q ~ 36를 갖는다) 액틴보다 (C의 Q 값과 보충 수준도 2b 참조)이다. 이것은 NAD3 및 PSBB 각각 이론적으로 167,181 및 47,790 배 농축 된 것과 같 으며, DC 샘플 ( 그림 2B , 계산을위한 그림 2 의 범례 참조)에서 ACTIN 에 비례합니다. 총 게놈 DNA 샘플에서 ACTIN에 비해 NAD3 및 PSBB 의 배 증가 는 각각 158 및 10,701에 불과합니다. organellar 게놈 핵 게놈 (37)보다 및 세포 소기관의 체육의 수 있음을 셀당 더 큰 복사본 번호에 있는지 주어, 전체 게놈 DNA의 핵의 증폭를 기준으로 organellar 증폭 산물의 높은 풍요 로움을 찾는 놀라운 일이 아니다r 세포는 조직 유형 또는 발달 단계에 따라 다를 수 있습니다 38 , 39 . 전반적으로, DC 방법은 원심 분리 속도가 미토콘드리아를 선택적으로 분리하고 핵 및 엽록체 "오염"을 줄이기 위해 최적화되므로 미토콘드리아를 우선적으로 풍부하게한다는 것을 나타낸다.MF 총 gDNA의 메틸화되지 않은 분획은 또한 두 세포 기관 모두의 상당한 농축을 나타내며, 이들 표적의 천연 상대적인 양을 유지할 것으로 예상된다. 메틸화 분획 액틴 NAD3 및 PSBB 상대적인 폴드 부화 각각 20,551 및 1,703,253이다 (도 2A 및 2B). 메틸화 분획에서, ACTIN에 비하여 NAD3 및 PSBB에 대한 배 증가 는 각각 31 및 823이고, indiMBD2-Fc 단백질이 메틸화 된 핵 DNA의 풀다운에서 매우 효율적이라고 주장했다. 엽록체 증폭 물은 총 게놈 DNA (~ 6 C q 초기), 메틸화 분획 (~ 5 C q 초기) 및 비 메틸화 분획 (~ 6 C q 초기)에서 미토콘드리아 증폭 물보다 더 풍부하기 때문에 이것은 이들 앰플 리콘의 천연 풍부는 MDB2 풀다운에 의해 실질적으로 변화되지 않는다. 우리는이 게놈을 구체적으로 시퀀싱하는 것에 관심을 갖고 있기 때문에 비 메틸화 된 세포막 (organellar) 분획물에 초점을 맞추고 있습니다. 그러나 핵 게놈이 주요 관심사라면, MF와 메틸화 분획의 연속 시퀀싱은 세포 내 DNA "오염"의 감소로 인해 총 게놈 DNA 시퀀싱보다 훨씬 높은 핵 게놈 범위를 산출 할 것이다.
qPCR이 이용 가능하지 않다면, 종단점 PCR (qPCR과 동일한 프라이머를 사용)이 qualita세포 기관 순도에 대한 평가. 이 경우, 순수한 세포 소기 DNA 샘플은 미토콘드리아 및 플라 스티드 앰플 리콘에 대한 증폭을 보여 주지만, 아가로 오스 겔에서의 핵산 증폭 물의 검출 가능한 증폭은 보이지 않는 반면, 전체 게놈 DNA는 이전 연구 11 , 12 .
두 가지 방법으로 분리 된 organellar DNA는 NGS에 적합합니다 :
Trimmed and cleaned PE sequencing reading (4.3 단계 참조)은 이전에 발표 된 wheat organellar reference genomes에 매핑되었고, 각 샘플을 매핑하는데 사용 된 read 량은 ~ 800,000에서 1,100,000까지의 범위였다 ( 그림 3I ). de novo Illumina sequencing에서 이용 가능한 밀 엽록체 및 미토콘드리아 게놈지도 결과는 qPCR res(미토콘드리아 (mt) 및 엽록체 (cp) 게놈 각각에지도의 그림 3A 및 3B , ~ 80 % 및 ~ 10 %가 맵핑 됨) 및 MF 방법 2 개의 세포 소기관 게놈의 천연 풍부 성을 반영하는 DNA를 생성한다 ( 그림 3A 및 3B , 판독의 ~ 20 % 및 ~ 80 %가 각각 mt 및 cp 게놈에 매핑 됨). 두 가지 방법 모두, 밀 소기형 게놈의 이론적 적용 범위 (계산을위한 그림 3 의 범례 참조)는 100X 적용 범위 (및 MF 방법의 비 메틸화 분획에서 엽록체 게놈에 대한 최대 2,000X 범위) 12 개의 라이브러리가 다중화되면 ( 그림 3C 및 3D ,이 분석에 포함 된 6 개의 라이브러리는 별도의 분석을 위해 추가로 6 개의 라이브러리와 풀링되어 총 12 개의 라이브러리단일 시퀀싱 레인에 모아짐). 적용 범위에 대한보다 상세한보기는 특정 깊이에서 커버 된 게놈의 분율뿐만 아니라베이스 당 커버리지 수준 ( 그림 3E -3I )을 조사함으로써 달성되었다. MF 방법의 경우 평균 1베이스 적용 범위는 mt 게놈의 경우 ~ 300 - 450X이고 cp 게놈의 경우 4,000 - 5,000X입니다. DC 방법의 경우, mt 및 cp 게놈의 평균베이스 당 적용 범위는 각각 ~ 900 - 1,300 및 ~ 500 - 700X입니다. 그러나, 매우 낮은 또는 높은 적용 범위를 갖는 mt 및 cp 게놈의 작은 분획이 있었고, 이는 어느 방법으로부터 도 유도 된 세포 소기관 DNA에서 보였다 ( 도 3I ). 평균 이상의 적용 범위는 organellar 게놈 사이의 상 동성 영역에 해당하며, 낮은 적용 범위는 서열화 된 품종과 출판 된 참고 문헌 간의 SNP 또는 기타 작은 변이를 나타낼 수 있습니다. 이 개념을 뒷받침하기 위해 이러한 스파이크의 높은 적용 범위는 MF 방법 (도 3E 및 3I )으로부터 유도 된 mt DNA에 대해 가장 두드러 졌는데 , 이는이 방법에서 cp 게놈의 높은 적용 범위로 인한 것일 수있다. 설명 할 수 없게 cp 게놈의 적용 범위는 cp DNA를 따라 MBD2-Fc 풀다운에서 약간의 편향으로 인한 DC 방법 ( 그림 3G 및 3H )보다 MF 방법에서 불균등합니다. 왜 이런 경우인지를 결정하기 위해 추가 실험이 필요할 것입니다. 그럼에도 불구하고, mt와 cp 게놈은 상대적으로 균등 한 적용 범위를 가지고 있었고 주어진 범위에서 서열화 된 게놈의 분획을 조사하여 증명할 수있는 누락 된 범위의 큰 영역도 없었다 ( 그림 3E -3H ). 또한 두 게놈에 대한 적용 범위 수준은 변형 분석과 같은 다운 스트림 분석에 충분하다고 간주됩니다. 희소 한 변이의 분석에 필요하다고 생각되는 경우,r 풀 된 샘플은 더 큰 적용 범위를 얻을 수 있습니다. HiSeq 라이브러리는 현재 PE300 MiSeq 라이브러리와 달리 PE150 길이로 제한되어 있기 때문에 훨씬 더 많은 수의 샘플을 HiSeq 레인에 풀링 할 수 있습니다. 시퀀스 길이를 희생 시키더라도 훨씬 더 큰 시퀀싱 깊이를 얻을 수 있습니다.
매핑 접근법을 사용하여 핵 게놈 오염 수준을 조사하기 위해 PE 판독 매핑 범주를 검사했습니다. PE 판독은 다양한 구성으로 기준 게놈에 매핑 할 수 있습니다. 읽기 1과 2가 두 개의 메이트간에 일정한 "예상"거리를두고 (라이브러리의 평균 삽입 크기를 기반으로하며 대개 매핑 소프트웨어의 입력 매개 변수로 지정됨) 헤드 - ),이 PE 읽기는 "일치하여"매핑됩니다. 대조적으로, "불일치"지도 작성은 동료가 예상보다 적거나 크게 예상 할 수없는 상황으로 매핑하는 상황입니다(head-to-tail 또는 tail-to-tail)로 참조 게놈이나 맵에 접근 할 수 있습니다. 하나의 동료 만이 참조 게놈과 정렬된다면, 그 PE 판독은 참조 게놈과 일치하지 않거나 불일치하게 매핑되지 않는다. 세 가지 읽기 - 매핑 범주 모두에서 PE 읽기는 하나 또는 여러 번 참조 게놈에 정렬 될 수 있습니다.
DC와 MF가 분리 된 organellar DNA의 경우, 미토콘드리아 게놈에 대한 판독 매핑은 우연히 정렬 된 일치하는 한 시간 카테고리 ( 그림 4A ) 였지만, 읽기는 엽록체 게놈에 대해 상대적으로 동일한 비율로 한 번 일치하고 일치하는 한 번 ( 그림 4B ), 아마도 엽록체 게놈에있는 큰 거꾸로 된 반복 및 매우 높은 적용 범위 수준 때문일 수 있습니다. 그러나 핵 게놈에 매핑 된 PE 읽기가 적고 한 번에 한 번 이상 매핑됩니다.일치하거나 불일치하는 방식 ( 즉, 오직 한 명의 친구 만지도 할 수 있음). 이들은 세포괴 게놈이나 잘못 조립 된 지역에 상동 인 핵 게놈의 서열에 "off-target"을 매핑 할 가능성이 가장 높습니다. ( 그림 4C ) DC 또는 MF 방법으로부터 분리 된 세포 기관 DNA의 낮은 수준의 핵 게놈 오염을 나타내는 작은 양의 판독 (<5 %)만이 핵 게놈에 일치하여 qPCR 결과에도 반영된다 ( 그림 4 2A ). Chinese Spring non-ethiolated tissue로부터의 MBD2-Fc 풀다운 후 핵 분획을 시퀀싱하여 pulmown이 메틸화되지 않은 DNA의 제거에서 얼마나 효율적인지를 결정 하였다. 세포 분별 유도 라이브러리에서 세포 내 참조 게놈에 매핑 된 읽기의 1 % 미만이지만 모든 읽기의 약 45 %가 핵 게놈에 매핑되었습니다 ( 그림 4 ). 그러나 대부분의 읽기는 불협화음으로 매핑됩니다.이는 밀 핵 참조 게놈에서 높은 수준의 오 조립과 단편화를 반영 할 가능성이있다. 그럼에도 불구하고, 결과는 MBD2-Fc 풀다운이 메틸화 된 핵 DNA로부터 메틸화되지 않은 세포 기관 DNA를 제거하는데 매우 효율적임을 시사한다. 이러한 방법으로 생성 된 세포 내 풍부한 DNA는 미토콘드리아와 엽록체 서열의 혼합물을 포함하고 있으며, 이들 세포 소기관 사이의 고대 유전자 전달로 인한 서열 유사성이 유전체에 남아 있기 때문에, 게놈은 생물 정보 학적으로 해결되어야합니다.
잎 조직의 표층화는 오르테 톨 풍부를 현저하게 변화시키지 않는다 :
전통적으로, 유리질 추출물을 방해 할 수있는 페놀 및 전분의 수준을 줄이기 위해 식물 미토콘드리아 DNA 분리에 유리 된 조직이 선호된다n 또는 다운 스트림 응용 프로그램 13 . organellar 게놈 농축 수준이 성장 조건에 의해 변경되거나 개선 될 수 있는지를 결정하기 위해, etiolated 조직과 non-etiolated 조직 모두 MF 프로토콜과 시퀀싱을 받았다. 흥미롭게도, etiolation은 감지 할 수 없게 non-etiolated 조건에 비해 organellar 참조 게놈 ( 그림 3A 및 3B ) 또는베이스 별 적용 범위 ( 그림 3I )에 매핑 된 판독의 비율을 변경하지 않았다. 우리는 또한 etiolated 및 non etiolated 조직 모두와 차동 원심 분리를 사용하여 organellar DNA를 분리하고 qPCR를 사용하여 다른 조직 사이에 농축에 약간의 차이가 발견되었습니다 (데이터는 표시되지 않음). 이는 생리 학적으로 관련성이있는 비 - 탈식 (not-etiolated) 조직이 세포 증식 연구에 사용될 수 있음을 시사한다.
품질 관리MF DNA는 장시간 연속 시퀀싱에 가장 적합합니다 :
장기 판독 시퀀싱이 연구자들에게보다 쉽게 접근 할 수있게되면 고 분자량 DNA의 분리가 점차 중요 해지고 있습니다. 손상되지 않은 상태와 품질에 대해 분리 된 세포 기관 DNA를 평가하기 위해 PFGE가 사용되었습니다. 총 게놈 DNA는 전형적으로 PFGE에서 확산 번짐으로 옮겨지며, 분자량은 프로토콜과 DNA가 어떻게 추출 후 저장되고 취급되는지에 의해 결정됩니다. 게놈 팁으로 분리 된 전체 게놈 DNA는 50kb를 초과해야하며 PFGE를 사용하여 확인되었습니다 ( 그림 5 , 레인 2). 유전체 팁의 총 게놈 DNA는 세포 내 DNA에서 핵을 분별하기 위해 Microbiome Enrichment Kit에 입력으로 사용됩니다. 분획 화 후에 얻어진 핵 분획은 크기가 감소하지만 약 50kb를 중심으로 남아있다 ( 도 5 , 레인 4). 이것은 su가 아니다.MBD2-Fc- 결합 비드로부터의 용출로서 핵 분획의 상대적으로 거친 취급이 열 및 단백질 분해 효소 K 분해를 필요로한다는 점을 감안할 때, 놀랍다. 제한된 질량으로 인해 organellar fraction은 PFGE에서 실행되지 않았지만 TapeStation으로 분석 한 결과 DNA> 50kb가 나타났습니다 (데이터는 표시되지 않음). 차동 원심 분리로 얻은 세포 기관 DNA는 평균 20kb의 질량을 가지며, 이는 확장 된 세포 기관 분리 프로토콜과 그 이후의 컬럼 기반 DNA 추출 및 농축에 기인 한 것 같다. 그라데이션 기반 세포 기관 분리 및 대체 DNA 추출 방법은 더 큰 DNA 단편 크기를 유지할 수 있습니다. 그럼에도 불구하고,이 프로토콜에서 얻은 크기의 DNA는 라이브러리 준비 과정에서주의를 기울이면 10 또는 15kb 시퀀싱 판독을 생성하는 데 사용될 수 있습니다.
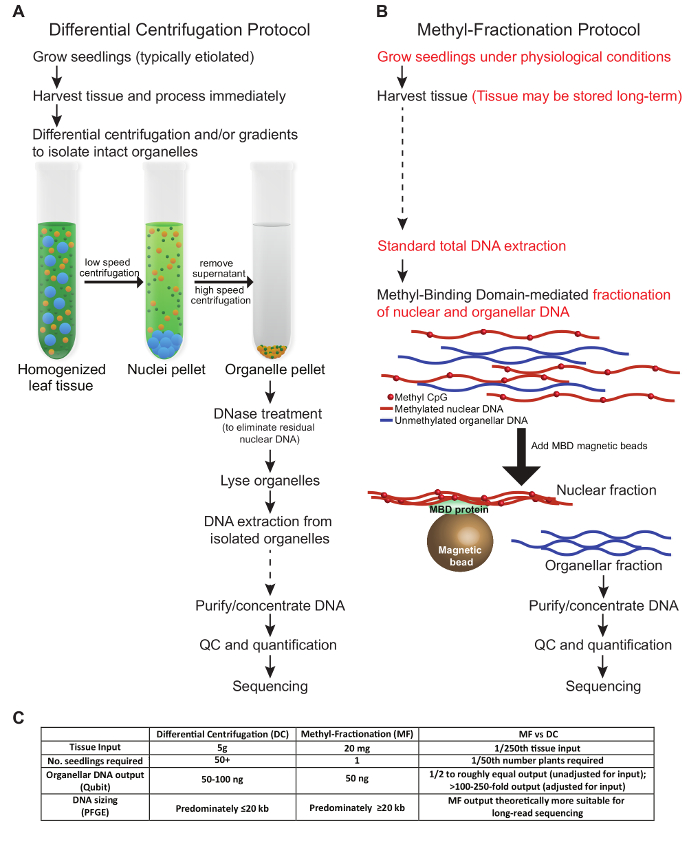
그림 1 : 두 개의 메 토의 비교보기Plant Organellar DNA에 대해 Enrich로 보내주십시오. 전통적인 DC 프로토콜 ( A )은 MF 프로토콜 ( B )과 대조됩니다. 샘플 얼기 및 해동을 피하는 것이 좋습니다. 그러나 샘플을 장기간 보관할 수있는 단계는 점선 화살표 ( A 및 B )로 표시됩니다. 프로토콜 간의 주요 차이점은 빨간색으로 강조 표시됩니다 ( B ). ( C )이 표는 조직 입력, 필요한 식물의 수, DNA 산출 및 결과 DNA 크기의 측면에서 방법을 비교한다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

그림 2 : 두 가지 방법으로 분리 된 세포질 DNA의 핵 DNA 오염 평가. (
(B)는 테이블 (A)의 그래프에 도시 된 C의 Q 값과 액틴에 organellar 증폭 상대적인 배 농축을 나타낸다. * Fold enrichment = 2 (Cq ACTIN - Cq Target) . 이 공식은 각 프라이머 세트에 대해 2의 완벽한 효율을 가정합니다.2에서 각 프라이머 세트의 이온은 무시할 수 있으며 계산 및 전반적인 경향에 거의 영향을 미치지 않습니다 ( ACTIN = 1.961, NAD3 = 1.95 및 PSBB = 1.989). 프라이머 효율은 총 게놈 DNA의 5 배 1:10 희석액으로 일련의 표준 곡선을 작성하여 평가 하였다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

그림 3 : 엽록체 및 미토콘드리아 게놈의 읽기 매핑 및 이론적 범위. 미토콘드리아 ( A ) 또는 엽록체 ( B ) 중국어 스프링 참조 게놈에 매핑 된 판독의 백분율. 중국 봄 미토콘드리아 ( C ) 또는 엽록체 ( D ) 참조 유전자의 상응하는 이론적 적용 범위mes, 각각 450 및 135 kb의 게놈 크기를 가정하고, 총 판독 수 및 상이한 게놈에 매핑 된 판독의 백분율을 사용하여 계산된다. MF 방법 ( E 및 G ) 또는 DC 방법 ( F 및 H )에서 세포 기관 DNA에 대한 범위의 게놈 분포. 패널 E - H 의 데이터는 중국 봄철 etiolated 샘플에서 나온 것이지만 다른 모든 샘플도 비슷한 경향을 보였다. ( Ⅰ ) 패널 A - D의 모든 샘플에 대한 평균, 최저 및 최고베이스 당 커버리지. "E"를 포함한 표본 라벨은 표본 추출 된 표본을 나타내며 "NE"는 비 표출 표본을 나타냅니다. DC는 차동 원심 분리법으로 분리 된 DNA를 나타내고, Unmethylated는 MBD2-Fc (MF 프로토콜)로 풀다운 후 비 메틸화 분획에있는 DNA를 나타낸다. "Chris"라고 표시된 샘플은 밀 Triticum aestivum을 지정합니다.'크리스.' CS는 밀 Triticum aestivum 'Chinese Spring의 샘플을 지정합니다. 참고 : 엽록체, 미토콘드리아 및 세포 내 소기관과 핵 게놈 사이의 고대 유전자 전달에 기인 한 핵 게놈과의 서열 상 동성으로 인해, 소량의 미가공 독서는 여러 게놈에 매핑 될 수 있습니다. 또한 organellar 참조 게놈에 매핑되지 않는 읽기는이 그림에 표시되지 않습니다. 따라서 여기에 표시된 백분율 ( A 및 B )은 총 100 %가 아닙니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

그림 4 : PE는 밀 핵 유전체에 매핑을 읽습니다. PE의 카테고리 백분율 미토콘드리아 (A) , 엽록체 (B) 또는 핵 (C) 중국 봄 참조 게놈으로 매핑 유형을 읽습니다. - E는 etiolated 샘플을 지정하고 - NE는 비 etiolated 샘플을 지정합니다. DC는 차동 원심 분리법으로 단리 된 DNA를 나타내고, 비 메틸화는 MF 프로토콜에서 MBD2-Fc로 풀다운 후 풀린 비 메틸화 분획에있는 DNA를 나타내고, 메틸화는 MBD2-Fc 풀다운 후 핵 분획을 나타낸다. "Chris"라고 표시된 샘플은 밀 Triticum aestivum 'Chris'를 나타냅니다. CS는 밀 Triticum aestivum 'Chinese Spring'의 샘플을 지정합니다. 맵핑되지 않은 읽기는 표시되지 않습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
oad / 55528 / 55528fig5.jpg "/>
그림 5 : PFGE를 이용한 DNA 품질 검사. 분화 원심 분리 (레인 3)로부터 얻은 밀 총 게놈 DNA (레인 2), MBD2-Fc 풀다운 접근법 (레인 4)을 갖는 MF 후 핵 분획을 1 % 아가 로스 겔에서 마커로 사용 된 1 kb 확장 사다리 (레인 1 및 5). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
| 버퍼 이름 | 레시피 | 노트 | 방법 |
| STE 버퍼 | 50 mM Tris pH 7.8, 20 mM EDTA pH 8.0, 0.6 % (w / v) 폴리 비닐 피 롤리 돈 (PVP), 0.2 % (w / v) 소 혈청 알부민 (BSA), 0.1 % (v / v) β- 머 캅토 에탄올 (BME) | 자당, 트리스 및 EDTA만을 함유하는 완충액 혼합물은 한 달 전에 제조하여 4 ℃에서 보관할 수있다. PVP, BSA 및 BME는 사용 직전에 필요한 양의 완충액에 새로 첨가해야합니다. | 방법 # 1 |
| ST 버퍼 | 400 mM 슈 크로스, 50 mM 트리스 pH 7.8, 0.6 % (w / v) 폴리 비닐 피 롤리 돈 (PVP), 0.1 % (w / v) 소 혈청 알부민 (BSA) | 자당과 트리스 만 함유 된 완충액 혼합물은 한 달 전에 제조하여 4 ℃에서 보관할 수 있습니다. ST 완충액은 EDTA 또는 BME를 함유하지 않으며,보다 낮은 농도의 BSA를 함유한다. | 방법 # 1 |
| DNase 주식 | 0.15 M NaCl 중의 2 mg / ml DNase를 2 mg / ml의 스톡 농도 | -20 ° C에서 200 ㎕ 분주를 보관하십시오. DNase 작업 솔루션 (샘플 당 200 μl의 DNase 용액)을 준비하려면아래 표 1. DNase 분해에 대한 자세한 내용은 아래의 전체 프로토콜을 참조하십시오. DNase 작업 용액은 신선한 상태로 준비해야합니다. DNase 반응을 멈추기 위해서는 400 mM EDTA pH 8.0 용액이 필요합니다 (반응을 멈추기 위해 필요한 최종 농도는 0.2 M EDTA입니다, 자세한 내용은 전체 프로토콜 참조). | 방법 # 1 |
| DNase 작업 솔루션 | ST 완충액 0.25 ㎎ / ㎖의 DNase과 20 mm의 MgCl 2 | 샘플 당 200ul을 새로 준비하십시오. 농도는 최종 반응 부피에 대한 것이므로 혼합 : 62.5 μl 2 mg / ml DNase (최종 500 μl 반응 부피 기준), 4 μl 1M MgCl 2 (200 μl DNase 용액 부피 기준) 및 ST 버퍼 133.5 μl 200 μl의 최종 부피. | 방법 # 1 |
| 용해 완충액 | 20 mM EDTA pH 8.0; 10 mM Tris pH 7.9; 500 mM 구아니딘 -HCl; 200 mM NaCl; 1 % 트리톤 X-100; 0.5 mg / ml 용해 효소홍역 | lysing 효소를 제외하고 모든 성분을 섞고 실내 온도에 저장하십시오. 용해 효소는 즉시 사용하기 위해 작은 분액으로 새로 첨가해야합니다. | 방법 # 2 |
도표 1 : 집에서 만드는 완충기 및 일하는 주식의 조리법.
| 농도 워크 시트 | |||||||
| 샘플 이름 | 빈 장치 무게 (g) | 충전 된 장치의 무게 (g) | 채워진 양 (ul, 채워진 빈 무게) | 1 회 회전 후 무게 (20 분 *, g) | 1 차 스핀 후 볼륨 (ul, filled빈 무게 빼기) | 무게 후 2 차 스핀 (X min *, g) | 2 차 스핀 후 볼륨 (ul, 채워진 빈 무게) |
| 실제 복구 된 볼륨은 계산 된 볼륨보다 몇 ul 작을 것입니다. | |||||||
표 2 : 농도 워크 시트.
| 이름 | 게놈 특이성 | 유전자 서열 근원 | 서열 (5 '- 3') |
| Ta_ACTIN - F | 핵무기 | 그래 미네 비계 IWGSC_CSS_1AS_scaff_3272162 : 10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | 핵무기 | 같은 상기와 | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | 미토콘드리아 | NCBI 가입 EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | 미토콘드리아 | 같은 상기와 | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | 엽록체 | NCBI 가입 KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | 엽록체 | 같은 상기와 | GCTGCCTGTTTCCTTGTAGTT |
표 3 : qPCR 프라이머 목록.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
지금까지 대부분의 organellar sequencing 연구는 특정 DC를 풍부하게하는 전통적인 DC 방법에 초점을두고 있습니다. 모스 ( 40)를 포함하여 다양한 식물로부터 세포 소기관을 분리하는 방법이 설명되었다. 밀 15 및 귀리 11 과 같은 단핵구; 아라비돕시스 11 , 해바라기 17 , 유채 14 등의 족자입니다. 대부분의 프로토콜은 잎 조직 13 , 14 , 15 , 16 , 17 에 중점을두고 있으며 일부는 종자 11을 비롯한 다양한 조직 유형에 맞게 조정되었습니다. 원형질체로부터 세포 소기관의 분리도 (41)을 보여왔다. 그러나 이것은 모든 시스템에 적용 할 수 없으며 관심 대상 조직이 제한적일 때도 가능하지 않습니다. 많은 이들 orga네 라르 (nellar) 분리 방법은 생리 학적 연구와 같은 특정 실험을 위해 손상되지 않은 세포 기관을 복구하도록 설계되었습니다. 이러한 프로토콜은 번거롭고 일반적으로 특정한 세포막 분획물을 분리하는데 매우 효율적이지만 큰 조직 입력을 필요로하는 자당 또는 퍼콜 구배와 같은 밀도 구배를 사용해야합니다 ( 즉, 5g 이상 킬로그램을 초과하여 조직 유형). 그러나, DC 방법은 회전 속도 및 밀도 구배를 변화시킴으로써 미토콘드리아 또는 엽록체와 같은 특정 세포 분획을 풍부하게하도록 최적화 될 수있다. 대조적으로, MF 접근법은 훨씬 적은 시작 물질 (20mg)을 필요로하지만 미토콘드리아 및 플라 스티드 DNA는 DNA 추출에 사용되는 조직의 상대적인 존재 당 존재합니다. 그럼에도 불구하고 MF 프로토콜은 혼합 된 세포 기관 DNA를 분리하는 대체 방법을 제공하며 소량의 조직으로 시작하는 경우 특히 유용합니다.
티 기관 분리 후 샘플 순도를 평가하고, 현재까지의 대부분의 연구는 종점 PCR 및 겔 전기 영동 11 , 12 만을 사용합니다. 이것은 샘플 순도를 정성스럽게 측정합니다. 그러나 낮은 수준의 증폭은 아가 로스 겔에서 시각화되지 않을 수 있습니다. qPCR 14 와 같은 품질 관리에 대한 더 많은 양적 측정이 포함 된 보고서는 거의 없습니다. 두 방법 모두에서 분리 된 DNA 표본 순도의 정량적 평가를 위해 qPCR 및 시퀀싱을 사용하여 핵산이 표본에 얼마나 남아 있는지와 미토콘드리아 대 엽록체 DNA의 상대적 비율을 결정했습니다. 여기에서 평가 된 두 가지 방법 모두 핵 DNA를 제거하는 데 효율적입니다. 두 가지 방법 모두 미토콘드리아 DNA와 엽록체 DNA가 혼합되어 있지만, 비율은 다르지만.
어둠 속에서 자란 식물 (고리 화)은 페놀 릭 (phenolics)의 감소로 인해 세포 기관 분리를 촉진하는 것으로보고되었다ref> 13 그러나이 비교에서 우리는 빛이 자란 샘플보다 Etiolated 한 조직으로 작업 할 때 상당한 이점을 발견하지 못했다. 비록 빛을 재배 할 때 특수한 엽록체의 비율이 더 높을지라도 총 plastid 수는 엽록체 게놈에 매핑 된 판독의 비율에 반영된 것은 빛 조건에 따라 변하지 않기 때문에, 상이한 조직 또는 상이한 스트레스 인자 하에서의 이종성의 평가 또는 발현 분석과 같은 하류 기능 분석을 위해, 게놈 시퀀싱을 생리적으로 관련된 조건 하에서 자란 식물.
짧은 판독 시퀀싱 기술을 사용하는 어플리케이션의 경우 여기에서 비교 한 두 기술 모두 적절한 DNA 양과 품질을 산출합니다. 그러나 단일 분자 시퀀싱 응용 분야에서 20kb를 초과하는 독도를 오래 읽으려면 더 많은 양의 고품질 DNA가 필요합니다. 예를 들어, 이상적으로는 순수한 orga 1 μg 이상분자량> 20 KB와 넬러 휘트 DNA 20 kb의 인서트 라이브러리 제제 (42) 내에서, 저 입력 프로토콜이 필요하다. 새로운 사용자가 개발 한 저 입력 프로토콜은 DNA 요구량 ( 즉, 50 ng 또는 심지어 20 미만)을 감소시킬 수 있지만 높은 품질의 고 분자량 DNA를 라이브러리 준비 단계로 보내는 것은 여전히 어려움이 있습니다. 더 많은 조각이 우선적으로 SMRTbell에 삽입되고 라이브러리 43 의 크기 분포를 없애기 때문에 DNA의 대부분이 20kb 이상인 것이 필수적입니다. 우리는 많은 수제 DNA 추출 프로토콜과 DNA 추출을위한 많은 상용 프로토콜을 시도했다 (표시되지 않음). 밀엽 조직의 경우, DNA 양과 품질, 특히 길이 간의 최적의 균형은 시판용 키트 27 , 29를 사용하여 얻어졌다. 관심의 식물 종 및 조직에 따라, alternati추출 프로토콜이 똑같이 적합하거나 더 유익 할 수 있습니다. 그럼에도 불구하고, 우리는 MBD2-FC 풀다운 방식 28 분별 하였다 크기 고 분자량 게놈 DNA> 50킬로바이트 총 추출, 제한된 원료로부터 긴 판독 순서에 순종이라고 결론 지었다. 장래의 연구는 롱 - 인서트 라이브러리 준비 및 후속 장시간 읽기 시퀀싱에 필요한 분획 화 이후에 요구되는 출발 물질의 한계를 시험해야한다. 비판적으로이 접근법은 전체 게놈 증폭없이 장기 판독 시퀀싱에 적합한 단일 잎의 하위 샘플에서 DNA를 분리하는 강력한 방법을 제공 할 수 있습니다. 우리는이 접근법이 추가 조직 유형에 쉽게 적응할 수 있고 다른 식물 종에 광범위하게 적용될 수있을 것으로 기대합니다. 그것은 교차 조직이나 희소 한 조직 유형의 개별 세대에서의 시퀀싱과 같은 조직 양이 제한적인 상황에서 특히 유용 할 것입니다.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
저자는 경쟁적 이익이 없다고 선언합니다.
이 간행물의 상호 또는 상업 제품에 대한 언급은 특정 정보를 제공하기위한 목적 일 뿐이며 미 농무부 (US Department of Agriculture)의 추천이나 보증을 의미하지는 않습니다. USDA는 기회 균등 제공자이자 고용주입니다.
Acknowledgments
우리는 미국 농무부 농업 연구청과 국립 과학 재단 (IOS 1025881 및 IOS 1361554)으로부터 자금을 지원하고 싶습니다. 우리는 R. Caspers에게 온실 유지 관리 및 식물 관리에 감사드립니다. 우리는 Illumina 라이브러리 준비 및 시퀀싱이 수행 된 Minnesota Genomics Center 대학에도 감사드립니다. 저널 편집자와 4 명의 익명의 평론가가 우리 원고를 더욱 강화시킨 것에 대해 감사드립니다. 우리는 또한 OECD가 일본의 동료들과 협력 프로젝트를 위해 이러한 프로토콜을 통합하는 SK와의 협력에 감사한다.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
