

Summary
Immunolabeling Methoden für die unterschiedliche Populationen von Mikrotubuli in das sich entwickelnde Gehirn Zebrafisch Analyse werden hier beschrieben, die sind breit anwendbar auf andere Gewebe. Das erste Protokoll beschreibt eine optimierte Methode für Immunolabeling stabile und dynamische Mikrotubuli. Das zweite Protokoll stellt eine Methode zum Bild und speziell im Entstehen begriffenen Mikrotubuli zu quantifizieren.
Abstract
Mikrotubuli (MTs) sind dynamisch und fragile Strukturen, die schwierig zu Bild in Vivo, insbesondere in vertebrate Embryonen sind. Immunolabeling Methoden werden hier beschrieben, um verschiedene Populationen von MTs in den Entwicklungsländern Neuralrohr des Zebrafish Embryos zu analysieren. Während der Fokus auf Nervengewebe befindet, ist diese Methodik breit anwendbar auf andere Gewebe. Die Verfahren sind für früh zur Mitte-Somitogenesis-Embryonen (1 Somiten zu 12 Somiten), aber sie zu einer Reihe von anderen Stufen mit relativ geringfügigen Anpassungen sind optimiert. Das erste Protokoll stellt eine Methode zur Bewertung der räumlichen Verteilung der stabile und dynamische MTs und eine Quantitative Analyse dieser Populationen mit Bildverarbeitungs-Software. Dieser Ansatz ergänzt vorhandene Tools Bild Mikrotubuli Dynamik und Verteilung in Echtzeit, mit transgenen Linien oder transiente Expression tagged Konstrukte. Solche Werkzeuge sind in der Tat sehr nützlich, aber sie nicht ohne weiteres zwischen dynamischen und stabilen MTs unterscheiden. Die Möglichkeit, Bild und analysieren diese ausgeprägte Mikrotubuli Populationen hat wichtige Implikationen für Verständnis Mechanismen Zelle Polarisation und Morphogenese. Das zweite Protokoll beschreibt eine Technik, um im Entstehen begriffene MTs gezielt zu analysieren. Dies wird erreicht durch die Erfassung der de Novo Wachstumseigenschaften der MTs im Laufe der Zeit nach Mikrotubuli Depolymerisation mit dem Medikament Nocodazole und eine Erholungsphase nach Medikament auswaschen. Diese Technik noch nicht auf das Studium der MTs in den Zebrafish Embryos angewendet, sondern ist ein wertvoller Test zur Untersuchung der in Vivo -Funktion von Proteinen in die Mikrotubuli Montage verwickelt.
Introduction
Mikrotubuli (MTs) sind Polymere von α und β-Tubulin, die in linearen Protofilaments zusammenbauen, von denen mehrere kombinieren, um eine hohle Röhre1,2bilden. MTs sind polarisierte Strukturen, mit schnell wachsenden plus enden und minus enden, die an die Centrosome oder andere Mikrotubuli-organizing Center (MTOC)3verankert sind langsamwüchsig. De novo MT-Bildung wird durch Keimbildung bei der γ-Tubulin-Ring-Komplex (γ-TURC), bietet eine Vorlage für MT Montage4eingeleitet. In einer bestimmten Zelle können zwei Populationen von MTs, die wiederum über unterschiedlich unterschieden werden. Dynamische MTs erforschen ihre zelluläre Umgebung durch den Wechsel zwischen Phasen von Wachstum und Schrumpfung in einem Prozeß bekannt als dynamische Instabilität5. Im Gegensatz zu dynamischen MTs stabile MTs nicht wachsen und haben eine längere Halbwertszeit als dynamische MTs6.
Jahrzehntelanger Forschung in Zellbiologie hat eine anspruchsvolle Reihe von Tools zur Untersuchung MT Struktur und Funktion zur Verfügung gestellt und führte zu einen großen Körper des Wissens über diese Zellskelett Elemente. Zum Beispiel MTs spielen eine zentrale Rolle bei der Einrichtung und Wartung der Zellpolarität, die nicht nur auf ihre innere Polarität, sondern auch für die differenzielle subzelluläre Verteilung der Stall im Vergleich zu dynamischen MTs7, ist 8. dagegen weit weniger versteht sich über MT Architektur und Funktion in komplexere dreidimensionale (3D) Umgebungen, z. B. vertebrate Embryos, teilweise wegen der Herausforderung von imaging-MT Zytoskeletts mit hoher Auflösung. Trotz dieser Einschränkung die jüngsten Generation der GFP exprimierenden transgene Linien dieses Label MTs oder transiente Expression fluoreszent markiert MT Marker erhöht hat unser Verständnis von der dynamischen Veränderungen, die MTs zu unterziehen und ihre zellulären und Rolle des Zebrafish Embryos. Das Gesamtnetz der MT kann abgebildet in transgenen Linien in die Tubulin ist entweder direkt beschriftet9 oder Tubulin Polymere mit MT-assoziierte Proteine indirekt gekennzeichnet sind Doublecortin-Like-Kinase (Dclk) oder Ensconsin (EMTB)10, 11. Andere Linien (und Konstrukte) erzeugt wurden, die Bewertung der MT innere Polarität durch gezielt beschriften MT plus enden aktivieren oder Centrosome verankert minus enden11,12,13, 14. die Macht dieser Werkzeuge liegt in der Fähigkeit, MT Dynamik im Leben, zu studieren Organismen zu entwickeln. Solche Studien haben gezeigt, zum Beispiel die räumliche und dynamische Verteilung der MTs in bestimmten Zellpopulationen, die Ausrichtung der mitotischen Spindeln im Gewebe Morphogenese (ein Indikator für die Ebene der Zellteilung), durchläuft die Polarität des MT-Polymers Bezug auf Prozesse wie Zelle Dehnung und Migration, und MT Wachstumsrate von Komet Geschwindigkeit9,13,15bestimmt. Die Begrenzung dieser Werkzeuge ist, dass sie nicht ohne weiteres zwischen stabiler und dynamischer MT Bevölkerungsgruppen diskriminieren.
Zeichnung aus der reichen Zelle Biologie Literatur, werden Immunolabeling Methoden, um stabile und dynamische MTs des Zebrafish Embryos Bild hier beschrieben, sind die Verwendung von transgenen Linien ergänzen. Der weit verbreiteten Einsatz solcher Immunolabeling Methoden in der Zebrabärbling wurde durch die Schwierigkeit MT Integrität zu bewahren, während des Verfahrens Fixierung etwas behindert. Protokoll Nr. 1 beschreibt eine optimierte Methode für Immunolabeling insgesamt, dynamisch, und stabile MTs in Querschnitte der entwickelnden Zebrafisches Hinterhirn. Darüber hinaus ist eine einfache Methode, die Verwendung von kommerziell verfügbaren Software beschrieben, um diese MT-Populationen zu quantifizieren. Stabile MTs unterscheiden sich von dynamischen MTs basierend auf mehrere post-translationalen Modifikationen von α-Tubulin, wie Acetylierung und Detyrosination, die auf stabile MTs über Zeit16,17ansammeln. Im Zebrafish Embryos tritt Acetylierung auf ciliary und axonalen MTs aber nicht auf stabile Interphase MTs18, die Nützlichkeit dieses Markers auf eine Teilmenge der stabilisierten MTs zu begrenzen. Im Gegensatz dazu scheint Detyrosination auf allen stabilen MTs im Zebrafish Embryos18auftreten. Diese Post-translationale Modifikation stellt die Carboxy-Terminal Glutaminsäure α-Tubulin (Detyrosinated Tubulin)18 und mit Anti-Glu-Tubulin19detektiert werden. Obwohl Detyrosination bevorzugt auf stabile MTs auftritt, zeigt experimentelle Beweise, dass dieser Post-translationale Modifikation zurückzuführen, sondern als eine Ursache für MT Stabilität16. Die gegenseitige MT Bevölkerung, bestehend aus dynamischen MTs zeichnet sich mit einem Antikörper, anti-Tyr-Tubulin, erkennt, dass speziell die Tyrosinated Form von α-Tubulin19. Nach Immunolabeling mit diesen Markern und konfokale Bildgebung kann die Quantitative Analyse von MTs (Länge, Anzahl und relative Häufigkeit) in definierten Regionen des entwickelnden Neuralrohrs durchgeführt werden. Eine optimierte Methode ist hier für die Durchführung dieser Analyse mit 3-d-Bildverarbeitung Software zur Verfügung gestellt. Diese Methode kann auf Fragen bezüglich Morphogenese und der Einrichtung oder Reifung der Zelle Polarität20angewendet werden. In der Tat, die Ausarbeitung von polarisierten Arrays von stabilen MTs begleitet viele entwicklungspolitische Veranstaltungen, darunter Photorezeptor Morphogenese21, Epithelisierung von Zellen in den Entwicklungsländern Neuralrohr18 und Axon Bildung8.
Protokoll Nr. 2 beschreibt eine in-Vivo -Adaption eines Zelle Biologie Assays MTs Analyse während ihrer Versammlung Phase (Keimbildung/Anchorage und Wachstum)22,23. Im Entstehen begriffenen MTs sind kernhaltige an die Centrosome und anschließend an subdistal Anhängsel der Mutter Zentriol23verankert. Eine Methode, um entstehende MT nachwachsen nach Depolymerisation analysieren wird beschrieben. Dieses Protokoll enthält Informationen über die Nocodazole Behandlung, MTs, das Medikament Auswaschung Verfahren und der Nachbehandlung Erholungsphase depolymerisieren. MT nachwachsen wird in regelmäßigen Abständen überwacht.s Post Auswaschung von Immunolabeling mit Markierungen für insgesamt MTs (anti-β-Tubulin) neben Marker für die Centrosome (anti-γ-Tubulin) und Kern (4', 6-Diamidino-2-Phenylindole (DAPI)), nach den allgemeinen Verfahren, die im Protokoll Nr. 1beschrieben. Der MT Depolymerisation Schritt dieses Protokolls ist wichtig, da es ermöglicht die Beurteilung der de Novo MT Wachstum als Erweiterung der bereits vorhandenen MTs. Diese Technik unterscheidet sich somit von anderen veröffentlichten Verfahren, MT Wachstumsraten (in Abwesenheit der Depolymerisation) zu messen, durch die Verwendung einer plus Spitze Markers wie Ende bindende Protein 3 verschmolzen zu grün fluoreszierendes Protein (EB3-GFP), wie im Tran Et Al.gezeigt, 2012-11. Darüber hinaus dieser Assay eignet sich besonders für die Analyse von Embryonen in de Novo MT Baugruppe defekt wie die zuvor gemeldeten NEDD1 Mutanten in der Rekrutierung von γ-Tubulin, das Centrosome beeinträchtigt ist, was zu unvollständig Bildung des Neuralrohrs und neuronale Defekte24.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Ethik-Anweisung: die Verfahren beschrieben folgen der University of Maryland Baltimore County Tierbetreuung Leitlinien.
1. Analyse des stabilen und dynamische MTs verwenden Immunolabeling (Protokoll 1)
- manuelle Dechorionation von Embryonen vor der Fixierung
- erhalten frisch Embryonen von überschüssigem Wasser gießt hervorgebracht und dann sammeln überzählige Embryonen in einem Kunststoff Petrischale (siehe Tabelle der Werkstoffe).
- Entfernen Sie jeglichen Schmutz aus dem System Wasser und Transfer Embryonen, ein neues Gericht mit Embryo Medium gefüllt (siehe Tabelle der Materialien) um sicherzustellen, dass die Embryonen in einer sauberen Umgebung zu entwickeln.
- Ermöglichen die Embryonen zu entwickeln, um die gewünschte Stufe in einem temperierten Inkubator auf 28,5 ° c
- Ort-Embryonen, die jünger als 24 h nach Befruchtung (hpf) in eine Glasschale vor Dechorionation.
Hinweis: Dechorionate Embryonen vor der Fixierung, schnelle Durchdringung der Fixiermittel und bewahren MT Integrität zu maximieren. Verwendung Embryo Medium anstatt Wasser zu den zusätzlichen Ca 2 + während der Dechorionation erforderlich. - Entfernen Sie manuell die Chorions aus Embryonen während in Petrischale, mit feinen Pinzette unter dem sezierenden Mikroskop.
- Kneifen ein kleines Gebiet in der Runde, transparente Chorion, die den Embryo mit ein paar Zangen und sanft auseinander ziehen Zange erstelle ich einen Bruch in der Membran umgibt.
- Vergrößern die Eröffnung von neugierigen zart auf den geplatzten Chorion mit Pinzette. Achten Sie darauf, den Embryo mit der Zange zu berühren, da es platzen könnte.
- Fixierung der inszenierten Embryonen
- Transfer inszeniert, Dechorionated Embryonen bis 1,5 mL Zentrifuge Röhren. Entfernen Sie so viel Embryo Medium wie möglich mit einem Glas Pasteurpipette.
Hinweis: Führen Sie Fixierung und Droge-Behandlungen auf jung (Mitte-Somitogenesis) Embryonen vor der Bildung der neuronalen Zentren Vermittlung Schmerzempfindung, erfordern keine Additonal Verfahren zur Schmerzlinderung während der Euthanasie. Entwicklungsstadien sind definiert im Kimmel Et Al., 1995 25. Die 4. und 5. und 11. / 12. Somiten Etappen wurden verwendet, um Bilder für die Zahlen 2 und 3 zu erhalten. - Bereiten Sie 4 % Paraformaldehyd (PFA) HT Baugruppe Puffer (MAB) Fixativ (siehe Tabelle der Materialien) durch die Kombination von 1 mL 8 % PFA pro 1 mL 2 X MAB und Zugabe von 2 µL 100 % Triton x-100 pro 1 mL Gesamtvolumen.
Achtung: Tragen Sie Handschuhe beim Umgang mit Lösungen mit PFA und Triton x-100, die hautreizende sind. - Fix Embryonen in 1 mL 4 % PFA/MAB Fixiermittel für 5 min bei 28,5 ° C. Aspirat Fixativ mit einer Pipette mit 1 mL frisch Fixativ zu ersetzen, und 3 h bei Raumtemperatur (RT) auf einer Wippe inkubieren.
Hinweis: Proben müssen behoben werden schnell auf ihre biologische Temperatur (28,5 ° C Zebrafisch) temperaturabhängig MT Depolymerisation zu verhindern.
- Transfer inszeniert, Dechorionated Embryonen bis 1,5 mL Zentrifuge Röhren. Entfernen Sie so viel Embryo Medium wie möglich mit einem Glas Pasteurpipette.
- Aspirieren Fixiermittel und 1 mL 1 x Tris gepufferte Kochsalzlösung mit NP40 (TBS-NP40) Puffer. Schütteln Sie sanft bei RT auf ein Rocker drei Mal für 5 Minuten. Speichern von Embryonen bei 4 ° C in 1 mL frisch 1 X TBS-NP40 für nicht mehr als 7 Tage.
Achtung: Tragen Sie Handschuhe beim Umgang mit Lösungen mit NP-40, hautreizend. - Berandungen Embryonen für Immunolabeling
- Hitze RT 4 % niedriger Schmelzpunkt (LMP) Agarose Einbettung Medium in einem geschlossenen Behälter, bis die Lösung klar mit einer Heizplatte wird eingestellt auf 50 ° C in der Nähe einen sezierenden Mikroskop . Halten Sie den Behälter geschlossen zwischen Proben und beheizt die einbettende Prozeß (Schritte 1.4.4-1.4.6).
- Transfer Embryonen von 1,5 mL Röhren in einer Petrischale mit einer Glaspipette Zentrifugieren und füllen Sie es mit 1 X TBS-NP40.
- Entfernen Sie die große Eigelb Zellen aus Somitogenesis Embryonen (4-5 und 11-12 Somiten) in der Petrischale mit feinen Pinzette unter die Vergrößerung des einen sezierenden Mikroskop- 26. Halten Sie den Embryo durch die Rute Knospe mit einem paar der Zange und ziehen Sie die Eigelb-Zellen mit dem anderen Paar um das Hinterhirn Gewebe zu erhalten. Die de-Dotter Embryonen auf eine Fläche von der Petrischale frei von Eigelb übertragen.
Hinweis: Einbetten Embryonen in der Agarose-gefüllte Form einzeln zu verhindern vorzeitige Verhärtung der LMP-Agarose. - Füllung ein 12 x 5 mm x 3 mm auch der speziellen Form mit 200 µL geschmolzen LMP Agarose mit einer Mikropipette. Führen Sie die Schritte 1.4.5.-1.4.6. schnell (innerhalb von 20 s die Form zu füllen), Embryo einzubetten, bevor die LMP-Agarose, RT abkühlt und erstarrt.
- Feine Pinzette verwenden, um eine de-Dotter durch den Tailbud aus der Petrischale mit der Agarose-gefüllte Form zu seinem konischen Ende unter dem sezierenden Mikroskop Embryotransfer.
- Einsatz feine Pinzette, den Embryo in die Form zu orientieren, so dass der Vibratome in der gewünschten Fläche schneidet. Erstellen Sie Querschnitte durch den Embryo orientieren, so dass das Hinterhirn Gewebe mit der dorsalen Oberfläche zugewandte Kante und seine Vorderfläche zugewandten Ende spitz zulaufenden Bereich parallel zur Länge der Form verläuft. Wiederholen Sie die Schritte 1.4.4-1.4.6 für die restlichen Embryonen.
- Zulassen der Agarose einbetten um für 5 min bei RT zu festigen
- Erzeugen 40 µm Abschnitte der höchsten Achse von der Agarose eingebettet Embryonen (Schritte 1.4.1-1.4.7) mit einem Vibratome mit der speziellen Schale gefüllt mit 1 x TBS-NP40. Übertragen Sie Abschnitte von Interesse auf einer 24-Well-Platte in 500 µL 1 x TBS-NP40 mit feinen Pinzette. Legen Sie in den Abschnitten von nur einem Embryo pro Bohrloch.
Hinweis: Siehe 18 für weitere Details verweisen. Sicherstellen Sie, dass Abschnitte hydratisiert auf alle Zeiten in mindestens 250 µL Puffer und Rock bei niedriger Drehzahl (10-25 u/min) für die verbleibenden Schritte zur Trennung bleiben von Agarose Einbettung zu verhindern. Waschmittel in die Sperrung zu präsentieren und Waschlösungen sollte verringern die Oberflächenspannung des flüssigen Mediums und Eintauchen der Abschnitte. Kontrollieren Sie die Abschnitte in den Vertiefungen während und nach allen Manipulationen bleiben. Seien Sie vorsichtig, versehentlich verwerfen Abschnitte bei Waschungen zu verhindern.
- Entfernen den Puffer und 500 µL Lösung zu blockieren. Rock für mindestens 1 h bei RT
Hinweis: Verwenden Sie eine blockierende Lösung, die enthält 5 % Seren von Wirtsarten von jedem Sekundärantikörper verwendet werden (siehe Tabelle der Werkstoffe). - Inkubation in 300 µL Primärantikörper verdünnt in blockierende Puffer für 36-72 h bei 4 ° C auf einer Wippe. Waschen zweimal in 600 µL 1 x TBS-NP40 auf einer Wippe für jeweils 30 min bei RT
Hinweis: Doppel-Label Abschnitte durch Inkubation im primären Antikörper gegen insgesamt MTs (anti-β-Tubulin oder anti-α-Tubulin) und stabile MTs (Anti-Glu-Tubulin) oder dynamische MTs (Anti-Tyr Tubulin). Wählen Sie primäre Antikörper, die aufgeworfenen Fragen in verschiedenen Wirtsarten, wenn α-Tubulin Bevölkerung doppelte Kennzeichnung für insgesamt und posttranslational modifiziert werden. Siehe Tabelle der Materialien für Antikörper-Verdünnungen. - Inkubation in 300 µL Fluorophor-konjugierten Sekundärantikörper verdünnt in blockierende Puffer auf ein Rocker für 16-24 h bei 4 ° C im Dunkeln. Waschen zweimal in 600 µL 1 x TBS-NP40 auf einer Wippe für jeweils 30 min bei RT
Hinweis: Wickeln Sie die Multi-gut Schale mit Sekundärantikörper in Folie ab diesem Zeitpunkt und nach jeder Manipulation abschrecken zu verhindern. Wählen Sie sekundäre Antikörper, die mit der Host-Immunglobulin an den Primärantikörper reagieren. Wählen Sie Sekundärantikörper Fluorophore, die separate, nicht überlappende Emissionsspektren haben. Siehe Tabelle der Materialien für Antikörper-Verdünnungen. - Die Embryonen in 500 µL DAPI-Lösung auf einer Wippe für 30 min bei RT. Wash dreimal in TBS-NP40 rocken bei RT 5 min inkubieren.
Hinweis: Nukleare Kennzeichnung bietet zellulären Kontext für die MT-Quantifizierung durchgeführt im Schritt 1.12. - Geben Sie einen Tropfen Eindeckmittel mit Anti-Fade-Agent in der Mitte einer Folie staubfrei. Verwenden Sie feine Pinzette Abschnitte auf der Montage mittlerer Tröpfchen übertragen. Statt einem staubfreien Deckgläschen auf die Probe. Speichern von Folien an einem trockenen, dunklen und kühlen Ort, eingewickelt in Alufolie, bis Bildgebung erfolgt.
Hinweis: Kreisen die Abschnitte auf der Rückseite der Folie mit einem Fine-bestückte permanent-Marker vor Bildgebung wird dazu beitragen, um Abschnitte zu identifizieren, wenn Sie das Mikroskop verwenden. - Mount Abschnitte auf einer invertierten Laserscanning confocal Mikroskop durch die Anbringung der Folie auf die Bühne mit dem Deckglas mit Blick auf das Ziel Confocal Imaging. Bestimmen der geeigneten Optics (Ziel, Laser und Kanaleinstellungen erlangen und offset) auf einen Steuerschieber und konsistent zwischen Proben 27 aufbewahren. Vermeiden Sie Übersättigung die Pixel um Datenverlust zu verhindern.
- Z-Stapel konfokale Bilder mit Kanal-Einstellungen für die ausgewählten Sekundärantikörper Fluorophore erfassen und Speichern der Bild-Dateien- 27. Z-Stapel für jeden Abschnitt zu erwerben.
Hinweis: Replizieren die Parameter verwendet, um die Bilder in den Abbildungen 2 und 3 zu erwerben, mithilfe der folgenden Erwerb Einstellungen: Modus = XYZ; Objektiv Vergrößerung = 63 X Öl Immersion-Objektiv; Objektive numerische Apertur = 1,4; Z-Schritt = 0,1 µm; Z-Tiefe = 16.23 µm. verwenden Sie die folgenden Kanaleinstellungen: DAPI Anregung mit 20 % UV-Bereich Laser, Emissionsbereich Filter = 430-480 nm, Photomultiplier (PMT) Gewinn = 525 V und PMT Offset =-1.72 %; 448 nm Fluorophor (siehe Tabelle der Materialien) Anregung mit 20 % 488 nm Laser, Emissionsbereich Filter = 493-573 nm, PMT Gain = 689 V und PMT Offset = -0,2 %; 594 nm Fluorophor Anregung mit 32 % 594 nm Laser, Emissionsbereich Filter = 608-706 nm, PMT Gain = 768 V und PMT Offset = -6,8 %. - Rohdatendateien mit einmaligen, beschreibenden Dateinamen speichern und erstellen Sie eine Kopie für die Bearbeitung in Bildanalysesoftware.
- Zusammenstellung von Z-Stacks für die Anzeige von maximal Projektionen
- mit öffentlichen Bereich 3-d-Bildanalyse-Software (z. B. ImageJ) Kopie der Daten-Datei zu öffnen. Scheck, der jeden Kanal als individuelle Bildsequenz (Z-Stapel) angezeigt wird.
- Bild Kanäle mithilfe der folgenden Menüfolge aufgeteilt: “ Bilder/Farbe/Split Kanäle ”.
- Schaffen eine zusammengefügte Bild durch die Überlagerung der Kanäle von Interesse mithilfe der folgenden Menüfolge: " Bilder/Farbe/Merge Kanäle. " wählen Sie die 594 nm, 488 nm und DAPI-Kanäle falsche rot, grün und blau, beziehungsweise sein. Überprüfen Sie " Create Composite " und wählen Sie " OK " 28.
Hinweis: Lassen Sie den DAPI-Kanal, um besser vermitteln Detail speziell für MTs in eine maximale Projektion wie in Abbildung 2 und 3, mit nur falsche Farben für die anderen beiden Kanäle auswählen. - Den zusammengeführten Z-Stapel prüfen und notieren Sie sich die Anfangs- und Endfarbe Positionen der inneren beste Z-Ebenen für alle sichtbaren Kanäle. Die äußeren Z-Ebenen, die in der Regel suboptimal Signal aufgrund der unebenen Oberflächen des Abschnitts zu entlassen. Beziehen sich auf 29 Einzelheiten verweisen.
- Visualisieren den zusammengeführten Z-Stapel als ein einziges 2-D-Bild durch eine maximale Intensität Projektion des Z-Stack mithilfe den folgenden 3D-Bild Analyse Menü Reihenfolge ausführen: " Bilder/Stack/Z-Projekt " geben Sie die Anfangs- und Endposition der inneren besten Z-Ebenen aus Schritt 1.11.3 als die " Slice Start " und " Stop-Scheibe, " bzw.. Wählen Sie " Max Intensität " als Projektionstyp und klicken Sie auf " OK ". Beziehen sich auf 28 für weitere Details verweisen.
- Kennzeichnung analysieren MT
- der kommerziellen 3D Bildanalyse-Software zu öffnen. Wählen Sie " Bibliothek erstellen " und geben Sie einen beschreibenden Namen für die Image-Bibliothek. Klicken Sie " anlegen. " ziehen Sie raw-Bilddateien aus dem confocal Mikroskop in die Bibliothek generiert. Größere Dateien erfordern mehr Zeit, um zu übertragen.
- Wählen Sie eine Datei zu analysieren. Wählen Sie " erweitert Fokus " aus der " Ansicht " Menü das Kanal zusammengeführt Bild im Hauptfenster anzeigen.
- Anpassen der Schnittstellenüberwachung, indem Sie die Schieberegler für jeden Kanal auf links oder rechts ziehen, bis das Hintergrundsignal reduziert wird und das echte Signal robust ist. Beachten Sie, dass jeder Kanal zeigt eine true-Signal für das Molekül gekennzeichnet (z. B. DAPI-Kanal zeigt längliche oder mitotische Kerne aber nicht Auto-Fluoreszenz aus dem Zytoplasma oder Agarose).
- Wählen Sie die " Freehand Region von Interesse (ROI) " Werkzeug und skizzieren des Interessenbereichs analysiert werden. Wählen Sie die " Aktionen " Registerkarte "gefolgt von " auf Auswahl zuschneiden " um das Bild zu beschneiden. Speichern Sie zugeschnittenes Bilddatei unter einem neuen Namen. Klicken Sie auf die " Messungen " Registerkarte, um das Protokoll für das Filtern von bestimmten Objekten relevant für die 3-d-Analyse zu erstellen.
- Drag " Objekte finden Sie " in das Protokoll-Fenster. Benennen Sie das erste Protokoll " DAPI. " den DAPI-Kanal in dem Dropdown-Menü auswählen. Ziehen Sie die folgenden Einstellungen auf das DAPI-Protokoll und legen Sie sie unten " Objekte finden " in der folgenden Reihenfolge (Tabelle 1): " füllen Sie Löcher in ObjectsŔ " Separate ObjectsŔ berühren " Objekte durch SizeŔ ausschließen " Ausschließen, nicht berühren, ROIs ".
Hinweis: Das Ziel der Einstellungen in Tabelle 1 sind zuerst festlegen einer Schwelle, die Signale verwirft dessen Verteilung und Größe sind nicht konsistent mit der Größe der Objekte, die analysiert wird. Zum Beispiel ein Signal nicht groß genug, um einen Kern, wenn Kerne zählen zu beseitigen. - Führen die Sequenz (Schritt 1.12.9), montieren Sie die restlichen Filter für β-Tubulin und andere Markierungen mit den Einstellungen in der Tabelle 1.
- Select " Maßnahme " am unteren Rand jedes Protokoll. Wählen Sie " Intensität und Volumen-Messung " und " Skelett Länge " für alle Tubulin Kennzeichnung, sondern nur die erstere für das DAPI-Signal.
- Zeichnen einen ROI in der Region gemessen werden. Beobachten Sie die Messungen unter den " Zusammenfassung " tab, nachdem die Software die Region verarbeitet. Kopieren Sie die Daten und speichern Sie sie auf eine praktikable Tabellenkalkulation, wie in Tabelle 2. Erstellen Sie eine Backup Kopie der Kalkulationstabelle für die spätere Analyse.
- Wählen Sie Messungen von Interesse (z. B. Länge der MT-Bündel, Anzahl der MT-Bundles/Kern, wie Sie mit verschiedene Markierungen) in der Tabellenkalkulation und analysieren, um die Durchschnittswerte für jede Gruppe zu bestimmen.
Hinweis: Die durchschnittliche MT-Bündel-Länge = die Summe der ' Skelett Länge für β-Tubulin bedeuten ' für jedes Embryo dividiert durch die Gesamtzahl der Embryonen. Finden Sie in Zeile 20 der Tabelle 2. Das Arbeitsblatt so formatieren, dass Variablen und experimentellen Gruppen leicht grafisch dargestellt werden.
2. De Novo MT-Montage-Assay (Protokoll Nr. 2)
- Konstrukt und Test der Multi-gut durchströmten Apparat ( Abbildung 1) 2 Tage vor dem Experiment.
Hinweis: Der Apparat ermöglicht gleichzeitige Auswaschung von mehreren experimentellen Gruppen nach Nocodazole Behandlung mit Lieferungen aus Tabelle der Materialien. Silikon-Versiegelung erfordert mindestens 24 Stunden Trocknungszeit bevor es keine Toxizität der Embryonen gefährdet.- Eine 50 mL Zentrifugenröhrchen in zwei Hälften geteilt längs, mit Hilfe einer Schablone oder Bandsäge.
- Cut 7 Halbkreise mit einem Radius von 3 cm aus 70 µm Nylon mesh und einpassen fest in eine Zentrifuge die Hälfte der Split Rohr. Kleben Sie Halbkreise in die Zentrifugenröhrchen parallel zu den 10 mL Abstufung Markierungen mit Aquarium-Safe Silikon-Versiegelung. Lassen Sie das Gerät für 2 Tage trocknen und spülen durch Einweichen in ein Becherglas mit Wasser für 2-3 h.
- Linie oberste (Gewinde) Ende der geschnittenen Zentrifugenröhrchen mit Knetmasse, so dass die Höhe der Flüssigkeit im Durchflussverfahren Gerät beibehalten eine Tiefe von ¼ Zoll ( Abbildung 1, 2 und 3 Ansichten hat).
- Vorbereitung der Auswaschung Apparat durch den Kolben aus einem 50-mL-Spritze entfernen und Einfügen von 12 Zoll von feinen Schlauch in die Spitze. Drücken Sie den Schlauch so weit wie es geht und um das Gelenk mit Modelliermasse versiegeln.
- Vornässen des Netzes mit Embryo Medium damit die Flüssigkeit durch das gesamte durchströmten Gerät laufen. Winkel des Geräts auf Eis so dass Flüssigkeit in allen Abteilen bündelt aber noch leert sich der Vorderseite befindet sich die Ton-Felge. Mit einem Ring-Ständer, aussetzen der Auswaschung Apparat über dem Durchfluss-Gerät auf dem Eis ( Abbildung 1).
- Chill 200 mL Embryo Medium auf Eis und gießen Sie genug in den Apparat auswaschen, damit alle Luftblasen gelöscht werden und der Durchfluss ca. 7 mL/min einstellen der Durchflussmenge durch Änderung der Höhe der Spritze ist.
- Enzymatisch Dechorionate Embryonen
- machen eine funktionierende Lösung von unspezifischen Protease durch Verdünnen von 1 mL 10 mg/mL nicht-spezifische Protease Lager in 20 mL Embryo Medium.
- Chemische Dechorionation Perform an Embryonen 1 h vor dem Timepoint wenn sie erwartet werden, um die gewünschte Entwicklungsstufe zu erreichen. Digest Chorions indem Embryo Medium aus 100 mm Petrischalen mit inszeniert, Embryonen und das Hinzufügen von 20 mL unspezifische Protease Arbeitslösung.
- Embryonen Inkubation bei 37 ° C für 5 min.
Hinweis: Nicht mehr als 5 min oder verwenden eine höhere Konzentration von unspezifischen Protease Lösung, sonst werden die Embryonen fallen auseinander einmal behandelt mit Nocodazole. - Schnell, unspezifische Protease pipette und Gerichte mit ca. 25 mL Embryo Medium nachfüllen. Einmal zu wiederholen.
- Mit einer 1-mL Glaspipette, Transfer Embryonen jünger als 24 hpf Glas Geschirr um sie vor Beschädigungen zu schützen.
- Komplette Dechorionation von Chorions mit einer feinen Pinzette, manuell zu entfernen, wie in Schritt 1.1.5 beschrieben.
- Legen Sie Glas Petrischalen mit Dechorionated Embryonen in einem Inkubator 28,5 ° C für mindestens 30 Minuten, bis sie das gewünschte Entwicklungsstadium erreichen.
- Depolymerize die vorhandenen MTs
- bereiten eine funktionierende Lösung von 5 µg/mL Nocodazole durch die Kombination von 50 µL 1 mg/mL Lager Nocodazole mit 10 mL Eis kalt Embryo Medium.
Achtung: Benutzen Sie Handschuhe beim Umgang mit Nocodazole, hautreizend. - Austausch der Embryo Medium der Nocodazole behandelten Gruppe mit 10 mL kalte Nocodazole funktionierende Lösung. Legen Sie Petrischalen auf Eis, für einen angemessenen Zeitraum für die Entwicklungsphase (z. B. 1 h für 4-5 Somiten Embryonen). Beiseite unbehandelten Kontrolle Embryonen in einer Petrischale auf Eis neben Auswaschung Proben im Schritt 2.3.4.1 behoben werden.
- Transfer Embryonen mit einer Feuer-poliert 1-mL-Glas-Pipette auf den Durchfluss-Apparat, mit getrennten Fächern für jede Versuchsgruppe. Starten der Nocodazole auswaschen durch strömenden Eis kalt Embryo Medium in die Top 50 mL Spritze.
Hinweis: Verwenden Sie mindestens 30 Embryonen pro Versuchsgruppe. Versuchsgruppen könnte darin bestehen, der Kontrolle Embryonen oder eine Vielzahl von Morpholino oder RNA injiziert Embryonen. Die Auswaschung wird insgesamt ca. 150 mL des Embryos Medium alle 8-10 min. auswaschen hinzugefügt werden der Nocodazole erfordern, während Sie weiterhin MT Wachstum mit Eis zu hemmen. Halten die Embryonen auf Eis ist essentiell für den Erfolg des Assays, denn MTs bei kalten Temperaturen instabil sind und Kälte die Entwicklung im frühen Embryo verzögert. - Erlauben MTs regrow nach 20 min der Auswaschung bei RT durch die Übertragung der Embryonen auf Glas Petrischalen mit warm (28,5 ° C) Embryo Medium mit einem Feuer poliert 1 mL Glaspipette. Einen Timer gestartet wird, sobald die Embryonen transferiert werden.
- Fixieren die Kontrolle und Auswaschung Embryonen bei 1 min, 5 min und 10 min von ca. 10 Embryonen in einem 1,5 mL Pipettieren Zentrifugieren Rohr gefüllt mit 1 mL 4 % PFA/MAB Fix (28,5 ° C) und gemäß den Anweisungen in Schritt 1.2.3.
- bereiten eine funktionierende Lösung von 5 µg/mL Nocodazole durch die Kombination von 50 µL 1 mg/mL Lager Nocodazole mit 10 mL Eis kalt Embryo Medium.
- Proben vorbereiten, für Immunolabeling, wie in beschrieben 1.3-1.5 Abschnitte.
- Immunolabel schwimmende Abschnitte und Bild Embryonen als in Abschnitten 1.6-1.10 mit folgenden Änderungen Primärantikörper Spezifikationen beschrieben: Einsatz 1: 500 Kaninchen Anti-γ-Tubulin und 1: 200 Maus Anti-β-Tubulin.
- Prozess und Bilder mit 3-d-Bildanalyse-Software zu analysieren, wie unter Schritt 1.12.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Analyse der stabile und dynamische MTs mit immunolabeling
In Protokoll Nr. 1ist die Verteilung der MT Sub-Populationen während der frühen (neuronale Kiel) und späten Neuralrohr (neuronale Stab) Entwicklungsstadien offenbart, mit Glu-Tubulin und Tyr-Tubulin als Marker für stabile und dynamische MTs, beziehungsweise. Dynamische MTs überwiegen in dem Hinterhirn Stadium der neuronalen Kiel (4-5 Somiten) (Abbildung 2A-D). Als der Kiel in der neuronalen Stab (11. / 12. Somiten), eine Stufe der erweiterten Epithelisierung entwickelt sind qualitativ weniger MTs immunreaktiven mit dem Tyr-Tubulin-Antikörper (Abbildung 2E-H), vor allem in der ventralen Stab. Im Gegensatz dazu Glu-Tubulin ist verstreut und punktförmige während der neuronalen Kiel (Abbildung 3A-D), aber ist in der ventralen neuralen Stab entlang MT Traktate (Abbildung 3E-H) angereichert. Pfeilspitzen zeigen spezifische MT-Bündel oder Strukturen wo Kennzeichnung erhöht.
Obwohl Anti-Glu-Tubulin und Tyr-Tubulin Antikörper in der gleichen Wirtsarten (verhindert eine doppelte Kennzeichnung Experiment) hergestellt wurden, zeigen diese Ergebnisse, dass stabile und dynamische MT Marker nur selten in der Zebrafisch Hinterhirn überschneiden. Erstens hat die ventrale neurale Rute stabiler (Abbildung 3F) als dynamische (Abbildung 2F) MTs. Die Trendwende in der dorsalen neuralen Rod, konsistent mit einem Modell der Zebrafisch gebildet Neurulation, in dem die dorsale Gewebe dynamische bleibt, bis das Neuralrohr ist,20. Zweitens während mitotische Spindeln voll und ganz mit dem Tyr-Tubulin-Antikörper in der neuronalen Kiel (Abbildung 2D, Pfeilspitzen), nur die Basis der Spindel, deckungsgleich mit dem Centrosome gekennzeichnet sind ist mit der Stabilität Marker beschriftet Glu-Tubulin ( Abbildung 3 D, Pfeilspitzen). Β-Tubulin Immunfluoreszenz, üblich, beide Assays, informiert den Experimentator der Verteilung der alle MTs und bietet eine Grundlage, um unspezifische Kennzeichnung zu entlassen.
Messobjekte mit 3-d-Bildanalyse-Software ergibt sich eine große Menge von Daten, die in eine praktische Tabelle (Tabelle 2) organisiert werden können. Länge, Anzahl und Flächenmessungen zu machen, verwenden wir nur eine Teilmenge der Daten, die zur Analyse zur Verfügung steht. Eine der Komponenten der Daten, die wir weiter nicht zu analysieren ist die Anzahl der identifizierten Objekte. Diese Nummer wird als eine interne Qualitätskontrolle verwendet, da die Zahl nicht zwischen unterschiedlich sollten wie Abschnitte und das Verhältnis der Kerne zu MTs in Einzelbehandlung Zustand ähnlich bleiben sollte. Ein Ausreißer ist ein Indikator, der entweder die Analyse mit angepassten Filter erneut ausgeführt werden soll oder das Bild ist zu schlecht mit der Bezeichnung um zu analysieren. Daher sollten alle Ausreißer Bilder mit angepassten Einstellungen Korsen werden. Die Ausreißer Abschnitt sollte geprüft werden, auf Anzeichen von schlechter Kennzeichnung oder physischen Schaden, die ungewöhnliche Objekt zählt führen können. Sobald die Analyse abgeschlossen ist, und kontrollierte Qualität, nützlicher Informationen aus wiederhergestellt werden kann die Rohdaten wie durchschnittliche Länge von insgesamt MTs und stabile MTs oder das Verhältnis der stabilen MTs zu total MTs (Tabelle 3). Zusätzlich zu diesen Messungen erhalten viele andere Kennzahlen mit der 3-d-Bildanalyse-Software, die verwendet werden, um Rückschlüsse auf die MTs oder ihre Beziehung zu anderen Zellstrukturen (Kern, Centrosome, etc.) zu zeichnen.
De novo MT-Montage-assay
Die Nocodazole Behandlung depolymerizes MTs was diffuse Kennzeichnung (Abbildung 4A, 4-D und 4 G). Wie die MTs nachwachsen, kann sie reichen von der Centrosome (Abbildung 4B, 4E, und 4 H), jedoch dies nicht offensichtlich in einer Ebene durch ihre nicht-planaren Flugbahnen (Abbildung 4C, 4F, und 4I). Dennoch, einige Bildanalyse-Software sind in der Lage Messlängen in 3-d, so dass eine Bewertung der MT Wachstum nach der Nocodazole auswaschen (Tabelle 4). Eine wichtige Beobachtung, die aus dem Dataset in Tabelle 4 gewonnen werden kann ist, dass die mittlere Länge der MTs scheint im Laufe der Zeit nach der Nocodazole-Auswaschung in allen Regionen des Neuralrohrs analysiert. Wie oben erwähnt, bieten andere Arten von Metriken von 3-d-Bildanalyse-Software erhalten zellularen Kontext um die MT-Daten (z. B. Verhältnis von MTs pro Kern) zu interpretieren.
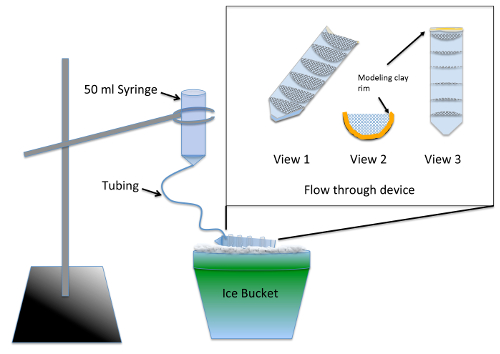
Abbildung 1 : Darstellung der Auswaschung Vorrichtung zur de novo MT Montage Assay. Der Einschub ist eine Nahaufnahme des durchströmten Gerätes gemacht aus Mesh in ein 50 mL Zentrifugenröhrchen, schneiden Sie längs eingeklebt. Das Netz teilt die Durchfluss-Gerät so, dass mehrere Gruppen gleichzeitig verarbeitet werden können. Während des Gebrauchs Embryo Medium wird hinzugefügt, um die Spritze und fließt langsam durch die Schläuche, durchströmten Gerät, die eine ständige Spülung für alle Versuchsgruppen zu füllen. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2: Verwendung von Immunolabeling zum dynamischen MTS Bild Dechorionated Embryonen wurden in geeigneten Phasen (4-5 in A-D und 12-13 Somiten E -h), quer geschnitten, durch das Hinterhirn und Immunolabeled mit Antikörpern gegen β-Tubulin (grün in A und E) behoben. alle MTs und Tyrosinated α-Tubulin (rot in B und F) markieren, um dynamische MT Populationen zu offenbaren. Hochdynamische MTs in den fusionierten Bildern (C, G) und ihre höheren Vergrößerungen (D, H) als Bereiche ersichtlich wo Gelbes Etikett sichtbar (Pfeilspitzen in D, H) ist. Skalieren von Balken = 25 µm (A-C und E-G) und 10 µm (D und H). Bitte klicken Sie hier für eine größere Version dieser Figur.

FIgure 3: Einsatz von Immunolabeling zu stabilen MTS. Bild Dechorionated Embryonen wurden behoben, durch das Hinterhirn und Immunolabeled auf geeigneten Stufen (4-5 Somiten in A -D und 12-13 Somiten E -h) geschnitten. Stabile MTs sind beschriftet mit Antikörpern gegen die Detyrosinated Form von α-Tubulin (Glu-Tubulin) (rot in B und F) während der gesamten MTs mit einem allgemeinen β-Tubulin-Antikörper (grün in A und E) visualisiert wurden. Rote und gelbe Signale in zusammengeführten Bildern (C, G) und ihrer höheren Vergrößerungen (D, H) repräsentieren Bereiche der hohen MT Stabilität (Pfeilspitzen in D, H). Skalieren von Balken = 25 µm (A-C und E-G) und 10 µm (D und H). Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 4: Verwendung von Immunolabeling zum Bild der im Entstehen begriffenen MTS. Dechorionated Embryonen wurden am 4. und 5. Somiten fixiert und quer durch das Hinterhirn geschnitten. Abschnitte wurden Immunolabeled mit β-Tubulin (D, E, und F) anlässlich der wachsenden MTs und γ-Tubulin (A, Bund C), die Keimbildung Punkt/Centrosome zu markieren. Eine dorsale Region des Neuralrohrs ist boxed in (A, D; B, E und C-F) und bei höherer Vergrößerung gezeigt (G, H, ich, beziehungsweise), Kerne (DAPI, blau), Centriolen (γ-Tubulin, rot) und insgesamt zu offenbaren MTs (β-Tubulin, grün). Weiße Pfeilspitzen: NS1 MTs und Centriolen; gelbe Pfeilspitzen: die zweite Zentriol einer Zelle sichtbar ist. Skalieren von Balken = 25 µm (A-F) und 10 µm (G-ich). Bitte klicken Sie hier für eine größere Version dieser Figur.

Tabelle 1: Standardeinstellungen Sie für das Filtern von Objekten in 3-d-Bildanalyse-Software.

Tabelle 2: repräsentative Rohdatensatz mit 3-d-Bildanalyse Software analysieren stabile MTs erzielt. Jede Spalte entspricht Messungen aus einem einzigen Abschnitt. Min: kleinste Maß; Max: größte Messung; SD: Standardabweichung; SE: Standardfehler.

Tabelle 3: Beispiele für Datasets, die 3-d beigezogen werden kann Bild Analysesoftware zur Quantifizierung von stabilen MTs. Wählen Sie Messungen der mittleren Länge von insgesamt (β-Tubulin) und stabil (Glu-Tubulin) MTs berechnet, indem der Durchschnitt der mittleren Skelett Länge für das entsprechende Schild aus allen Proben (siehe Tabelle 2) und das Verhältnis der Stall um MTs (Gesamt Glu-Tubulin Streifen pro β-Tubulin Streifen) berechnet, indem die Anzahl der durchschnittlichen β-Tubulin dividiert durch die durchschnittliche Anzahl der Glu-Tubulin.

Tabelle 4: Beispiele für Datasets, die 3-d-Bildanalyse-Software zur Analyse entnommen werden kann de novo MT-Montage. Repräsentative Ergebnisse aus der de Novo MT Assembly experimentieren, Vergleich der Datensätze erhalten für drei Mal Wiederherstellungspunkte (1, 5 und 10 min) nach Nocodazole auswaschen. Für jeden Zeitpunkt sind Messungen für nukleare Graf, Centriolen (γ-Tubulin Puncta), Anzahl der insgesamt MTs (β-Tubulin Streifen) gezeigt für ausgewählte Regionen von den abgebildeten (Querschnitt des entwickelnden Neuralrohrs) analysiert.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Derzeit gibt es viele Methoden für imaging-MT-Dynamik in der frühen Entwicklung der Zebrafisch, von live Imaging tagged Moleküle bis hin zu Immunolabeling der Gewebe11,12,13,14behoben. MTs in einer einzelnen Zelle in dynamischen oder stabile Staaten existieren können, zwar Epithelisierung ein Prozess in dem MTs schrittweise im Laufe der Zeit stabilisiert werden. Mit Hilfe von Markern für stabile und dynamische MTs bietet eine Möglichkeit, dieses Phänomen zu visualisieren. Die hier vorgestellte Methode nutzt die Macht der 3-d imaging-Software, den Übergang von dynamischen, stabile MT-Populationen in einem Querschnitt von Zebrafish embryonale Gewebe zu quantifizieren. Im Protokoll Nr. 2wird die Methode verwendet eine verschiedene Bevölkerung der im Entstehen begriffenen MTs beschriften und die Keimbildung und das Wachstum Überstunden zu folgen.
MTs sind notorisch schwierig zu Bild in ihrem nativen Zustand aufgrund ihrer Neigung zur depolymerisieren. Somit ist die Schlüsselkomponente dieser Methode schnelle Fixierung der MTs in der gesamten Embryo. Dies geschieht durch die Fixierung bei physiologischen Temperaturen starten und verwenden einen Puffer, der sowohl die MTs stabilisiert und die Durchlässigkeit des Embryos erhöht. Die Fixierzeit ist auch wichtig, da die verkürzten Fixierung zu verhaften MTs Fehler auftritt, während übermäßige Fixierung Epitope, Antikörperbindung stören überdecken kann. Die vorgeschlagenen Fixierzeit von 3-4 h arbeitet mit Embryonen, die in der Mitte des Gastrulation bis zu 24 h nach Befruchtung sind. Embryonen gegen jüngere Ende der Zeitskala für näher zu 3 h festzusetzen während ältere Embryonen die gesamte 4 h brauchen können. Sogar mit richtigen Fixierung werden die MTs mit der Zeit depolymerisieren, so schneiden und Immunolabeling innerhalb einer Woche der Festsetzung auftreten müssen.
Sobald das Gewebe richtig fixiert ist, können Probleme mit Immunolabeling auftreten. Das häufigste Problem gestoßen wurde armen Durchdringung durch das Zentrum des Gewebes, vor allem, wenn zu viele Abschnitte in den gleichen Brunnen ausgebrütet werden. Erhöhung der Konzentration von den primären Antikörper und Inkubation Zeit für primäre und sekundäre Antikörper, zusammen mit den steigenden Reinigungsmittel um Permeabilisierung der Embryonen zu verbessern, werden die meisten Immunolabeling Probleme lindern. Wenn die Antikörper-Kennzeichnung durch Fixierung Probleme oder Immunolabeling fehlschlägt, ist es möglich, die Ursache zu ermitteln, durch die Untersuchung des Antikörpers labeling Muster. Schlechte Fixierung führt zu intensiven Kennzeichnung in der Membran und Kennzeichnung im Zytoplasma diffundieren, während übermäßige Fixierung führt schwach bezeichnen, die MT Architektur behält. Schlechte Penetration des Antikörpers, erscheint jedoch als Bereiche in der Mitte des Gewebes ohne Kennzeichnung.
MT Bildern in einer sinnvollen Weise zu analysieren ist abhängig von hoher Bildqualität. MT Länge in 3-d zu erfassen, sollte die Z-Schritt-Mindestgröße für das Ziel und die numerische Apertur möglich verwendet werden. Hier gezeigten Bilder wurden mit einem 63 X Öl auftauchen Ziel mit 1,4 numerischer Apertur produzieren die folgenden erfasst: Pixel = 240 nm, Z-Schritt = 0,1 µm, Z-Stack-Größe = 16.252 µm. Denn die Breite der einzelnen MT 25 nm, etwa 10 mal unten die Grenze der Auflösung für ein Lichtmikroskop, diesem metrischen lässt sich nicht genau gemessen mit dieser Technik. Stattdessen können nur MT Längen gleich oder größer als die minimale Pixelgröße erreichbar in allen drei Dimensionen gemessen werden. Linie und/oder Rahmen im Durchschnitt kann die MT-Signal-Definition verbessern. MT-Analyse sollte für hochwertige Abschnitte reserviert werden. Während Gewebe mit schlechter Fixierung abgebildet und analysiert werden kann, milde Overfixation können ausgeglichen werden, indem man sorgfältig die Laserintensität und um das schwache Signal zu erkennen und gleichzeitig eine gute Dynamik gewinnen. Schlechte Antikörper eindringen, kann zwar nicht optimal, durch Begrenzung der Bildaufnahme auf gut gekennzeichneten Regionen, was zu imaging einen dünneren Abschnitt (ca. 5-10 µm) korrigiert werden. Hoher Hintergrund von der Etikettierung kann kompensiert werden, indem Sie die Filtereinstellungen anpassen. Wenn irgendwelche dieser Anpassungen vorgenommen werden, ist es jedoch notwendig, um sicherzustellen, dass die Filter-Schwelle akzeptabel auf jeder Ebene des Z-Stack.
3-d-Bildanalyse-Software ermöglicht den Experimentator, MT Länge, Fläche, Winkel, Fülle und andere Metriken im 3D-Raum fixierte Gewebe Abschnitte zu quantifizieren. Die hier beschriebene Methode bietet Richtungen für solche Daten mit im Handel erhältlichen Software zu erhalten. Jedoch können die Filterung Module Public Domain-Software erweitert mit entsprechenden Plugins/Makros, Analyse für alle zugänglich zu machen oder angepasst. Raw-Bilder muss vor der Analyse thresholded zu vermeiden, einschließlich Hintergrund und unspezifische Signale in der Quantifizierung. Sobald die Analyse abgeschlossen ist, und Daten werden in eine praktikable Kalkulationstabelle übertragen, können viele Rückschlüsse aus dem Datensatz erfolgen. Die hier vorgenommenen Berechnungen gehörte Glu-Tubulin Streifen pro β-Tubulin Streifen oder das Verhältnis der stabilen MTs zu total MTs wo ' 1 ' steht, dass die gesamte MT Zytoskeletts in einen ROI stabilisiert wurde. Wenn der Experimentator möchte ihre quantitative Daten ergänzen erzeugen eine polierte tagged Image-Dateiformat (TIFF) ist Bild mit Maßstabsleisten mühelos mit 3-d-Bildanalyse-Software.
Dieser Test erlaubt die funktionelle Analyse von Proteinen in MT Versammlung, in Vivoverwickelt. Wenn Immunolabeling auf Schnittserien abwechselnd durchgeführt wird, könnte dieses Protokoll verwendet werden, um dynamische und stabile MTs in der gleichen Embryo zu studieren. In der Zukunft ermöglichen Modifikationen wie erhöhte Reinigungsmittel oder veränderte einbetten Winkel den Einsatz dieser Methoden für ältere Embryonen und ein breiteres Spektrum von anatomischen Fragen.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben nichts preisgeben.
Acknowledgments
Das konfokale Mikroskop wurde mit Mitteln von US National Science Foundation (NSF), Grant #DBI-0722569 gekauft. Die Forschung wurde durch die US nationale Institute der Gesundheit/National Institute of General Medical Sciences (NIH/NIGMS) Grant #GM085290 und US-Department of Defense (DOD) Grant #W81XWH-16-1-0466 verliehen an r.m. Brewster unterstützt. E. Vital wurde unterstützt durch einen Zuschuss zu UMBC vom Howard Hughes Medical Institute durch die Pre-College und Undergraduate Science Education Program, #52008090 zu gewähren. S.p. Brown wurde unterstützt durch eine US of Education GAANN Fellowship, ein Meyerhoff Graduate Fellowship von NIH/NIGMS Grant #GM055036 und eine Forschung Assistentenstelle finanziert durch die US DOD Grant #W81XWH-16-1-0466 finanziert.
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
