

Summary
Immunomarquage des méthodes pour analyser des populations distinctes des microtubules dans le cerveau en développement du poisson zèbre sont décrites ici, qui sont largement applicables à d’autres tissus. Le premier protocole décrit une méthode optimisée d’immunomarquage des microtubules stables et dynamiques. Le deuxième protocole fournit une méthode pour l’image et de quantifier les microtubules naissants spécifiquement.
Abstract
Les microtubules (MTs) sont des structures dynamiques et fragiles qui sont difficiles à image in vivo, en particulier chez les embryons de vertébrés. Immunomarquage méthodes sont décrites ici pour analyser des populations distinctes de MTs dans le développement du tube neural de l’embryon de poisson zèbre. Alors que l’accent est mis sur les tissus nerveux, cette méthodologie est largement applicable à d’autres tissus. Les procédures sont optimisés pour les dès le début à mi-somitogenèse-stade embryons (1 somite à 12 somites), cependant ils peuvent être adaptés à un éventail d’autres stades avec des ajustements relativement mineurs. Le premier protocole fournit une méthode pour évaluer la distribution spatiale des MTs stables et dynamiques et d’effectuer une analyse quantitative de ces populations avec logiciel de traitement d’image. Cette approche vient compléter les outils existants à la dynamique des microtubules image et de la distribution en temps réel, en utilise des lignées transgéniques ou expression transitoire de constructions étiquetées. En effet, ces outils sont très utiles, mais ils ne distinguent pas facilement MTs dynamiques et stables. La capacité de l’image et d’analyser ces populations distinctes de microtubules a des implications importantes pour la compréhension des mécanismes qui sous-tendent la polarisation de la cellule et la morphogenèse. Le deuxième protocole décrit une technique pour analyser spécifiquement les MTs naissantes. Ceci est accompli en capturant les propriétés de croissance de novo de MTs au fil du temps, suite à la dépolymérisation des microtubules avec la nocodazole de drogue et une période de récupération après le lavage de la drogue. Cette technique n’a pas encore été appliquée à l’étude des MTs chez les embryons de poisson-zèbre, mais est un test utile pour étudier la fonction in vivo des protéines impliquées dans l’assemblage des microtubules.
Introduction
Les microtubules (MTs) sont des polymères de α - et β-tubuline qui s’assemble en protofilaments linéaire, dont plusieurs se combinent pour former un tube creux1,2. MTs sont des structures polarisées, avec l’essor de plus fins et croissance lente moins les extrémités qui sont ancrées à centrosome ou autres organisation des microtubules center (MTOC)3. De novo Formation de MT est initiée par nucléation à l’anneau γ-tubuline complexe (γ-TURC), qui fournit un modèle pour les MT Assemblée4. Dans une cellule donnée, deux populations de MTs se distinguées ce tour-ci au fil à des vitesses différentes. Dynamiques MTs explorent leur environnement cellulaire par la commutation entre les phases de croissance et de rétrécissement dans un processus appelé instabilité dynamique5. Contrairement aux MTs dynamiques, stables MTs sont non productrices et ont une demi-vie plus longue que la dynamique MTs6.
Des décennies de recherche en biologie cellulaire a fourni une sophistiqué panoplie d’outils pour étudier la fonction et la structure de la MT et a donné lieu à un vaste corpus de connaissances sur ces éléments du cytosquelette. Par exemple, MTs jouent un rôle central dans l’établissement et le maintien de la polarité cellulaire, qui est attribuable non seulement à leur polarité intrinsèque, mais aussi à la distribution subcellulaire différentielle des stable versus dynamique MTs7, 8. en revanche, beaucoup moins est entendu sur l’architecture de MT et de fonctionner dans des environnements de (3-d) en trois dimensions plus complexes, tels que les embryons de vertébrés, en partie en raison de relever le défi de l’imagerie du cytosquelette MT à haute résolution. Malgré cette limitation, la récente génération de GFP-transgénique exprimant les lignes que MTs de l’étiquette ou une expression transitoire de marqueurs de MT fluorescent-étiquetées a augmenté notre compréhension des changements dynamiques qui subissent une MTs et leur cellulaire et rôle de développement de l’embryon de poisson zèbre. L’ensemble du réseau MT peut être photographié sur des lignées transgéniques dans laquelle tubuline est soit directement marqué9 ou tubuline polymères sont indirectement étiquetés à l’aide de protéines associées à la MT Doublecortin-like-kinase (Dclk) ou Ensconsin (EMTB)10, 11. Autres lignes (et constructions) ont été générées qui permet d’évaluer des polarité intrinsèque MT par marquage spécifiquement MT plus fins ou ancrées à un centrosome moins les extrémités11,12,13, 14. la puissance de ces outils réside dans la possibilité d’étudier la dynamique de la MT en direct, les organismes de développement. Ces études ont révélé, par exemple, la distribution spatiale et dynamique de MTs dans les populations de cellules spécifiques, l’orientation du mitotique broches en tissus subissant la morphogenèse (indicateur du plan de division cellulaire), la polarité du polymère MT en ce qui concerne les processus tels que l’élongation cellulaire et de la migration et les taux de croissance MT déterminé par comète vitesse9,13,15. La limitation de ces outils est qu’ils ne sont pas facilement discriminatoires entre les populations stables et dynamiques de MT.
Puisant dans la littérature de biologie cellulaire riche, méthodes d’immunomarquage à l’image des MTs stables et dynamiques dans l’embryon de poisson zèbre sont décrites ici, qui sont complémentaires à l’usage des lignées transgéniques. La généralisation de ces méthodes d’immunomarquage dans le poisson-zèbre a été quelque peu gênée par la difficulté à préserver l’intégrité de la MT au cours de la procédure de fixation. 1 Protocole décrit une méthode optimisée d’immunomarquage total, dynamique, et MTs stables dans les sections du développement postérieur de poisson-zèbre efficaces. En outre, une méthode simple utilisant commercialement logiciels disponibles sont décrite afin de quantifier ces populations de MT. MTs stables sont distinguent des MTs dynamiques basés sur plusieurs modifications post-traductionnelles de α-tubuline, tels que l’acétylation et détyrosination, qui s’accumulent sur les MTs stables au fil du temps16,17. Chez l’embryon de poisson zèbre, acétylation apparaît au MTs ciliaires et axonales alors pas stable interphase MTs18, limiter l’utilité de ce marqueur à un sous-ensemble de MTs stabilisées. En revanche, détyrosination semble se produire sur toutes les MTs stables dans l' embryon de poisson zèbre18. Cette modification post-traductionnelle expose l’acide glutamique carboxy-terminale de le α-tubuline (tubuline détyrosinée)18 et peut être détectée à l’aide d’anti-Glu-tubuline19. Bien que détyrosination se produit préférentiellement sur MTs stables, des preuves expérimentales indiquent que cette modification post-traductionnelle est un résultat de, plutôt qu’une cause de, MT stabilité16. La population de MT réciproque, composée de MTs dynamiques, se distingue à l’aide d’un anticorps, anti-Tyr-tubuline, qui reconnaît spécifiquement la forme tyrosinée de tubuline α19. Après l’immunomarquage avec ces marqueurs et imagerie confocale, l’analyse quantitative des MTs (longueur, nombre et abondance relative) est possible dans des régions définies du tube neural en voie de développement. Une méthode simplifiée est fournie ici pour réaliser cette analyse à l’aide de logiciels de traitement d’images 3D. Cette méthode peut être appliquée pour répondre aux questions au sujet de la morphogénèse et la création ou la maturation des cellules polarité20. En effet, l’élaboration de tableaux polarisées de MTs stables accompagne de nombreuses manifestations du développement, y compris les photorécepteurs morphogenèse21, épithélialisation des cellules dans le développement du tube neural18 et axon formation8.
Protocole n° 2 décrit une adaptation in vivo d’une épreuve de biologie cellulaire pour analyser des MTs au cours de leur Assemblée phase (/ l’ancrage de la nucléation et croissance)22,23. Naissantes MTs sont nucléés à centrosome et par la suite fixés au subdistal appendices de la mère centriole23. On décrit une méthode pour analyser naissant MT repousse après la dépolymérisation. Ce protocole fournit des détails sur le traitement de la nocodazole pour dépolymériser MTs, la procédure de lavage des drogues et la période de récupération après le traitement. MT repousse est contrôlée à intervalles régulierslavage post s par immunomarquage avec des marqueurs pour MTs totales (anti-β-tubuline) aux côtés de marqueurs pour le centrosome (anti-γ-tubuline) et noyau (4', 6-diamidino-2-phénylindole (DAPI)), selon les procédures décrites dans le protocole 1. L’étape de dépolymérisation MT du présent protocole est essentielle car elle permet d’évaluation de la croissance de MT de novo plutôt qu’extension de MTs préexistants. Cette technique est donc distincte des autres procédures publiées pour mesurer les taux de croissance de MT (en absence de dépolymérisation) à l’aide d’un marqueur plus comme l’ailier liaison protéine 3 fondue à la protéine fluorescente verte (GFP-EB3), comme illustré dans la Tran et al., 11de 2012. En outre, ce test est particulièrement utile pour analyser les embryons défectueux en Assemblée de novo MT, tels que les mutants de NEDD1 rapportées antérieurement, dans lequel le recrutement de la γ-tubuline à centrosome est entravé, ayant pour résultat incomplet la formation du tube neural et défauts neuronale24.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
déclaration d’éthique : les procédures décrites ci-dessous suivre l’Université des directives de soins aux animaux le comté de Baltimore Maryland.
1. analyse de Stable et dynamique MTs à l’aide d’immunomarquage (protocole 1)
- dechorionation manuelle des embryons avant fixation
- obtenir fraîchement pondus embryons par excès système décanter et puis à recueillir les embryons restants dans une boîte de Pétri de plastique (voir Table des matières).
- Enlevez tous les débris de l’embryon de l’eau et le transfert de système à un nouveau plat rempli de milieu de l’embryon (voir Table des matières) pour s’assurer que les embryons se développent dans un environnement propre.
- Autoriser les embryons se développer jusqu'à l’étape désirée dans une étuve à température contrôlée à 28,5 ° C.
- Embryons lieu moins de 24 h après la fécondation (hpf) dans un plat en verre avant dechorionation.
Remarque : Dechorionate embryons avant fixation pour maximiser la pénétration rapide de l’intégrité de MT fixateur et de préserver. Moyen d’embryon utilisation au lieu de l’eau du système pour fournir la supplémentaires Ca 2 + requis au cours de la dechorionation. - Supprimer manuellement les chorions des embryons en boîte de Pétri, à l’aide de pinces fines sous un microscope à dissection.
- Pincer une petite zone dans le chorion rond et transparent qui entoure l’embryon avec une paire de pinces et doucement tirer pinces dehors pour créer une rupture dans la membrane.
- Agrandir l’ouverture en l’écartant délicatement sur le chorion rupture avec une pincette. Veillez à ne pas toucher l’embryon avec la pince, car il pourrait entraîner une rupture.
- Fixation d’embryons mis en scène
- transfert mis en scène, des embryons de déchorionés pour tubes de centrifugeuse de 1,5 mL. Enlevez autant moyen embryon possible à l’aide d’une pipette Pasteur en verre.
Remarque : Effectuer des traitements de fixation et de la drogue sur des embryons de jeunes (mi-somitogenèse), avant la création des centres neurales médiation la sensation de douleur, nécessitant aucune procédure supplémentaire pour soulager la douleur pendant l’euthanasie. Stades de développement sont définis dans Kimmel et al., 1995 25. Les stades somite 4-5 et 11-12 ont été utilisées pour obtenir des images de Figures 2 et 3. - Préparer l’Assemblée/Mt de paraformaldéhyde (PFA) 4 % fixateur de tampon (MAB) (se reporter à la Table des matières) en combinant 1 mL 8 % PFA / 1 mL 2 X MAB et en ajoutant 2 µL 100 % Triton X-100 par 1 mL de volume total.
ATTENTION : Porter des gants lors de la manipulation des solutions contenant de l’IFP et le Triton X-100, qui sont irritants pour la peau. - Fix embryons dans 1 mL 4 % PFA/MAB fixateur pendant 5 min à 28,5 ° C. aspirer le fixateur avec une pipette, remplacez-le par fixateur frais 1 mL et incuber pendant 3 h à température ambiante (RT) sur une base berçante.
NOTE : Les échantillons doivent être fixées rapidement à leur température biologique (28,5 ° C pour le poisson-zèbre) pour empêcher la dépolymérisation MT dépendant de la température.
- transfert mis en scène, des embryons de déchorionés pour tubes de centrifugeuse de 1,5 mL. Enlevez autant moyen embryon possible à l’aide d’une pipette Pasteur en verre.
- Aspirer le fixateur et ajouter 1 mL 1 x tampon Tris salin avec NP40 tampon (SCT-NP40). Agiter doucement à ta sur un rocker trois fois de 5 min chacun. Conservation des embryons à 4 ° C en 1 mL frais 1 X TBS-NP40 pour pas plus de 7 jours.
ATTENTION : Porter des gants lorsque vous manipulez des solutions contenant des NP-40, un irritant de la peau. - 4 % de chaleur RT bas point de fusion (LMP) les embryons de sectionnement pour immunomarquage agarose enrobage moyen dans un récipient fermé jusqu'à ce que la solution devienne claire à l’aide d’une plaque chauffante réglé à 50 ° C, placé à proximité d’un microscope à dissection . Garder le récipient fermé entre échantillons et chauffée pendant tout le processus d’incorporation (mesures 1.4.4-1.4.6).
- Transfert des embryons de la 1,5 mL centrifuger les tubes à une boîte de Pétri à l’aide d’une pipette de verre et remplissez-le avec 1 X TBS-NP40.
- Enlever les cellules de gros jaunes d’embryons au stade somitogenèse (4-5 et 11-12 somites) dans la boîte de Pétri à l’aide de pinces fines sous le grossissement d’une dissection de microscope 26. Tenez l’embryon par le bourgeon de la queue avec une paire de pinces et décollez les cellules jaunes avec l’autre paire afin de préserver le tissu de cerveau postérieur. Transférez les embryons de vitellus à une zone de la boîte de Pétri exempt de débris de vitellus.
NOTE : Incorporer des embryons dans le moule rempli de gel d’agarose individuellement pour éviter un durcissement prématuré de l’agarose LMP. - Remplir un 12 x 5 mm x 3 mm bien du moule sectionnement avec 200 µL fondu agarose LMP à l’aide d’une micropipette. Effectuez les étapes 1.4.5.-1.4.6. rapidement (moins de 20 s de remplissage du moule) pour incorporer l’embryon avant que l’agarose LMP se refroidit à la RT et solidifie.
- Pince fine permet de transférer un embryon de vitellus par le tailbud de la boîte de Pétri au moule rempli d’agarose vers son extrémité effilée sous un microscope à dissection.
- Pince fine utilisation d’orienter l’embryon dans le moule, tel que le vibratome coupes dans le plan désiré. Créer des sections transversales en orientant l’embryon telle que le tissu de cerveau postérieur est parallèle à la longueur du moule avec son faisant face au bord de la face dorsale et sa face antérieure, face à la fin de la région conique. Répétez les étapes 1.4.4-1.4.6 pour les embryons restants.
- Autoriser l’incorporation d’agarose pour solidifier pendant 5 min à température ambiante.
- Générer 40 µm tronçons de l’axe plus haut de l’agarose incorporé embryons (mesures 1.4.1-1.4.7) utilisant un vibratome avec le plat de sectionnement rempli avec 1 x TBS-NP40. Transfert des sections d’intérêt à une plaque 24 puits dans 500 µL 1 x TBS-NP40, à l’aide de pinces fines. Déposer les éléments d’un seul embryon par puits.
Remarque : Se référer pour faire référence à 18 pour plus de détails. S’assurer que les sections restent hydratées à tout moment au moins 250 µL de tampon et rock à basse vitesse (10 à 25 tr/min) pour les étapes restantes empêcher la séparation de l’incorporation d’agarose. Détergents présent dans le blocage et solutions de lavage devraient réduire la tension superficielle du milieu liquide et permettre submersion des sections. Vérifier que les sections restent dans les puits pendant et après toutes les manipulations. Soyez prudent pour éviter les sections accidentellement rejets lors de lavages.
- Retirer le tampon et ajouter 500 µL de solution de blocage. Rock pendant au moins 1 h à température ambiante.
Remarque : Utiliser une solution de blocage qui contient 5 % des sérums provenant de l’espèce hôte de chaque anticorps secondaire à utiliser (voir Table des matières). - Incuber en anticorps primaires de 300 µL dilués dans un tampon bloquant pour 36 à 72 heures à 4 ° C sur une bascule. Laver deux fois à 600 µL 1 x TBS-NP40 sur une bascule de 30 min chacun, à ta.
NOTE : Double marquage des sections par incubation dans des anticorps primaires contre MTs totales (anti-β-tubuline, ou anti-α-tubuline) et MTs stables (anti-Glu-tubuline) ou MTs dynamiques (tubuline anti-Tyr). Sélectionnez des anticorps primaires qui ont été soulevées dans différentes espèces hôtes lorsqu’un double marquage pour total et sera modifié les populations α-tubuline. Se référer à la Table des matières pour les dilutions anticorps. - Incuber dans 300 µL d’anticorps secondaires conjugué à un fluorophore dilué dans un tampon bloquant sur un balancier pendant 16 à 24 heures, à 4 ° C dans l’obscurité. Laver deux fois à 600 µL 1 x TBS-NP40 sur une bascule de 30 min chacun, à ta.
Remarque : Enroulez le plat multipuit contenant un anticorps secondaire dans du papier à partir de là et après chaque manipulation afin d’éviter l’extinction. Sélectionnez des anticorps secondaires qui réagissent avec l’immunoglobuline de l’hôte de l’anticorps primaire. Choisissez des fluorophores anticorps secondaire qui ont des spectres d’émission distincts, sans chevauchement. Se référer à la Table des matières pour les dilutions anticorps. - Incuber les embryons dans 500 µL de solution de DAPI sur un balancier pendant 30 min, à RT. Wash trois fois au SCT-NP40 bascule à ta de 5 min chacun.
NOTE : Marquage nucléaire fournit un contexte cellulaire pour la quantification de MT effectuée à l’étape 1.12. - Déposer une goutte de milieu de montage avec agent anti-fondu sur le centre d’une lame non poussiéreux. Pince fine permet de transférer des sections de la gouttelette moyen de montage. Placez une lamelle de poussière sur le dessus de l’échantillon. Stocker des diapositives dans un endroit sec, sombre et frais, enveloppé dans du papier, jusqu'à ce que l’imagerie est réalisée.
NOTE : Encerclant les sections sur le dos de la lame à l’aide d’un marqueur permanent pointe fine avant l’imagerie aidera à identifier les sections lors de l’utilisation du microscope. - Imagerie confocale
- Mont sections sur un microscope confocal de laser inversé en apposant la diapositive à la scène avec la lamelle face à l’objectif. Déterminer l’optique approprié (objectif, laser et canaux de communication tels que le gain et offset) sur une lame de contrôle et gardez-les cohérente entre les échantillons 27. Éviter de sursaturer les pixels pour éviter toute perte de données.
- Capturer images confocales Z-piles à l’aide de paramètres de canal pour les fluorophores anticorps secondaire sélectionnés et enregistrer des fichiers image 27. Acquérir des Z-piles pour chaque section.
NOTE : Reproduire les paramètres utilisés pour acquérir les images dans les Figures 2 et 3 en utilisant les paramètres d’acquisition suivants : mode = XYZ ; grossissement objectif = 63 X lentille d’immersion d’huile ; ouverture numérique objective = 1,4 ; Z-étape = 0,1 µm ; Profondeur Z = 16.23 µm. utiliser des canaux de communication suivants : excitation DAPI avec laser UV-portée de 20 %, gamme de filtre d’émission = 430-480 nm, photomultiplicateur (PMT) gain = 525 V et décalage PMT =-1.72 % ; 448 nm fluorophore (voir Table des matières) excitation avec laser à 488 nm 20 %, gamme de filtre d’émission = 493-573 nm, gain PMT = 689 V et décalage PMT = -0,2 % ; excitation de fluorophore 594 nm avec laser nm 594 de 32 %, gamme de filtre d’émission = 608-706 nm, gain PMT = 768 V et décalage PMT = -6,8 %. - Enregistrer des fichiers de données brutes avec des noms de fichiers uniques, descriptive et créer une copie pour l’édition de logiciel d’analyse image.
- Compilation de Z-piles pour l’affichage des maximums
- ouvrir la copie de fichier de données à l’aide du logiciel image en 3D du domaine public analyse (p. ex., ImageJ). Vérifiez que chaque canal apparaît comme une séquence d’images individuelles (Z-stacks).
- Split des canaux de l’image en utilisant la séquence de menu suivantes : “ canaux Images/couleur/Split ”.
- Créer une image fusionnée en superposant les canaux d’intérêt en utilisant la séquence de menu suivantes : " Images/couleur/fusion channels. " sélectionner le 594 nm, 488 nm et canaux DAPI de fausse couleur rouge, vert et bleu, respectivement. Vérifier " Create composite " et sélectionnez " OK " 28.
NOTE : Omettre le canal DAPI pour mieux faire en détail spécifique à MTs dans une projection maximum comme dans la Figure 2 et 3, seule sélection de fausses couleurs pour les deux autres chaînes. - Examiner la pile-Z fusionnée et prendre note de début et de fin des postes des Z-plans intérieurs de meilleurs pour tous les canaux visibles. Rejeter les Z-plans extérieurs qui ont généralement un signal sous-optimal en raison de la surface inégale de la section. Se reporter à la référence 29 pour plus de détails.
- Visualiser la pile-Z fusionnée comme une seule image 2D en effectuant une projection de l’intensité maximale de la Z-pile à l’aide de la séquence de menu analyse 3D image suivante : " Images/pile/Z-projet. " entrer de début et fin des postes des meilleurs intérieurs Z-plans de l’étape 1.11.3 comme la " tranche de démarrage " et " arrêt de tranche, " respectivement. Sélectionnez " intensité Max " comme type de projection et cliquez " OK ". Se référer pour faire référence à 28 pour plus de détails.
- Analyse MT étiquetage
- Ouvrez le logiciel d’analyse commerciale image en 3D. Sélectionnez " créer une bibliothèque de " et de fournir un nom descriptif pour la bibliothèque d’images. Cliquez sur " créer. " faire glisser des fichiers image raw générés à partir du microscope confocal sous la bibliothèque. Gros fichiers requièrent plus de temps pour transférer.
- Sélectionner le fichier à analyser. Choisissez " étendu la mise au point " de la " vue " menu pour afficher l’image de canal : fusion dans la fenêtre principale.
- Ajuster le seuil en faisant glisser la grille de calcul pour chaque canal à gauche ou à droite jusqu'à ce que le signal de fond est réduit et le vrai signal est robuste. Observer que chaque canal présente un véritable signal pour la molécule marquée (par exemple, chaîne DAPI montrant des noyaux oblongs ou mitose mais pas auto-fluorescence dans le cytoplasme ou agarose).
- Sélectionner le " Freehand région d’intérêt (ROI) " outil et décrire la région intéressante à analyser. Sélectionnez le " Actions " suivie d’onglet " Crop à sélection " de recadrer l’image. Enregistrez le fichier de l’image recadrée sous un nouveau nom. Cliquez sur le " mesures " onglet pour créer le protocole pour le filtrage des objets spécifiques pertinents pour l’analyse 3D.
- Glisser " trouver des objets " à la fenêtre de protocole. Renommez le premier protocole " DAPI. " sélectionner le canal DAPI dans le menu déroulant. Faites glisser les paramètres suivants au protocole DAPI et placez-les ci-dessous " trouver des objets " dans l’ordre suivant (tableau 1) : " remplir les trous à ObjectsŔ " Séparé de toucher ObjectsŔ " Exclure des objets par SizeŔ " Exclure sans toucher les ROIs ".
Remarque : L’objectif des paramètres dans le tableau 1 sont tout d’abord définir un seuil qui ignore les signaux dont la distribution et la taille sont incompatibles avec la taille des objets analysés. Par exemple, éliminer un signal pas assez grand pour être un noyau lors du comptage des noyaux. - Effectuer la séquence (étape 1.12.9) pour assembler les autres filtres pour la β-tubuline et d’autres marqueurs en utilisant les paramètres dans le tableau 1.
- Select " mesure " au bas de chaque protocole. Choisissez " intensité et Volume mesure " et " squelettique longueur " pour tout marquage de tubuline, mais seul le premier pour le signal DAPI.
- Dessiner un retour sur investissement autour de la région à mesurer. Observer les résultats sous la " Résumé " onglet après le logiciel traite de la région. Copier les données et les enregistrer dans une feuille de calcul réalisable, comme dans le tableau 2. Créer une copie de sauvegarde de la feuille de calcul pour une analyse ultérieure.
- Sélectionnez mesures d’intérêt (par exemple, la longueur du bundle MT, nombre de faisceaux de MT/noyau, comme l’a révélé avec différents marqueurs) dans la feuille de calcul et les analyser afin de déterminer les moyennes pour chaque groupe.
NOTE : La MT bundle longueur moyenne = la somme de la ' longueur squelettique de moyenne pour la β-tubuline ' pour chaque embryon divisé par le nombre total d’embryons. Se reporter à la ligne 20 du tableau 2. Format de la feuille de calcul afin que les variables et les groupes expérimentaux sont représentées graphiquement facilement.
2. De Novo MT Assemblée Assay (Protocole N° 2)
- construction et test le multipuits accréditives appareil ( Figure 1) 2 jours avant l’expérience.
Remarque : L’appareil permet un lavage simultané de plusieurs groupes expérimentaux après traitement nocodazole utilise des conduites de Table des matières. Scellant de silicone exige au moins 24 heures de temps de séchage avant qu’il ne présente aucun risque de toxicité pour les embryons.- Diviser un tube à centrifuger 50 mL en deux sur la longueur, à l’aide d’une scie sauteuse ou scie à ruban.
- Demi-cercles cut 7, avec un rayon de 3 cm, de 70 µm nylon mesh et élaguer pour s’adapter étroitement dans une moitié de ce schisme centrifuger tube. Coller les demi-cercles dans le tube à centrifuger parallèle les marquages de gradation de 10 mL à l’aide de scellant silicone d’aquarium-safe. Laissez l’appareil sécher pendant 2 jours et rincer par trempage dans un bécher d’eau pendant 2-3 h.
- Ligne de l’extrémité supérieure (filetée) du tube à centrifuger coupées avec le modelage d’argile tels que la hauteur du liquide conservé dans le dispositif intermédiaire a une profondeur de ¼ po ( Figure 1, vue 2 et 3).
- Préparer l’appareil de lavage en retirant le piston d’une seringue de 50 mL et insertion de 12 pouces de tubulure fine dans la pointe. Pousser le tube autant que possible et sceller autour de l’articulation à l’aide de glaise à modeler.
- Pré mouillage du maillage à l’aide de moyen d’embryon pour permettre le liquide à courir à travers le dispositif entier de cheminement. Angle de l’appareil sur la glace donc ce liquide avec Betclic dans tous les compartiments mais toujours vide à l’avant où se trouve le bord de l’argile. Utiliser un support de bague, suspendre l’appareil de lavage au-dessus du dispositif de cheminement sur la glace ( Figure 1).
- Chill 200 mL du milieu de l’embryon sur la glace et verser suffisamment dans l’appareil de lavage pour s’assurer que toutes les bulles d’air sont désactivées et que le débit est environ 7 mL/min. ajuster le débit d’eau en changeant la hauteur de la seringue.
- Enzymatiquement dechorionate embryons
- faire une solution de protéase non-spécifique de diluer 1 mL de 10 mg/mL stock non-spécifiques protéase dans 20 mL de milieu d’embryon.
- Dechorionation chimique effectuer sur des embryons 1 h avant le validant quand ils devraient atteindre le stade de développement souhaité. Digest chorions par retrait d’embryon support de 100 mm de Pétri contenant en scène embryons et ajouter 20 mL de solution de travail non-spécifiques protéase.
- Embryons incuber à 37 ° C pendant 5 min.
Remarque : Ne pas dépasser les 5 min ou utilisez une plus forte concentration de la solution de protéase non spécifique, comme cela se traduira dans les embryons tomber dehors une fois traités avec la nocodazole. - Rapidement pipeter des protéases non-spécifiques et remplir les plats avec environ 25 mL de milieu d’embryon. Répéter une fois.
- à l’aide d’une pipette de verre de 1 mL, transfert embryons âgés de moins de 24 hpf au verre plats afin des pour protéger contre les dommages.
- Dechorionation complète en supprimant manuellement chorions à l’aide d’une paire de pinces fines, comme indiqué au point 1.1.5.
- Placer le verre de Pétri contenant des embryons de déchorionés dans un incubateur à 28,5 ° C pendant au moins 30 min avant d’avoir atteint le stade de développement souhaité.
- Depolymerize le système commercial multilatéral existant
- préparer une solution de 5 µg/mL nocodazole en combinant 50 µL 1 mg/mL stock nocodazole avec 10 mL de milieu d’embryon froid glace.
ATTENTION : Utilisez des gants lorsque vous manipulez la nocodazole, un irritant de la peau. - Échanger le milieu de l’embryon de la nocodazole groupe de traitement avec 10 mL de solution de travail la nocodazole froid. Placer les boîtes de Pétri sur glace pendant un temps suffisant pour le stade de développement (par exemple, 1 h pour les embryons de somite 4-5). Mettre de côté embryons témoins non traités dans une boîte de Petri sur glace fixée aux côtés d’échantillons d’affouillement à l’étape 2.3.4.1.
- Transfert embryons à l’aide d’une pipette de verre poli de 1 mL de feu à l’appareil intermédiaire, utilisant des compartiments séparés pour chaque groupe expérimental. Commencer le sevrage de la nocodazole par moyen d’embryon froid glace coulée dans le haut de la seringue de 50 mL.
Remarque : Utilisez au moins 30 embryons par groupe expérimental. Groupes expérimentaux pourraient consister en des embryons témoins ou diverses morpholino ou d’embryons de RNA-injecté. L’effondrement du pont exigera un total d’environ 150 mL de milieu d’embryon à ajouter chaque 8-10 min. lavage la nocodazole tout en continuant à inhiber la croissance de MT avec de la glace. Garder les embryons sur glace est essentielle pour la réussite de ce test parce que MTs sont instables à froid et froid retarde le développement de jeunes embryons. - Permettre à MTs de repousse après 20 min d’affaissement au RT par transfert d’embryons de verre plats de Pétri contenant chaud moyen d’embryon (28,5 ° C) à l’aide d’une pipette de verre poli mL 1 feu. Dès que les embryons sont transférés, démarrer un minuteur.
- Les embryons contrôle et lavage de fixer à 1 min, 5 min et 10 min en pipettant également environ 10 embryons dans un 1,5 mL Centrifuger le tube rempli de fix PFA/MAB 1 mL 4 % (28,5 ° C) et en suivant les instructions à l’étape 1.2.3.
- préparer une solution de 5 µg/mL nocodazole en combinant 50 µL 1 mg/mL stock nocodazole avec 10 mL de milieu d’embryon froid glace.
- Préparer les échantillons pour immunomarquage tel que décrit dans les sections 1.3-1.5.
- Sections flottantes de marquer et d’embryons d’image comme décrites dans les sections 1.6-1.10 avec les modifications suivantes aux spécifications de l’anticorps primaire : utilisation 1/500 lapin anti-γ-tubuline et 1 : 200 de souris anti-β-tubuline.
- Processus et analyser les images à l’aide de logiciels d’analyse image 3-d, comme indiqué au point 1.12.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Analyse des MTs stables et dynamiques à l’aide d’immunomarquage
Dans le Protocole N° 1, la répartition des sous-populations MT au cours de début (neural quille) et stades avancés (tige neurales) du développement du tube neural se révèle, à l’aide de Glu-tubuline et Tyr-tubuline comme marqueurs pour MTs stables et dynamiques, respectivement. Dynamiques MTs prédominent dans le cerveau postérieur au stade de la quille neuronal (4-5 somites) (Figure 2A-D). La quille se développe dans la tige neurale (11-12 somites), une phase d’épithélialisation accrue, qualitativement moins MTs sont immunoréactives avec l’anticorps anti-Tyr-tubuline (Figure 2E-H), surtout dans la tige ventrale. En revanche, Glu-tubuline est épars et ponctuée tout au long de la quille neuronale (Figure 3A-D), mais est enrichie dans la tige neurale ventrale le long du tractus MT (Figure 3,E-H). Pointes de flèche pointent vers spécifiques MT faisceaux ou structures où l’étiquetage est augmentée.
Bien que les anti-Glu-tubuline et anti-Tyr-tubuline anticorps ont été produits dans la même espèce hôte (empêchant une double expérience d’étiquetage), ces résultats indiquent que les marqueurs de MT stables et dynamiques se chevauchent rarement dans le cerveau postérieur de poisson-zèbre. Tout d’abord, la tige neurale ventrale est plus stable (Figure 3F) que dynamique (Figure 2F) MTs. La tendance est inversée dans la tige neurale dorsale, conforme à un modèle de poisson-zèbre neurulation dans laquelle le tissu dorsal reste dynamique jusqu'à ce que le tube neural est formé de20. Deuxièmement, alors que les fuseaux mitotiques est entièrement étiquetés avec l’anticorps de Tyr-tubuline dans la quille neuronale (Figure 2D, pointes de flèches), uniquement sur la base de la tige, qui coïncide avec le centrosome, sont étiquetés avec le marqueur de stabilité (Glu-tubuline Figure 3 D, pointes de flèches). Β-tubuline immunofluorescence, commun à ces deux épreuves, informe l’expérimentateur de la distribution de toutes les MTs et fournit une base pour faire disparaître l’étiquetage non spécifiques.
Objets avec image en 3D logiciel d’analyse de mesure se traduit par une grande quantité de données qui peuvent être organisées dans une commode table (tableau 2). Pour rendre la longueur, comte et mesures de surface, nous utilisons uniquement un sous-ensemble des données qui n’existe pas d’analyser. Une des composantes des données que nous ne ne pas analyser davantage est le nombre d’objets identifiés. Ce numéro est utilisé comme un contrôle de qualité interne, car le nombre ne devrait pas largement varier entre comme sections et le ratio des noyaux à MTs devraient rester semblable à une condition de traitement unique. Une valeur aberrante est un indicateur qui soit l’analyse doit être réexécutés avec filtres ajustés ou trop mal étiquetée pour analyser l’image. Ainsi, toutes les images de valeur aberrante devraient être ré-analysés avec les paramètres ajustés. La section atypiques doit être examinée pour des signes de mauvais étiquetage ou dommage qui pourrait résulter dans la numération objet insolite. Une fois l’analyse terminée et contrôlée de qualité, des informations utiles peuvent être récupérées depuis les données brutes telles que durée moyenne des MTs totales et stable MTs ou le ratio de MTs stables à MTs totales (tableau 3). En plus de ces mesures, de nombreux autres paramètres peuvent être obtenus en utilisant le logiciel d’analyse image en 3D qui permet de tirer des inférences sur les MTs ou leur relation à d’autres structures cellulaires (noyau, centrosome, etc.).
De novo Dosage de l’Assemblée de MT
Le traitement de la nocodazole depolymerizes MTs ayant pour résultat un marquage diffus (Figure 4A, 4D et 4 G). Comme le système commercial multilatéral repousse, ils s’étendent du centrosome (Figure 4B, 4E et 4 H), cependant, c’est peut-être pas évident dans un seul plan en raison de leurs trajectoires non plane (Figure 4C, 4F et 4I). Néanmoins, certains logiciels d’analyse image sont capables de mesurer des longueurs en 3-d, permettant l’évaluation de la croissance de MT après le lavage de la nocodazole (tableau 4). Une observation importante qui peut être obtenue à partir du dataset dans le tableau 4 , c’est que la longueur moyenne des MTs semble augmenter au fil du temps après le lavage de la nocodazole dans toutes les régions du tube neural analysés. Comme mentionné ci-dessus, autres types de paramètres obtenus à partir des logiciels d’analyse image 3D peuvent fournir le contexte cellulaire pour interpréter les données de la MT (ratio de MTs par noyau, par exemple,).
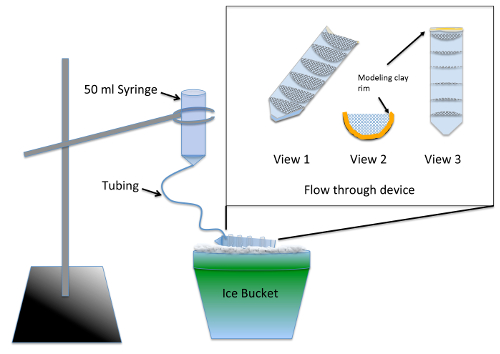
Figure 1 : Illustration des appareils de lavage pour de novo Dosage Assemblée MT. L’encart est un gros plan du dispositif intermédiaire filet collé dans un tube à centrifuger de 50 mL coupé sur la longueur. Le maillage compartimente le périphérique intermédiaire tel que plusieurs groupes expérimentaux peuvent être traitées simultanément. Pendant l’utilisation, support de l’embryon est ajouté à la seringue et s’écoule lentement dans la tubulure pour remplir l’appareil intermédiaire, fournissant un rinçage constant à tous les groupes expérimentaux. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 2: utilisation d’immunomarquage à l’image dynamique MTS. Déchorionés embryons ont été fixés aux stades appropriés (somites 12-13 et de 4-5 A-D E -h), sectionnées transversalement à travers le cerveau postérieur et immunomarquées avec des anticorps contre la β-tubuline (vert en A et E) pour marquer toutes les MTs et tyrosinée α-tubuline (rouge B et F) pour révéler les populations MT dynamiques. Très dynamiques MTs sont visibles dans les images fusionnées (C, G) et leurs grossissements (D, H) dans les zones où l’étiquette jaune est visibles (pointes de flèche en D, H). Barreaux de l’échelle = 25 µm (A-C et E.-G.) et 10 µm (D et H). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

FFigure 3: utilisation d’immunomarquage à l’image stable MTS. Déchorionés embryons ont été résolus, sectionnés à travers le cerveau postérieur et immunomarquées aux stades appropriés (4-5 somites en A -D et 12-13 somites dans E-H). Stables MTs sont étiquetés avec des anticorps contre la forme de détyrosinée de α-tubuline (Glu-tubuline) (rouges B et F) tandis que MTs totales ont été visualisés avec un anticorps générales β-tubuline (vert pour A et E). Des signaux rouges et jaunes dans les images fusionnées (C, G) et leurs grossissements (D, H) représentent des zones de forte stabilité de MT (pointes de flèche en D, H). Barreaux de l’échelle = 25 µm (A-C et E.-G.) et 10 µm (D et H). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 4: utilisation d’immunomarquage à l’image des MTS naissante. Déchorionés embryons ont été fixés à 4-5 somites et sectionnés transversalement à travers le cerveau postérieur. Sections ont été immunomarquées avec la β-tubuline (D, E et F) à l’occasion de plus en plus de MTs et de la γ-tubuline (A, Bet C) pour marquer le point de nucléation/centrosome. Une région dorsale du tube neural est convertie (boxed) en (A, D ; B, E et C-F) et montré à un grossissement plus élevé (G, H, j’ai, respectivement) pour révéler des noyaux (DAPI, bleu), centrioles (γ-tubuline, rouge) et total MTs (β-tubuline, verte). Blanc de pointes de flèches : colocalisation de MTs et de centrioles ; jaune de pointes de flèches : le centriole deuxième d’une cellule est visible. Barreaux de l’échelle = 25 µm (A-F) et 10 µm (G-I). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Tableau 1 : Paramètres par défaut pour le filtrage des objets dans le logiciel d’analyse image en 3D.

Tableau 2 : représentant ensemble de données brute obtenue par analyse d’image en 3D logiciel pour analyser stable MTs. Chaque colonne représente les mesures provenant d’une seule section. Mn : plus petite mesure ; Max : plus grande mesure ; SD : écart-type ; SE : écart-type.

Tableau 3 : exemples d’ensembles de données qui peuvent être obtenues en 3-d image logiciel d’analyse pour quantifier les MTs stables. Sélectionner les mesures de la longueur moyenne du total (β-tubuline) et stable (Glu-tubuline) MTs calculés en prenant la moyenne de la longueur moyenne de squelette pour le label pertinent de tous les échantillons (voir tableau 2) et le ratio d’étable pour un total de MTs) Glu-tubulin traînées par des stries de la β-tubuline) établi en calculant le nombre moyen de β-tubuline divisé par le nombre moyen de Glu-tubuline.

Tableau 4 : Exemples d’ensembles de données qui peuvent être obtenus au logiciel d’analyse image en 3D pour analyser de novo Assemblée de MT. Des résultats représentatifs de l’ensemble de novo MT experiment, en comparant les données obtenues pour les trois points de temps de récupération (1, 5 et 10 min) après le lavage de la nocodazole. Pour chaque point dans le temps, mesures obtenues pour comte nucléaire, centrioles (γ-tubuline examen), le nombre de totales MTs (stries de la β-tubuline), sont indiquées pour sélectionné régions de l’image analysée (section transversale du tube neural en développement).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Actuellement, il existe de nombreuses méthodes d’imagerie dynamique de MT dans le développement précoce de poisson-zèbre, allant de l’imagerie live de molécules marqués à immunomarquage de fixe tissu11,12,13,14. Bien que MTs dans une seule cellule peuvent exister dans les États stables ou dynamiques, épithélialisation est un processus dans lequel les MTs sont stabilisés progressivement au fil du temps. À l’aide de marqueurs pour MTs stables et dynamiques vous propose une façon de se représenter ce phénomène. La méthode présentée ici exploite la puissance des logiciels d’imagerie 3-d pour quantifier la transition entre dynamique et une population stable de MT dans une section transversale des tissus de l’embryon de poisson zèbre. Dans le Protocole N° 2, la méthode est utilisée pour étiqueter une population distincte de MTs naissantes et suivre leurs heures supplémentaires de nucléation et de croissance.
MTs sont notoirement difficiles à l’image dans leur état natif en raison de leur propension à dépolymériser. Ainsi, l’élément clé de cette méthode est une fixation rapide du SICT tout au long de l’embryon entier. Ceci est réalisé en démarrant la fixation aux températures physiologiques et en utilisant un tampon qui stabilise le système commercial multilatéral et augmente la perméabilité de l’embryon. Le temps de fixation est également important comme fixation écourtée ne parvient pas à arrêter MTs tandis que fixation excessive peut masquer des épitopes, interfère avec la liaison de l’anticorps. Le temps de fixation suggéré de 3-4 h fonctionne avec les embryons qui sont en milieu gastrulation jusqu'à la fécondation après 24h. Embryons vers la fin plus jeune de l’échelle de temps devraient être fixés pour rapprocher à 3 h alors que les embryons âgés peuvent avoir besoin de l’ensemble de 4 h. Même avec la fixation adéquate, le système commercial multilatéral sera dépolymériser avec le temps pour sectionnement et immunomarquage doivent avoir lieu dans la semaine de la fixation.
Une fois le tissu est fixé correctement, des problèmes peuvent survenir avec l’immunomarquage. Le problème le plus fréquent rencontré a été faible pénétration à travers le centre du tissu, en particulier, si trop de sections sont incubées dans le même puits. Augmentation de la concentration du temps d’incubation et les anticorps primaire pour des anticorps primaires et secondaires, combinée avec l’augmentation des détergents pour améliorer la perméabilisation des embryons va améliorer la plupart des problèmes immunomarquage. Si l’anticorps échoue en raison de problèmes de fixation ou d’immunomarquage problèmes d’étiquetage, il est possible de déterminer la cause en examinant l’anticorps modèle d’étiquetage. Mauvaise fixation provoquera un marquage intense dans la membrane et diffuse l’étiquetage dans le cytoplasme, tandis que la fixation excessive entraînera faible marquage qui conserve architecture MT. Faible pénétration de l’anticorps, toutefois, s’afficheront comme des zones dans le centre du tissu sans étiquetage.
La capacité d’analyser des images MT d’une manière significative est tributaire de l’imagerie de haute qualité. Pour capturer longueur MT en 3-d, la taille minimale de Z-étape possible pour l’objectif et l’ouverture numérique doit être utilisée. Images montrées ici ont été capturés avec un 63 X objectif émersion huile 1.4 ouverture numérique, produisant ce qui suit : pixel = 240 nm, Z-étape = 0,1 µm, taille de la pile-Z = 16.252 µm. Parce que la largeur d’un seul marqueur est 25 nm, à peu près ci-dessous 10 fois la limite de résolution d’un microscope optique, cette métrique ne peut pas être exactement mesurée à l’aide de cette technique. Au lieu de cela, les seules longueurs MT égales ou supérieures à la taille minimale de pixels réalisable dans les trois dimensions peuvent être mesurées. Ligne ou cadre en moyenne peut améliorer la définition de signal de MT. L’analyse MT devrait être réservée aux sections de haute qualité. Alors que le tissu avec mauvaise fixation ne peut pas être photographié et analysé, overfixation douce peut être contrebalancée en augmentant avec soin l’intensité du laser et gagner pour détecter le signaux faibles tout en conservant une bonne gamme dynamique. Pénétration d’anticorps pauvres, alors que c’est pas optimal, peut être corrigée en limitant l’acquisition d’images à des régions bien étiquetées, résultant dans une section plus mince (5 à 10 µm) de l’imagerie. Ambiants de l’étiquetage peut être compensée en ajustant les paramètres de filtre. Toutefois, si aucun de ces ajustements sont faits, il est nécessaire de vérifier que le seuil de filtres acceptable sur chaque avion de la Z-pile.
Logiciel d’analyse image 3D permet à l’expérimentateur de quantifier MT longueur, superficie, angle, abondance et autres mesures dans l’espace 3D des sections de tissu fixe. La méthode décrite ici fournit des directives pour l’obtention de ces données à l’aide de logiciels disponibles dans le commerce. Toutefois, les modules de filtrage peuvent être adaptés au logiciel du domaine public agrémenté de plugins pertinentes et/ou les macros, rendant l’analyse accessible à tous. Avant l’analyse, les images raw doivent être seuillées pour éviter d’inclure des fond et des signaux non spécifiques dans la quantification. Une fois que l’analyse est terminée et les données sont transférées vers une feuille de calcul réalisable, inférences beaucoup peuvent être faits de l’ensemble de données. L’un des calculs effectués ici a été traînées de Glu-tubuline par les stries de la β-tubuline, c'est-à-dire le ratio des MTs stables à MTs totales où 1 représente que le cytosquelette MT entière s’est stabilisé à un ROI. Si l’expérimentateur souhaite compléter leurs données quantitatives, générer un format de fichier d’image étiqueté poli (TIFF) image avec barres d’échelle se fait sans effort avec logiciel d’analyse image en 3D.
Ce test permet l’analyse fonctionnelle des protéines impliquées dans l’Assemblée de MT, in vivo. Si l’immunomarquage est effectuée sur l’alternance de coupes sériées, ce protocole pourrait servir à étudier MTs stables et dynamiques dans l’embryon même. À l’avenir, modifications comme détergents accrues ou altéré des angles encastrement permettra l’utilisation de ces méthodes pour les embryons plus vieux et un plus large éventail de questions anatomiques.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs n’ont rien à divulguer.
Acknowledgments
La microscopie confocale a été achetée avec des fonds de l’US National Science Foundation (NSF), subvention #DBI-0722569. La recherche a été financée par le U.S. National instituts de santé/National Institute of General Medical Sciences (NIH/NIGM) grant #GM085290 et le département américain de la défense (DOD) subvention #W81XWH-16-1-0466 décerné à Mr Brewster. E. Vital a été financée par une subvention de UMBC du Howard Hughes Medical Institute à travers le niveau préuniversitaire et programme d’enseignement de premier cycle sciences, subvention #52008090. S.P. Brown a été pris en charge par un département américain de l’éducation GAANN Fellowship, une bourse d’études supérieures Meyerhoff financé par subvention de NIH/NIGM, #GM055036 et un assistanat de recherche financé par le DOD américain grant #W81XWH-16-1-0466.
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
