

Summary
Inmunomarcación para analizar poblaciones distintas de los microtúbulos en el cerebro en desarrollo del pez cebra se describen los métodos, que son ampliamente aplicables a otros tejidos. El primer protocolo describe un método optimizado de inmunomarcación microtúbulos estables y dinámicos. El segundo protocolo proporciona un método para la imagen y cuantificar nacientes microtúbulos específicamente.
Abstract
Microtúbulos (MTs) son dinámicas y frágiles estructuras que son un reto a la imagen en vivo, particularmente en embriones de vertebrados. Aquí se describen los métodos de inmunomarcación para analizar poblaciones distintas de MTs en el tubo neural en desarrollo del embrión de pez cebra. Mientras el foco está en tejido neural, esta metodología es ampliamente aplicable a otros tejidos. Los procedimientos están optimizados para temprano a embriones en fase mediados somitogenesis (1 somite a 12 somitas), sin embargo se pueden adaptar a una amplia gama de otras etapas con ajustes relativamente menores. El primer protocolo proporciona un método para evaluar la distribución espacial de MTs estables y dinámicos y realizar un análisis cuantitativo de estas poblaciones con software de procesamiento de imagen. Este enfoque complementa las herramientas existentes para imagen microtúbulos dinámica y distribución en tiempo real, usa líneas transgénicas o la expresión transitoria de construcciones etiquetadas. De hecho, estas herramientas son muy útiles, sin embargo no distingue fácilmente entre MTs dinámicos y estables. La capacidad de la imagen y analizar estas poblaciones distintos microtúbulos tiene importantes implicaciones para los mecanismos de comprensión polarización celular y la morfogénesis. El segundo protocolo describe una técnica para analizar específicamente nacientes MTs. Esto se logra mediante la captura de las propiedades de crecimiento de novo de MTs en el tiempo, siguiendo la despolimerización de microtúbulos con el nocodazole drogas y un período de recuperación después de lavado de drogas. Esta técnica no se ha aplicado al estudio de MTs en embriones de pez cebra, pero es un valioso análisis para investigar la función en vivo de proteínas implicadas en el montaje del microtubule.
Introduction
Los microtúbulos (MTs) son polímeros de α - y β-tubulina que montar en protofilaments lineal, varios de los cuales se combinan para formar un tubo hueco1,2. MTs son estructuras polarizadas, con rápido crecimiento más extremos y de crecimiento lento menos extremos que están anclados en el centrosoma o de organización de microtúbulos centro (MTOC)3. De novo Formación de MT es iniciada por nucleación en el complejo de anillo de γ-tubulina (γ-TURC), que proporciona una plantilla para montaje MT4. En cualquier célula dada, dos poblaciones de MTs se pueden distinguir a su vez sobre a diferentes velocidades. Dinámicas MTs exploran su entorno celular por la conmutación entre las fases de crecimiento y contracción en un proceso conocido como inestabilidad dinámica5. A diferencia de dinámicas MTs, MTs estables son adultos y tienen una vida media más larga que la dinámica de MTs6.
Décadas de investigación en biología celular ha proporcionado un conjunto sofisticado de herramientas para el estudio de la función y la estructura de MT y dio lugar a un gran cuerpo de conocimiento sobre estos elementos citoesqueléticos. Por ejemplo, MTs juegan un papel central en el establecimiento y mantenimiento de la polaridad celular, que es atribuible no sólo a su polaridad intrínseca, sino también a la distribución subcelular diferencial estable versus dinámica MTs7, 8. en cambio, mucho menos se entiende sobre MT arquitectura y función en entornos más complejos tridimensionales (3D), como el embrión de vertebrados, en parte debido al desafío de citoesqueleto MT en alta resolución de imagen. A pesar de esta limitación, la reciente generación de transgénico GFP-expresando las líneas eso etiqueta MTs o expresión transitoria de fluorescencia etiquetada MT marcadores ha aumentado nuestra comprensión de los cambios dinámicos que MTs y su celular y papel del desarrollo en el embrión de pez cebra. Toda la red de MT puede ser reflejada en líneas transgénicas en que tubulina es directamente etiquetados polímeros9 o tubulina indirectamente están etiquetados con las proteínas asociadas a MT Doublecortin como cinasa (Dclk) o Ensconsin (EMTB)10, 11. Se han generado otras líneas (y construcciones) que permiten una evaluación de polaridad intrínseca MT al etiquetado específicamente MT más extremos o centrosoma anclado menos extremos11,12,13, 14. el poder de estas herramientas reside en la capacidad para estudiar la dinámica de la MT en vivo, desarrollo de organismos. Estos estudios han puesto de manifiesto, por ejemplo, la distribución espacial y dinámica de MTs en poblaciones celulares específicas, la orientación de mitotic husos en tejidos sometidos a morfogénesis (un indicador del plano de división celular), la polaridad del polímero MT lo que se refiere a procesos tales como la elongación celular y la migración y tasa de crecimiento de MT determinada por cometa velocidad9,13,15. La limitación de estas herramientas es que ellos no fácilmente discriminar entre poblaciones de MT estables y dinámicas.
A partir de la literatura de biología celular rico, inmunomarcación imagen de MTs estables y dinámicos en el embrión del pez cebra se describen los métodos, que son complementarios al uso de líneas transgénicas. El uso generalizado de tales métodos de inmunomarcación en el pez cebra ha sido un poco obstaculizado por la dificultad de preservar la integridad de MT durante el procedimiento de fijación. 1 Protocolo describe un método optimizado para inmunomarcación total, dinámica, y estables MTs en las secciones del cerebelo en desarrollo del pez cebra. Además, un método sencillo usando comercialmente el software disponible se describe para cuantificar las poblaciones de MT. Estables MTs se distinguen de MTs dinámicos basados en varias modificaciones poste-de translación de la α-tubulina, como la acetilación y detyrosination, que se acumulan de MTs estables largo tiempo16,17. En el embrión de pez cebra, la acetilación se produce MTs ciliares y axonales pero no estable interfase MTs18, limitando la utilidad de este marcador a un subconjunto de estabilizado MTs. En cambio, detyrosination parece ocurrir sobre todo estables MTs en el embrión de pez cebra18. Esta modificación poste-de translación expone el ácido glutámico de carboxi-terminal de α-tubulina (tubulina detyrosinated)18 y puede ser detectada utilizando anti-Glu-tubulina19. Aunque detyrosination se produce preferentemente en MTs estables, la evidencia experimental indica que esta modificación poste-de translación es el resultado de, en lugar de una causa de estabilidad MT16. Se distingue la población MT recíproca, compuesto por dinámicos MTs, usando un anticuerpo, anti-Tyr-tubulina, que reconoce específicamente la forma de tyrosinated de α-tubulina19. Después de inmunomarcación con estos marcadores y la proyección de imagen confocal, el análisis cuantitativo de MTs (longitud, número y abundancia relativa) puede realizarse en regiones definidas del tubo neural en desarrollo. Para realizar este análisis utilizando software de procesamiento de imágenes 3D se facilita un método racionalizado. Este método puede ser aplicado para resolver las preguntas con respecto a la morfogénesis y el establecimiento o la maduración de la célula polaridad20. De hecho, la elaboración de matrices polarizados de MTs estables acompaña a muchos eventos del desarrollo, incluyendo fotorreceptor morfogénesis21, epitelización de las células en el desarrollo neural tubo18 y axón formación8.
Protocolo n ° 2 describe una adaptación en vivo de un ensayo de Biología de la célula para analizar MTs durante su Asamblea fase (nucleación/anclaje y crecimiento)22,23. Nacientes MTs están nucleadas en el centrosoma y posteriormente ancladas a subdistal apéndices del centriolo madre23. Se describe un método para analizar el incipiente rebrote MT después de despolimerización. Este protocolo proporciona detalles sobre el tratamiento del nocodazole a despolimerizar MTs, el procedimiento de lavado de la droga y el período de recuperación después del tratamiento. MT re-crecimiento se controla a intervalos regulareslavado de post s por inmunomarcación con marcadores para MTs totales (anti β-tubulina) junto con marcadores para el centrosoma (anti γ-tubulina) y núcleo (4', 6-diamidino-2-phenylindole (DAPI)), según los procedimientos generales descritos en el Protocolo 1. El paso de la despolimerización de MT de este protocolo es esencial ya que permite la evaluación del crecimiento de MT de novo en lugar de extensión de MTs preexistentes. Esta técnica es por lo tanto distinta de los otros procedimientos publicados para medir las tasas de crecimiento de MT (en ausencia de despolimerización) mediante el uso de un marcador de punta más como fin vinculante proteínas 3 fusionado a la proteína verde fluorescente (EB3-GFP), como se muestra en Tran et al., 201211. Además, este análisis es particularmente útil para el análisis de embriones defectuosos en Asamblea de novo MT, como los mutantes previamente divulgados de NEDD1 en el que se deteriora el reclutamiento de γ-tubulina en el centrosoma, dando por resultado incompleto formación del tubo neural y defectos neuronales24.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
declaración de ética: los procedimientos describen a continuación sigue la Universidad de las pautas de cuidado de los animales de Maryland Baltimore County.
1. Análisis de estable y dinámico MTs uso de inmunomarcación (protocolo 1)
- dechorionation Manual de embriones antes de la fijación
- obtener recién generado embriones vertiendo el agua de exceso del sistema y luego recoger embriones restantes en un plato de Petri de plástico (consulte la Tabla de materiales).
- Eliminar cualquier residuo de los embriones de agua y la transferencia del sistema a un nuevo plato lleno con medio embrión (véase Tabla de materiales) para asegurar que los embriones se desarrollan en un ambiente limpio.
- Permitir que los embriones destinados a desarrollar la etapa deseada en una incubadora con control de temperatura a 28.5 ° C.
- Embriones de lugar menos de 24 h después fertilización (hpf) en un recipiente de vidrio antes de dechorionation.
Nota: Embriones de Dechorionate antes de la fijación para maximizar la rápida penetración de fijador y preservar la integridad del MT. Medio de embrión de uso en lugar de agua del sistema para proporcionar la Ca adicional 2 + durante la dechorionation. - Retire manualmente las chorions de embriones en placa de Petri, con unas pinzas finas con un microscopio de disección.
- Pellizque un área pequeña en el corion redondo, transparente que rodea el embrión con un par de pinzas y con cuidado tirar pinzas aparte para crear una ruptura en la membrana de.
- Ampliar la apertura por apalancamiento delicadamente en el corion roto con unas pinzas. Tenga cuidado de no tocar el embrión con el fórceps como podría romperse.
- Fijación de embriones etapas
- transferencia en escena, dechorionated embriones a tubos de centrífuga de 1.5 mL. Quitar como mucho medio embrión posible utilizando un pipeta Pasteur de cristal.
Nota: Realizar tratamientos de fijación y de la droga en embriones jóvenes (mid-somitogenesis), antes de la formación de los centros neuronales median la sensación de dolor, que no requiere ningún procedimiento adicional para aliviar el dolor durante la eutanasia. Etapas de desarrollo están definidos en Kimmel et al., 1995 25. Las etapas 4-5 y 11-12 del somite se utilizaron para obtener imágenes de las figuras 2 y 3. - Preparar 4% paraformaldehido (PFA) /MT Asamblea fijador de búfer (MAB) (véase Tabla de materiales) combinando 1 mL 8% PFA por 1 mL 2 X y añadir 2 μl 100% Tritón X-100 por 1 mL de volumen total.
PRECAUCIÓN: Use guantes durante la manipulación de soluciones que contengan PFA y Tritón X-100, que son irritantes de la piel. - Fix embriones en fijador de PFA/MAB 1 mL de 4% durante 5 minutos a 28.5 ° C. aspirado el fijador con una pipeta, reemplazarlo con fijador fresco 1 mL e incubar 3 h a temperatura ambiente (RT) en un eje de balancín.
Nota: Las muestras deben fijarse rápidamente en la temperatura del biológica (28,5 ° C de pez cebra) para prevenir la despolimerización de temperatura MT.
- transferencia en escena, dechorionated embriones a tubos de centrífuga de 1.5 mL. Quitar como mucho medio embrión posible utilizando un pipeta Pasteur de cristal.
- Aspire fijador y añadir 1 mL de 1 x solución salina con tampón Tris con NP40 (TBS-NP40) buffer. Frote suavemente en RT en un eje de balancín de tres veces por 5 minutos cada uno. Almacenar embriones a 4 ° C en 1 mL fresco 1 X TBS-NP40 durante no más de 7 días.
PRECAUCIÓN: Use guantes al manipular soluciones que contengan NP-40, un irritante de la piel. - Embriones de seccionamiento para inmunomarcación
- calor RT 4% bajo punto de fusión (LMP) agarosa incrustar medio en un recipiente cerrado hasta que la solución esté clara utilizando un plato caliente a 50 ° C colocada cerca de un microscopio de disección . Mantenga el envase cerrado entre muestras y climatizada durante todo el proceso de inclusión (medidas 1.4.4-1.4.6).
- Transferencia de embriones de 1,5 mL centrifugar los tubos a una placa Petri con una pipeta de vidrio y llenarlo con 1 X TBS-NP40.
- Eliminar las células de la yema grande de embriones en fase de somitogenesis (4-5 y 11-12 somitas) en la caja Petri con unas pinzas finas con el aumento de una disección microscopio 26. Sostener el embrión por la yema de la cola con un par de pinzas y retire las células de la yema con el otro par para preservar el tejido del cerebelo. Transferencia de los embriones de yolked a un área de la placa Petri libre de restos de yema.
Nota: Insertar embriones en el molde lleno de agarosa individualmente para evitar endurecimiento prematuro de la agarosa de la LMP. - Relleno uno 12 x 5 mm x 3 mm el molde seccionamiento con 200 μL de había derretido LMP agarosa usando una micropipeta. Realizar los pasos 1.4.5.-1.4.6. rápidamente (dentro de 20 s de llenar el molde) para incrustar el embrión antes de la agarosa de la LMP se refresca a RT y se solidifica.
- Usar pinzas finas para transferir un embrión de yolked por la tailbud de la caja Petri para el molde lleno de agarosa hacia su extremo cónico con un microscopio de disección.
- Fórceps fino de uso para orientar el embrión en el molde tal que el vibratome corte en el plano deseado. Orientación del embrión que el tejido del cerebelo corre paralelo a la longitud del molde con su superficie dorsal hacia el borde y su superficie anterior apuntando hacia la región cónica para crear secciones transversales. Repita los pasos 1.4.4-1.4.6 para los embriones restantes.
- Permitir la incorporación de agarosa para solidificar durante 5 min a TA.
- Generar 40 μm secciones del eje más alto de la agarosa incrustada embriones (medidas 1.4.1-1.4.7) con un vibratome el seccionamiento plato llenado con 1 x NP40 TBS. Transferencia de las secciones de interés para una placa de 24 pozos en 500 μL 1 x TBS-NP40 con unas pinzas finas. Coloque las secciones de un único embrión por bien.
Nota: Consulte la referencia 18 para más detalles. Asegurar que las secciones permanecen hidratadas en todo momento en al menos 250 μl de tampón y roca a baja velocidad (10-25 rpm) para los pasos restantes evitar la separación de la incrustación de agarosa. Detergentes presentan en el bloqueo y soluciones de lavado deben reducir la tensión superficial del medio líquido y permitir la inmersión de las secciones. Verificar que las secciones permanecen en los pozos durante y después de todas las manipulaciones. Tenga cuidado para evitar que accidentalmente descartar secciones durante lavados.
- Eliminar el buffer y agregar 500 μl de solución de bloqueo. Roca por al menos 1 h a RT.
Nota: Utilice una solución de bloqueo que contiene 5% de suero de la especie de cada anticuerpo secundario que se utilizará (véase Tabla de materiales). - Incubar en anticuerpos primarios de 300 μL diluidos en solución amortiguadora de bloqueo durante 36-72 h a 4 ° C en un eje de balancín. Lavar dos veces en 600 μL 1 x TBS-NP40 en un eje de balancín de 30 minutos cada uno, en RT.
Nota: Doble-secciones por incubación de anticuerpos primarios contra MTs totales (anti β-tubulina, o anti α-tubulina) y estables MTs (anti-Glu-tubulina) o dinámicas MTs (tubulina contra Tyr). Seleccionar anticuerpos primarios que se han planteado en diferentes especie cuando doble etiquetado total y postraduccional modifica las poblaciones de α-tubulina. Consulte la Tabla de materiales para las diluciones del anticuerpo. - Incubar en 300 μL de anticuerpos secundarios conjugados fluoróforo diluido en solución amortiguadora de bloqueo en un eje de balancín de 16-24 h a 4 ° C en la oscuridad. Lavar dos veces en 600 μL 1 x TBS-NP40 en un eje de balancín de 30 minutos cada uno, en RT.
Nota: Envolver el plato varios pocillos con anticuerpo secundario en papel desde este punto hacia adelante y después de cada manipulación para evitar enfriamiento. Seleccione secundarios anticuerpos que reaccionan con la inmunoglobulina de anfitrión del anticuerpo primario. Elegir el anticuerpo secundario fluoróforos que tienen espectros de emisión separados, no superpuestos. Consulte la Tabla de materiales para las diluciones del anticuerpo. - Incubar los embriones en 500 μl de DAPI solución en un rockero durante 30 min, a RT. Wash 3 veces en TBS NP40 oscilante a temperatura ambiente durante 5 minutos.
Nota: Etiquetado Nuclear proporciona contexto celular para la cuantificación de MT en paso 1.12. - Coloque una gota de medio de montaje con el agente anti-fade en el centro de la diapositiva libre de polvo. Use pinzas finas para transferir las secciones a la gota de medio de montaje. Coloque el cubreobjetos sobre la muestra libre de polvo. Almacenar diapositivas en un lugar seco, oscuro y fresco, envuelto en papel, hasta que se realiza la proyección de imagen de.
Nota: Dando vueltas las secciones en la parte posterior de la diapositiva usando un marcador permanente punta fina antes de la proyección de imagen ayudarán a identificar las secciones cuando se utiliza el microscopio. - Proyección de imagen confocal
- secciones de montaje de un láser invertido confocal microscopio estampando la diapositiva a la etapa con el cubreobjetos hacia el objetivo. Determinar la óptica adecuada (objetivo, láser y ajustes del canal como ganancia y offset) en un portaobjetos de control y mantenerlos constantes entre muestras 27. Evitar oversaturating los píxeles para evitar pérdida de datos.
- Capturar imágenes confocales Z-pilas utilizando ajustes del canal para el anticuerpo secundario seleccionado fluoróforos y guardar los archivos de imagen 27. Adquirir pilas de Z para cada sección.
Nota: Repetir los parámetros utilizados para la adquisición de las imágenes de las figuras 2 y 3 mediante el uso de las siguientes opciones de adquisición: modo = XYZ; Ampliación objetiva = 63 X lente de inmersión de aceite; apertura numérica objetiva = 1.4; Paso Z = 0.1 μm; Profundidad Z = 16.23 μm. Utilice la siguiente configuración de canal: excitación de DAPI con láser UV-gama de 20%, gama de filtro de emisión = 430-480 nm, ganancia del fotomultiplicador (PMT) = 525 V y el desplazamiento de la PMT =-1.72%; 448 nm fluoróforo (consulte la Tabla de materiales) excitación con 488 nm láser de 20%, gama de filtro de emisión = 493-573 nm, ganancia PMT = 689 V y PMT offset = -0.2%; excitación de fluoróforo de 594 nm con láser 594 32%, gama de filtro de emisión = 608 706 nm, ganancia PMT = 768 V y el desplazamiento de la PMT =-6.8%. - Guardar los archivos de datos en bruto con el nombre único y descriptivo y crear una copia para la edición de software de análisis de imagen.
- Compilación de Z - para la visualización de proyecciones de máxima
- Abra la copia del archivo de datos utilizando el software de análisis de imagen 3-d dominio público (por ejemplo, ImageJ). Compruebe que cada canal aparece como una secuencia de imágenes individuales (Z-stacks).
- Dividir canales de imagen utilizando la siguiente secuencia de menú: “ canales de imágenes/Color/Split ”.
- Crear una imagen fusionada por superposición de los canales de interés utilizando la siguiente secuencia de menú: " canales de combinación de Color de imágenes. " seleccionar el 594 nanómetro, 488 nm y canales DAPI a falso color rojo, verde y azul, respectivamente. Compruebe " crear compuestos " y seleccione " OK " 28.
Nota: Omitir el canal DAPI para mejor transmitir detalles específicos de MTs en una proyección máxima como en la figura 2 y 3, con sólo seleccionar colores para los otros dos canales. - Examinar la pila de Z fusionada y anote el comienzo y el final posiciones de mejor interiores Z-planos para todos los canales visibles. Despedir el exteriores planos de Z que tienen señal subóptima debido a los desniveles de la sección. Consulte la referencia 29 para más detalles.
- Visualizar la pila de Z combinada como una sola imagen 2-D mediante la realización de una proyección de intensidad máxima de la pila de Z utilizando la siguiente secuencia de menú de análisis de imagen 3-d: " imágenes/pila/Z-proyecto " entrar en el inicio y final de posiciones de los mejores interiores Z-planos de paso 1.11.3 como el " rebanada de inicio " y " sector de parada, " respectivamente. Seleccione " intensidad Max " como el tipo de proyección y haga clic " OK ". Consulte la referencia 28 para más detalles.
- Etiquetado análisis MT
- Abra el software de análisis de imagen 3D comercial. Seleccione " crear biblioteca " y proporcione un nombre descriptivo para la biblioteca de imágenes. Haga clic en " crear. " arrastrar archivos de imagen raw generados a partir del microscopio confocal en la biblioteca. Archivos más grandes requieren más tiempo para transferir.
- Seleccionar un archivo para analizar. Elegir " extendido foco " de la " vista " menú para mostrar la imagen fusionada de canal en la ventana principal.
- Ajustar el umbral arrastrando la herramienta deslizador para cada canal a la izquierda o la derecha hasta que la señal de fondo se reduce y la verdadera señal es robusta. Observar que cada canal muestra una verdadera señal de la molécula marcada (por ejemplo, canal DAPI mostrando núcleos oblongos o mitóticos, pero no auto fluorescencia desde el citoplasma o agarosa).
- Seleccione la " a mano alzada región de interés (ROI) " de la herramienta y delinear la región de interés a analizar. Seleccione la " acciones " pestaña seguido de " cultivo a la selección " para recortar la imagen. Guarde el archivo de la imagen recortada con un nuevo nombre. Haga clic en la " medidas " tab para crear el protocolo para el filtrado de objetos concretos relevantes para el análisis 3-d.
- Arrastre " encontrar objetos " a la ventana de protocolo. Cambie el nombre el primer protocolo " DAPI. " seleccionar el canal DAPI en el menú desplegable. Arrastre los siguientes ajustes en el protocolo DAPI y colocarlas debajo " encontrar objetos " en el siguiente orden (tabla 1): " llenar los agujeros en ObjectsŔ " Separadas tocando ObjectsŔ " Excluir los objetos de SizeŔ " Excluir sin tocar ROIs ".
Nota: El objetivo de los ajustes en la tabla 1 son para primero establecer un umbral que descarta las señales cuya distribución y tamaño son incompatibles con el tamaño de los objetos que están analizando. Por ejemplo, eliminar una señal no lo suficientemente grande como para ser un núcleo cuando contando núcleos. - Realizar la secuencia (paso 1.12.9) para ensamblar el resto de filtros para la β-tubulina y otros marcadores de parámetros en la tabla 1.
- Select " medida " en la parte inferior de cada protocolo. Elegir " intensidad y medida del volumen " y " longitud esquelética " de tubulina todas las etiquetas, pero sólo el primero para la señal DAPI.
- Dibujar un ROI de la región a medir. Observar las mediciones bajo el " extracto " ficha después de que el software de procesos de la región. Copiar los datos y guardarlos en una hoja de cálculo realizable, como en la tabla 2. Crear una copia de respaldo de la hoja de cálculo para su posterior análisis.
- Seleccionar las mediciones de interés (por ejemplo, longitud de paquete MT, número de MT paquetes de núcleo, como se revela con diferentes marcadores) en la hoja de cálculo y analizar para determinar los promedios para cada grupo.
Nota: La MT paquete longitud promedio = suma de los ' significa longitud esquelética de β-tubulina ' por cada embrión dividido por el número total de embriones. Consulte la fila 20 del cuadro 2. Formato de la hoja de cálculo para que las variables y grupos experimentales fácilmente se graficaron.
2. De Novo TA Asamblea ensayo (protocolo 2)
- construcción y prueba múltiples bien fluir-por aparato ( figura 1) 2 días antes del experimento.
Nota: El aparato permite lavado simultáneo de varios grupos experimentales nocodazole tratamiento usando fuentes de Mesa de materiales. Sellador de silicona requiere al menos 24 h de tiempo de secado antes que no plantea ningún riesgo de toxicidad para los embriones.- Partido de un tubo de centrífuga de 50 mL a la mitad longitudinalmente, con una caladora o sierra de banda.
- Cut 7 semicírculos con un radio de 3 cm, de 70 μm nylon malla y recortarlo para caber firmemente en una mitad de la fractura tubo de centrífuga. Pegue los semicírculos en el tubo de centrífuga paralelo a las marcas de graduación de 10 mL utilizando sellador de silicona de acuario-caja de seguridad. El dispositivo secar por 2 días y enjuague por inmersión en un vaso de agua para 2-3 h.
- Línea el extremo superior (roscado) del tubo de centrífuga corte con plastilina tal que la altura del líquido retenido en el dispositivo de flujo tiene una profundidad de 1/4 pulgada ( figura 1, vista 2 y 3).
- Preparar los aparatos de lavado retirar el émbolo de una jeringa de 50 mL y 12 pulgadas de la tubería fina en la punta. Empuje el tubo cuanto se pueda y sellar alrededor de la articulación utilizando arcilla de modelado.
- Humedezca previamente la malla usando medio embrión para permitir que el líquido recorra todo el fluir-por dispositivo. Ángulo del dispositivo en hielo así ese líquido piscinas en todos los compartimientos pero aún vacía en la parte delantera donde se encuentra el borde de la arcilla. Utilizando un soporte de anillo, suspender el aparato del lavado sobre el dispositivo a través de flujo de hielo ( figura 1).
- 200 mL de medio de embrión en el hielo y vierta suficiente en los aparatos de lavado para asegurar que se eliminan todas las burbujas de aire y que el caudal es aproximadamente 7 mL/min ajustar la tasa de flujo cambiando la altura de la jeringa de.
- Enzimático dechorionate embriones
- hacer una solución de trabajo de la proteasa no específicos diluyendo 1 mL de 10 mg/mL caldo no específica proteasa en medio del embrión de 20 mL.
- Dechorionation químico realizan en embriones 1 h antes del punto de tiempo cuando se esperan llegar a la etapa de desarrollo deseada. Chorions Digest quitando medio embrión de platos de Petri de 100 mm que contiene etapas embriones y agregar 20 mL de solución de proteasa no específico de trabajo.
- Embriones de incubar a 37 ° C por 5 min
Nota: No exceder 5 minutos o usar una mayor concentración de la solución de proteasa no específicos, como resultado los embriones que aparte una vez tratados con nocodazole. - Pipeta a proteasas no específicas y llenar platos con medio de embrión de aproximadamente 25 mL rápidamente. Repetir una vez.
- Con una pipeta de vidrio de 1 mL, transferencia embriones menores de 24 hpf para vidrio platos para protegerlos de daños.
- Dechorionation completa mediante la extracción manual chorions con un par de Pinzas finas, como se describe en el paso 1.1.5.
- Colocar cristal Petri que contenían dechorionated de embriones en una incubadora de 28,5 ° C durante un mínimo de 30 min hasta llegar a la etapa de desarrollo deseada.
- Depolymerize MTs existentes
- preparar una solución de trabajo de 5 μg/mL nocodazole combinando nocodazole stock de 50 μL 1 mg/mL con medio de embrión frío de hielo 10 mL.
PRECAUCIÓN: Use guantes cuando maneje nocodazole, un irritante de la piel. - Intercambio medio embrión del grupo nocodazole tratamiento con solución de trabajo de nocodazole frío de 10 mL. Coloque los platos Petri en hielo durante el tiempo necesario para la etapa de desarrollo (por ejemplo, 1 h para 4-5 embriones de somite). Apartar a embriones de control no tratados en una placa Petri sobre hielo fijará junto a las muestras de lavado en el paso 2.3.4.1.
- Transferencia de embriones utilizando una pipeta de vidrio pulido de 1 mL de fuego al aparato a través de flujo, con compartimientos separados para cada grupo experimental. Comience el nocodazole lavado verter medio embrión frío de hielo en la parte superior de la jeringa de 50 mL.
Nota: Use por lo menos 30 embriones por grupo experimental. Grupos experimentales podrían consistir en embriones de control o una variedad de morfolino o embriones inyectados de RNA. El lavado requieren un total de aproximadamente 150 mL de medio de embrión a agregar cada 8-10 minutos lavado el nocodazole mientras continúa inhibir el crecimiento de MT con hielo. Mantener los embriones en el hielo es esencial para el éxito de este ensayo porque MTs son inestables en temperaturas frías y frío retrasa el desarrollo de embriones tempranos. - MTs permite volver a crecer después de 20 minutos de lavado a temperatura ambiente mediante la transferencia de embriones a cajas Petri de vidrio que contiene caliente medio de embrión (28,5 ° C) usando una pipeta de vidrio de pulido 1 mL de fuego. Tan pronto como se transfieren los embriones, iniciar un temporizador.
- Fijar los embriones control y lavado en 1 min, 5 min y 10 min mediante pipeteo aproximadamente 10 embriones en un 1,5 mL centrifugar el tubo con solución de 1 mL de 4% PFA/MAB (28,5 ° C) y siguiendo las indicaciones de paso 1.2.3.
- preparar una solución de trabajo de 5 μg/mL nocodazole combinando nocodazole stock de 50 μL 1 mg/mL con medio de embrión frío de hielo 10 mL.
- Preparar muestras para el inmuno-etiquetado como se describe en las secciones 1.3-1.5.
- Immunolabel secciones flotantes y embriones de la imagen como se describen en secciones 1.6-1.10 con las siguientes modificaciones a las especificaciones del anticuerpo primario: anti-β-tubulina de ratón de uso 1: 500 conejo anti-γ-tubulina y 1: 200.
- Proceso y analizar imágenes utilizando el software de análisis de imagen 3-d, como se describe en el paso 1.12.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Análisis de MTs estables y dinámicos mediante inmunomarcación
En el Protocolo 1, se revelaron la distribución de las subpoblaciones de MT durante temprano (quilla neural) y últimas etapas (barra neural) del desarrollo del tubo neural, con Glu-tubulina y Tyr-tubulina como marcadores para MTs estables y dinámicas, respectivamente. Dinámicas MTs predominan en el cerebelo en el estadio de la quilla neural (4-5 somitas) (figura 2A-D). Como la quilla se convierte en el vástago neuronal (11-12 somitas), una etapa de epitelización mejorada, cualitativamente menos MTs son immunoreactive con el anticuerpo anti-Tyr-tubulina (figura 2E-H), especialmente en la barra ventral. En cambio, Glu-tubulina es dispersa y punteada a lo largo de la quilla neural (figura 3A-D), pero se enriquece en la barra de nervios ventral a lo largo de tramos de MT (figura 3E-H). Puntas de flecha señalan a paquetes específicos de MT o estructuras donde el etiquetado es aumentado.
Aunque se producen anticuerpos anti-Glu-tubulina y anti-Tyr-tubulina en la misma especie hospedadora (previniendo un doble experimento etiquetado), estos resultados indican que marcadores MT estables y dinámicos se traslapan raramente en el cerebelo de pez cebra. En primer lugar, la barra nervios ventral tiene más estable (figura 3F) que la dinámica (figura 2F) MTs. La tendencia se invierte en la barra dorsal neural, consistente con un modelo de pez cebra neurulation en que el tejido dorsal queda dinámico hasta que el tubo neural formado20. En segundo lugar, mientras que los husos mitóticos se etiquetan completamente con el anticuerpo de la Tyr-tubulina en la quilla neural (figura 2D, puntas de flecha), solamente la base del husillo, coincidente con el centrosoma, se etiqueta con el marcador de estabilidad (Glu-tubulina Figura 3 D, puntas de flecha). Inmunofluorescencia de la β-tubulina, común a ambos ensayos, informa al experimentador de la distribución de los MTs y proporciona una base para despedir no específicos de etiquetado.
Medir objetos usando software de análisis de imagen 3-d resulta en una gran cantidad de datos que pueden ser organizados en una tabla conveniente (cuadro 2). Para hacer mediciones de área, longitud y cuenta, estamos utilizando sólo un subconjunto de los datos que analizar. Uno de los componentes de los datos que tenemos no analizar es el número de objetos identificados. Este número se utiliza como un control de calidad interno, como el número no debe variar ampliamente entre como secciones y la proporción de núcleos a MTs deben permanecer similar en una condición de tratamiento único. Un outlier es un indicador que sea el análisis necesita volver a ejecutar con los filtros ajustados o que la imagen es demasiado mal etiquetada para analizar. Por lo tanto, todas las imágenes atípicas deben ser reanalizadas con ajustes. La sección de aislados debe examinarse signos de mal etiquetado o daño físico que pueda resultar en cuentas objeto inusual. Una vez terminado el análisis y controlada de calidad, información útil puede ser recuperada de los datos sin procesar tales como promedio de longitud de MTs total y estable MTs o la proporción de MTs estables a MTs total (tabla 3). Además de estas medidas, muchas otras métricas pueden obtenerse utilizando el software de análisis de imagen 3-d que puede utilizarse para sacar conclusiones acerca de MTs o su relación a otras estructuras celulares (núcleo, centrosoma, etc.).
De novo Ensayo conjunto de MT
El tratamiento del nocodazole depolymerizes MTs en etiquetado difusa (figura 4A, 4 D y 4 G). Como crecer el MTs, se extienden desde el centrosoma (figura 4B, 4E y 4 H), sin embargo, esto puede no ser obvio en un solo plano debido a sus trayectorias no-planar (figura 4C, 4F y 4I). Sin embargo, algunos software de análisis de imagen son capaces de medir longitudes en 3-d, lo que permite una evaluación del crecimiento de MT tras el lavado nocodazole (tabla 4). Una observación importante que se puede obtener del conjunto de datos en la tabla 4 es que la longitud promedio de MTs parece aumentar con el tiempo después del lavado nocodazole en todas las regiones del tubo neural analizados. Como se mencionó anteriormente, otros tipos de métricas de software de análisis de imagen 3-d pueden proporcionar contexto celular para interpretar los datos de la MT (por ejemplo, proporción de MTs por núcleo).
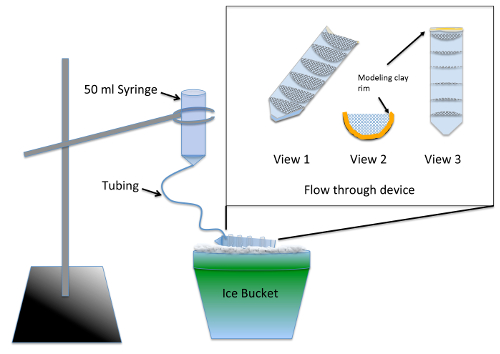
Figura 1 : Ilustración del aparato de lavado para de novo Ensayo de ensamblaje de MT. El margen es un primer plano del dispositivo a través de flujo de malla pegado en un tubo de centrífuga de 50 mL corte longitudinal. La malla compartmentalizes el dispositivo a través de flujo tal que se pueden procesar simultáneamente varios grupos experimentales. Durante el uso, medio embrión se agrega a la jeringa y fluye lentamente a través del tubo para llenar el dispositivo de flujo, proporcionando un enjuague constante a todos los grupos experimentales. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: uso de inmunomarcación para obtener imágenes dinámicas mts. Dechorionated embriones se fijaron en las etapas apropiadas (4-5 en A, D y 12-13 somitas en E-H), seccionadas transversalmente a través del cerebelo y immunolabeled con anticuerpos contra la β-tubulina (verde en A y E) para marcar todas las MTs y tyrosinated α-tubulina (rojo en B y F) para revelar la dinámicas poblaciones de MT. Altamente dinámicos MTs pueden verse en las imágenes combinadas (C, G) y sus aumentos más altos (D, H) como áreas donde la etiqueta amarilla es visible (flecha en D, H). Barras de escala = 25 μm (A-C y E-G) y 10 (D y H). Haga clic aquí para ver una versión más grande de esta figura.

FFigura 3: uso de inmunomarcación imagen estable mts. Dechorionated embriones se fijaron, seccionados por el cerebelo, immunolabeled en etapas apropiadas (4-5 somitas en A -D y 12-13 somitas en E-H). Estables MTs están marcados con anticuerpos contra la forma de detyrosinated de α-tubulina (Glu-tubulina) (rojos en B y F) mientras que MTs total fueron visualizados con un anticuerpo general β-tubulina (verde en A y E). Las señales rojas y amarillas en imágenes fusionadas (C, G) y sus aumentos más altos (D, H) representan áreas de alta estabilidad de MT (flecha en D, H). Barras de escala = 25 μm (A-C y E-G) y 10 (D y H). Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: uso de inmunomarcación imagen naciente mts. Dechorionated embriones eran fijos en 4-5 somitas y seccionados transversalmente por el cerebelo. Secciones immunolabeled con β-tubulina (D, E y F) para marcar el crecimiento MTs y γ-tubulina (A, By C) para marcar el punto de nucleación y el centrosoma. Región dorsal del tubo neural está encajonada en (A, D; B, E y C-F) y se muestra a mayor aumento (G, H, I, respectivamente) para núcleos (DAPI, azul), centríolos (γ-tubulina, rojo) y su total MTs (β-tubulina, verde). Blanco las puntas de flecha: colocalización de MTs y centríolos; amarillo las puntas de flecha: el centriolo segunda de una celda es visible. Barras de escala = 25 μm (A-F) y 10 (G-I). Haga clic aquí para ver una versión más grande de esta figura.

Tabla 1: Configuración predeterminada para el filtrado de objetos en el software de análisis de imagen 3D.

Tabla 2: Representante conjunto de datos raw obtenido mediante análisis de imágenes 3D software para analizar estable MTs. Cada columna representa las medidas de una sola sección. Min: medición más pequeña; Máximo: medida mayor; SD: desviación; SE: error de estándar.

Tabla 3: ejemplos de conjuntos de datos que pueden obtenerse en 3-d de imagen software de análisis para cuantificar MTs estables. Seleccionar mediciones de la longitud media total (β-tubulina) y estable (Glu-tubulina) MTs calculadas tomando el promedio de la longitud media esquelética para el sello pertinente de todas las muestras (ver Tabla2) y la relación estable para total () MTs Glu-Tubulin rayas por las rayas de la β-tubulina) calcula tomando el número promedio de β-tubulina dividido por la media cuenta de Glu-tubulina.

Tabla 4: Ejemplos de conjuntos de datos que pueden obtenerse del software de análisis de imagen 3-d para analizar de novo Asamblea de MT. Resultados representativos del conjunto de novo MT experimentan, comparar conjuntos de datos obtenidos para tres puntos de tiempo de recuperación (1, 5 y 10 min) después de lavado nocodazole. Para cada punto del tiempo, las mediciones obtenidas para conteo nuclear, centriolos (puncta de γ-tubulina), número de MTs total (rayas de la β-tubulina), aparecen para seleccionar las regiones de la imagen analizan (sección transversal del tubo neural en desarrollo).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Actualmente existen muchos métodos para la proyección de imagen dinámica del MT en el desarrollo temprano del pez cebra, que van desde imágenes directo de moléculas etiquetadas inmunomarcación de fijo tejido11,12,13,14. Aunque MTs en una sola célula pueden existir en Estados estables o dinámicos, epitelización es un proceso en el cual MTs se estabilizan progresivamente con el tiempo. Uso de marcadores para MTs estables y dinámicas ofrece una manera de visualizar este fenómeno. El método presentado aquí aprovecha el poder del software tratamiento de imágenes 3-d para cuantificar la transición de dinámica de poblaciones estables de MT en una sección de tejido del embrión de pez cebra. En el Protocolo 2, el método se utiliza para etiquetar una población distinta de la nacientes MTs y seguir su nucleación y crecimiento de las horas extraordinarias.
MTs son notoriamente difíciles de la imagen en su estado natal debido a su propensión a despolimerizar. Así, el componente clave de este método es la fijación rápida de MTs en todo el embrión entero. Esto se logra a partir de la fijación a temperaturas fisiológicas y utilizando un tampón que estabiliza el MTs y aumenta la permeabilidad del embrión. El tiempo de fijación es también importante fijación acortada no detención MTs mientras que la fijación excesiva puede enmascarar epitopos, interfiriendo con la Unión de anticuerpos. El tiempo de fijación sugerido de 3-4 h trabaja con los embriones que están en medio gastrulation a la fertilización después de 24 h. Embriones hacia el extremo menor de la escala de tiempo se deben fijar para más cerca 3 h mientras que los embriones mayores pueden necesitar todo el 4 h. Incluso con fijación adecuada, el MTs se despolimerizar con tiempo para que el seccionamiento y la inmunomarcación deben ocurrir dentro de una semana de fijación.
Una vez que el tejido se fija correctamente, pueden surgir problemas con inmunomarcación. El problema más común encontrado ha sido pobre penetración a través del centro del tejido, especialmente si también muchas secciones se incuban en el mismo pozo. Aumento de la concentración del tiempo principal anticuerpo e incubación para los anticuerpos primarios y secundarios, combinado con el aumento de los detergentes para mejorar la permeabilización de los embriones se mejorar la mayoría de los problemas de inmunomarcación. Si el anticuerpo etiquetado falla debido a cuestiones de fijación o inmunomarcación problemas, es posible determinar la causa examinando el anticuerpo etiquetado patrón. Fijación deficiente resultará en etiquetado intensa en la membrana y difuso de etiquetado en el citoplasma, mientras que la fijación excesiva dará lugar etiquetado débil conserva arquitectura MT. Pobre penetración del anticuerpo, sin embargo, aparece como áreas en el centro del tejido sin etiquetar.
La capacidad de analizar imágenes de MT de una manera significativa depende de imágenes de alta calidad. Para captar la longitud de MT en 3-d, Z-paso mínimo posible para el objetivo y la apertura numérica puede usarse. Imágenes que se muestran aquí fueron capturados con un 63 X objetivo de emersión de aceite con abertura numérica 1.4 produciendo los siguientes: pixel = 240 nm, paso de Z = 0.1 μm, tamaño de la pila Z = 16.252 μm. Porque el ancho de un MT solo es de 25 nm, aproximadamente 10 veces por debajo del límite de resolución de un microscopio de luz, este métrico no puede ser exactamente medido usando esta técnica. En cambio, se pueden medir sólo longitudes de MT iguales o mayores que el tamaño de píxel mínimo alcanzable en las tres dimensiones. Línea o marco promedio puede mejorar la definición de señal de MT. Análisis de MT deben ser reservado para las secciones de alta calidad. Tejido con fijación pobre no puede ser reflejada y analizado, overfixation suave puede ser contrapesado por cuidadosamente aumentar la intensidad de laser y ganar para detectar la débil señal manteniendo una buena gama dinámica. Penetración del anticuerpo pobre, aunque no óptima, puede corregirse limitando la adquisición de la imagen en regiones bien marcadas, resultando en una sección más fina (5-10 μm) la proyección de imagen. Alto fondo de etiquetado puede compensarse mediante el ajuste de la configuración del filtro. Sin embargo, si cualquiera de estos ajustes se realizan, es necesario comprobar que el umbral de filtros aceptablemente en cada plano de la pila de Z.
Software de análisis de imagen 3-d permite que el experimentador cuantificar MT longitud, área, ángulo, abundancia y otras métricas en el espacio 3D de las secciones de tejido fijo. El método descrito aquí proporciona instrucciones para la obtención de dichos datos utilizando el software disponible en el mercado. Sin embargo, se pueden adaptados a software de dominio público mejorado con plugins pertinentes o macros, hacer análisis a todos los módulos de filtrado. Antes del análisis, imágenes raw deben ser thresholded para evitar como fondo y las señales no específicas en la cuantificación. Una vez se completa el análisis y los datos son transferidos a una hoja de cálculo realizable, pueden hacer muchas inferencias del conjunto de datos. Uno de los cálculos realizados aquí era rayas Glu-tubulina por vetas de β-tubulina, o la proporción de MTs estables a MTs total donde 1 representa que el citoesqueleto de MT todo se estabilizó en un ROI. Si el experimentador desea complementar sus datos cuantitativos, generar un formato de archivo de pulido de imagen etiquetado (TIFF) imagen con barras de escala es fácil con software de análisis de imágenes 3D.
Este análisis permite el análisis funcional de proteínas implicadas en la Asamblea de MT, en vivo. Si se realiza inmunomarcación en alterna secciones seriales, este protocolo podría ser utilizado para estudiar MTs dinámicos y estables en el mismo embrión. En el futuro, modificaciones como detergentes mayor o ángulos alterados de inclusión permitirá el uso de estos métodos para embriones mayores y una gama más amplia de cuestiones anatómicas.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
El microscopio confocal fue adquirido con fondos de Estados Unidos National Science Foundation (NSF), grant #DBI-0722569. La investigación fue apoyada por los Estados Unidos nacional institutos de salud nacional Instituto de General ciencias médicas (NIH/NIGMS) grant #GM085290 y los Estados Unidos Departamento de defensa (DOD) grant #W81XWH-16-1-0466 adjudicado a R.M. Brewster. E. Vital fue apoyado por una beca a UMBC de la Howard Hughes Medical Institute a través del preuniversitario y licenciatura Ciencias Educación, grant #52008090. S.P. Brown fue apoyado por un departamento de los Estados Unidos de educación GAANN becas, una beca de postgrado de Meyerhoff financiado por subvenciones de NIH/NIGMS #GM055036 y una ayudantía de investigación financiado por el DOD de Estados Unidos grant #W81XWH-16-1-0466.
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
