

Summary
ここでは、他の組織に広く適用される、ゼブラフィッシュ脳の発達における微小管の異なる集団を分析するメソッドについて反応について説明します。最初のプロトコルでは、反応の安定性と動的微小管の最適化手法について説明します。第二議定書は、画像し、初期微小管を具体的に数値化する方法を提供します。
Abstract
微小管 (MTs) は、イメージ体内、特に脊椎動物の胚に挑戦して、ダイナミックで壊れやすい構造です。反応メソッドは、ゼブラフィッシュの胚の成長の神経管の MTs の異なる集団を分析するためここで記述されます。神経組織にフォーカスがある、この方法論は他の組織に広く適用されます。手順は、早くに半ば形成段階の胚 (12 体節に 1 体節)、しかし彼らに適応できる一連の比較的マイナーな調整と他のステージのために最適化されています。最初のプロトコルは、安定性と動的な MTs の空間分布を把握し、画像処理ソフトウェアでこれらの集団の定量的分析を行うメソッドを提供します。このアプローチは、既存のツールにリアルタイムを使用して形質転換線のタグ構造体の一過性の発現分布と画像微小管動態を補完します。確かに、このようなツールは、ダイナミックで安定した MTs とは容易に区別しないしかし、非常に便利。イメージを作成し、これらの異なる微小管集団を分析することで、細胞の極性形成と形態形成メカニズムの理解のための重要な含意。2 番目のプロトコルでは、新生 MTs を具体的に明らかにする手法について説明します。これは、次の薬ノコダゾールと薬物ウォッシュ後の回復期の微小管脱重合時間をかけて、MTs のde novoの成長特性をキャプチャで。この手法は、MTs のゼブラフィッシュ胚におけるにまだ適用されていない、微小管重合に関与する蛋白質の生体内機能を調査するための貴重なアッセイをします。
Introduction
微小管 (MTs) は、ポリマーの α- および β-チューブリン線形原繊維、いくつかを組み合わせて、中空の管1,2を形成にまとめることです。MTs は、終了プラス成長著しい中心体または他の微小管重合のセンター (脂質)3で固定終了マイナス成長が遅いとの偏波構造です。De novoMT 形成は (γ-テュルク) に複雑な γ-チューブリン環核 MT アセンブリ4のテンプレートを提供するによって開始されます。任意の指定されたセルに MTs の二集団区別できるそのターン以上異なるレートで。動的 MTs は、成長の段階と動的不安定5として知られているプロセスの収縮の間の切り替えによって、細胞環境を探る。動的 MTs とは異なり安定した MTs 非成長しているし、動的 MTs6より長い半減期があります。
細胞生物学の研究の十年は MT の構造と機能を研究するためのツールの高度な配列を提供、これらの細胞骨格要素に関する知識の大きいボディの結果します。例えば、MTs は確立と維持だけでなく安定動的 MTs7,対の差動細胞内分布にも、本質的な極性に起因する細胞極性の中心的な役割を再生8. 対照的に、まで以下について理解される MT アーキテクチャと脊椎動物の胚などのより複雑な三次元 (3 D) 環境で機能の一部高解像度で MT 骨格のイメージングへの挑戦のため。この制限にもかかわらず gfp 発現するトランスジェニックの最近の生成は行ラベル MTs または MT マーカーの蛍光タグの一過性の発現は、MTs を受けるダイナミックな変化と自分の携帯電話の私達の理解を増加しているとゼブラフィッシュ胚の発達の役割。どちらかは、ラベル付きの9またはチューブリンのポリマーは、直接 MT 関連タンパク質を使用してを分類しているが直接のチューブリンの形質転換線で視覚化される MT ネットワーク全体 Doublecortin ようなキナーゼ (Dclk) または Ensconsin (EMTB)10、 11。他の線 (および構造体) 生成されている MT プラス終了の具体的分類によって MT 本質的な極性の評価を有効にするまたは終了11,12,13,マイナス中心体固定14です。 これらのツールの力を成長の有機体ライブ、MT のダイナミクスを学習する能力であります。このような研究は明らかに、たとえば、特定の細胞集団に MTs の空間と動的配布、形態形成 (細胞分裂面のインジケーター) を受けている組織で細胞分裂の方向のスピンドルは、MT ポリマーの極性細胞の伸長など、移行プロセスに関連と MT 成長率彗星速度9,13,15によって決まります。これらのツールの制限は、彼らが容易に安定性と動的 MT 集団間に行いません。 ことです。
豊富な細胞生物学文献からの描画、反応メソッド ゼブラフィッシュ胚における安定性と動的な MTs のイメージを説明ここでは、形質転換線の使用を補完するものであります。ゼブラフィッシュのような反応方法の普及はやや固定プロシージャ中に MT の整合性を維持する難しさによって妨げられています。プロトコル 1の反応の合計、ダイナミックな最適化手法の概要を説明し、発展途上のゼブラフィッシュ脳の断面積で安定した Mt。さらに、商業的に使用する直接的な方法利用可能なソフトウェアは、これらの MT 集団を定量化に記載されます。安定した MTs は、α-チューブリンのアセチル化、detyrosination、時間16,17をかけて安定した MTs の蓄積などのいくつかの翻訳後修飾に基づく動的 MTs と区別されます。ゼブラフィッシュの胚のアセチル化は、毛様体と軸索の MTs の安定した界面 MTs18、安定した MTs のサブセットにこのマーカーの有用性を制限することではなく、発生します。対照的に、detyrosination は、ゼブラフィッシュ胚18のすべての安定した MTs で発生するが表示されます。この翻訳後修飾は、α-チューブリン (tubulin detyrosinated)18のカルボキシ末端のグルタミン酸を公開し、抗グルタミン酸チューブリン19を使用して検出することができます。Detyrosination は、安定した MTs に優先的に発生しますが、実験的証拠は MT 安定16の原因ではなく結果、この翻訳後修飾であることを示します。、抗-Tyr-チューブリン, α-チューブリン19の tyrosinated フォームを特異的に認識する抗体を用いた動的な MTs の構成相互の MT の人口は区別されます。次のこれらのマーカーと共焦点レーザー顕微鏡の反応、MTs (長さ、番号、および相対的な豊かさ) の定量分析は成長の神経管の定義された領域で実行できます。合理化されたメソッドは、3次元画像処理ソフトウェアを使用して分析を実行するためここで提供されます。このメソッドは、形態形成や設立成熟細胞極性20の質問はアドレスに適用できます。確かに、安定した MTs の偏光配列の精緻化に伴う光受容形態21、開発途上神経管18と軸索形成の8細胞の上皮化を含む多くの発達的イベント。
プロトコル 2では、そのアセンブリ フェーズ (アンカレッジ/核生成と成長)22,23中 MTs を分析する細胞生物学試験の生体の適応について説明します。新生 MTs は中心体を核、その後母中心小体23の subdistal 肢に固定します。次の解重合初期の MT 再生を分析する方法を説明します。このプロトコルは、MTs、薬物のウォッシュ アウト プロシージャおよび治療後の回復期間を耳下腺ノコダゾール処理に関する詳細を提供します。MT の再成長が一定間隔で監視します。合計 MTs のためのマーカーとの反応によって s post ウォッシュ アウト (抗 β-チューブリン) 中心体のマーカーと一緒に (抗 γ-チューブリン) と核 (4', 6-diamidino-2-phenylindole (DAPI))、プロトコル 1で説明する一般的な手順によると。De novo MT の評価ではなく既存の MTs の拡張が可能、このプロトコルの MT 解重合ステップは欠かせません。この手法は結合タンパク質 3 チャンらに示すように、緑色蛍光タンパク質 (EB3 GFP) に融合エンドなどプラスのヒント マーカーを使用して、(解重合の不在) の MT の成長率を測定するためその他のパブリッシュされた手順とは異なるため201211。この試金は胚・ デ ・ ノボMT アセンブリの欠陥を分析する場合に役立ちますまた、不完全なものに終って中心体には γ-チューブリンの募集が障害者である以前に報告したNEDD1変異など神経管形成と神経欠陥24。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
倫理ステートメント: 手順は下記に従って大学メリーランド州ボルチモア郡動物診療ガイドラインの
。1 です安定解析と動的 MTs を使用して反応 (プロトコル 1)
- 固定する前に胚のマニュアル dechorionation
- 取得新鮮なシステムが余分な水を注ぐことによって胚を生み出したと。プラスチック シャーレに残りの胚を収集 (材料の表 を参照してください).
- 胚中でエアダスター システム水と転送胚から新しい料理いっぱい削除 (材料の表 を参照)、胚をクリーンな環境で開発することを確認する 。
- 28.5 で低温インキュベーターの目的の段階に開発する胚を許可する ° C Dechorionation の前にガラスの皿で
- 24 h より若い場所胚後受精 (hpf).
定着剤・保護区 MT 整合性の急速な浸透を最大化するために固定する前に注意: Dechorionate 胚。追加 Ca 2 + dechorionation 時に必要なを提供するシステムの水の代わりに使用して胚中です 。
- は、胚ペトリ皿、解剖顕微鏡下での微細鉗子を使用してから、絨毛を手動で削除します。
- 膜の破裂を作成する鉗子とそっと離れてプル鉗子のペアを持つ胚を取り囲む円形で透明な絨毛膜内の小さな領域をピンチします 。
- は、鉗子を用いた破裂の絨毛膜の繊細な詮索好きな開口部を拡大します。それが破裂させ、鉗子と胚に触れないように注意してください 。
- 段階の胚の固定
- 転送、dechorionated 1.5 mL 遠心チューブに胚を上演します。ガラス パスツール ピペットを使用して、できるだけ多くの胚の中を削除します
。 メモ: 前の安楽死の中に痛みを軽減するために追加の手順を必要としない、痛み感覚を媒介する神経センターの形成の若い (半ば形成) 胚の固定・薬物治療を行います。発達段階は、キンメル ら 定義します。、1995 年 25。4-5、11-12 体節段階を用いて 図 2 および 3 の画像を取得します 。
- 準備 4% パラホルムアルデヒド (PFA)/MT アセンブリ バッファー (MAB) 定着剤 (材料の表 を参照してください) 8 %1 mL を組み合わせることで X MAB と 100 %2 μ L を加えて 1 mL 2 あたり PFA 総量の 1 mL あたりトリトン X-100
。 注意: は、皮膚刺激性である PFA とトリトン X-100 を含む溶液を処理しながら手袋を着用します 。
- 28.5 で 5 分間 1 mL 4 %pfa/MAB 定着剤で修正胚 ° C. 吸引ピペットと定着剤 1 mL 新鮮な液に置き換えるし、ロッカーに室温 (RT) で 3 時間インキュベートします
。 注: サンプルに修正してくださいすぐに彼らの生物学的温度 (ゼブラフィッシュの 28.5 ° C) で温度依存した MT 解重合を防ぐ 。
- 転送、dechorionated 1.5 mL 遠心チューブに胚を上演します。ガラス パスツール ピペットを使用して、できるだけ多くの胚の中を削除します
- 固定液を吸引し、x NP40 とトリス緩衝生理食塩水 1 mL 1 を追加 (TBS NP40) バッファー。3 回各 5 分のロッカーに RT で振ちましょう。1 mL の新鮮な 1 に 4 ° C で胚を保存 7 日の X TBS NP40
。 注意: が NP-40、皮膚刺激物質を含む溶液を処理するとき、手袋を着用します 。
ソリューションはホット プレートを使用して明確になるまで、密閉容器に媒体を埋め込み - 反応を押します胚
- 熱 RT 4% 低い融点 (LMP) agarose を解剖顕微鏡の近くに配置されて 50 ° C に設定.サンプル間閉じられ、埋め込みプロセス (手順 1.4.4-1.4.6) を通して暖房コンテナーを保持します 。
- 1.5 mL から移植胚ガラス ピペットを使用してシャーレにチューブを遠心し、1 でそれを埋める X TBS NP40 。
- は、解剖顕微鏡 26 の倍率下で微細鉗子を使用してシャーレの形成段階の胚 (4-5、11-12 体節) から大きな卵黄細胞を削除します。尾芽によって胚を 1 組の鉗子押し後脳組織を維持するために他のペアと卵黄細胞をはがします。シャーレの卵黄の破片の自由の領域にデ卵黄の胚を転送します
。 注: は、LMP agarose の早期硬化を防ぐために個別にアガロースで満たされた金型で胚を埋め込む 。
- 塗りつぶし 1 つ 12 x 5 mm × 3 mm 200 μ L と断面のカビのことをよく溶かし LMP アガロース マイクロ ピペットを使用しています。手順 1.4.5.-1.4.6 を実行します。すぐに (20 内充填型 s) LMP agarose RT に冷却し、固化する前に胚を埋め込む 。
- シャーレから解剖顕微鏡の下で、テーパー年末に向けてアガロースで満たされた金型に、尾で・ デ ・卵黄の胚を転送する微細鉗子を使用します 。
- 、Vibratome を目的の平面で切るように金型内に胎児の向きを使用罰金鉗子。その背面のエッジとその前方面がテーパーのリージョンの終了後脳組織実行金型の長さに平行その胚を定位して横断を作成します。残りの胚の手順 1.4.4-1.4.6 を繰り返します 。
- 室温 5 分を固めるための agarose 埋め込み許可
- 生成 40 μ m agarose の高い軸部で満ちている 1 断面の料理、vibratome を用いた胚 (手順 1.4.1-1.4.7) を埋め込まれた TBS NP40 x。利子のセクションを 500 μ L 1 で 24 ウェル プレートに転送 TBS NP40 微細鉗子を使用して x。ウェルあたり 1 つだけ胚のセクションに配置します
。 注: 詳細については、 18 を参照を参照します。セクションままバッファーおよび低速度 (10-25 rpm) agarose の埋め込みからの分離を防ぐために残りの手順でロックの少なくとも 250 μ L のすべての回で水和を確認します。ブロックに存在する洗剤と洗浄ソリューション必要があります液体培地の表面張力を減らすし、セクションの水没を許可します。中に、すべての操作の後に井戸にセクションが残っていることを確認します。洗浄中に誤って破棄セクションを防ぐために注意を使用します 。
- バッファーを削除し、ソリューションをブロックの 500 μ L を追加。室温 1 時間、少なくともロック
注: 使用される各二次抗体のホスト種から 5% 血清を含むブロック ソリューション (材料の表 を参照してください). - ロッカーの 4 ° C で 36 72 h のブロック バッファーで希釈した 300 μ L 一次抗体で加温します。600 μ L 1 で二回洗って 30 分ごとに、ロッカーに TBS NP40 x 室温
注: 合計 MTs (β-チューブリン抗または抗 α-チューブリン) や安定した MTs (Glu 抗チューブリン) や動的な MTs (反 Tyr チューブリン) に対して一次抗体の孵化によって二重ラベル] セクション。Α-チューブリンの人口合計とファーストクリーニング二重ラベル変更されたとき別のホスト種で提起されている一次抗体を選択します。抗体希釈液の 材料表 を参照してください 。
16-24 時間、暗闇の中で 4 ° C でのロッカーのブロック バッファーで希釈した蛍光標識二次抗体の - 加温 300 μ L。600 μ L 1 で二回洗って 30 分ごとに、ロッカーに TBS NP40 x 室温
注: は、箔この時点以降から、焼入を防ぐために各操作後の二次抗体を含むマルチよく皿をラップします。一次抗体のホスト免疫グロブリンと反応選択の二次抗体.二次抗体の同時独立した、非重複の発光スペクトルを持つを選択します。抗体希釈液の 材料表 を参照してください 。
- RT TBS NP40 を 5 分間常温揺動で 3 回洗浄での 30 分のロッカーに DAPI 溶液 500 μ l で胚をインキュベートします。
。 注: 核ラベリング手順 1.12 MT 定量化のため携帯電話のコンテキストを提供します 。
- は、埃のないスライドの中央に耐フェード エージェントでメディアをマウントのドロップを配置します。微細鉗子を使用すると、マウント中液滴のセクションを転送します。サンプルの上にほこり無料 coverslip の場所。イメージングが実行されるまで、箔に包まれて乾燥、暗くて涼しい場所にスライドを保存します
。 注: イメージ作成前の細い油性ペンを使用してスライドの背面にセクションを旋回顕微鏡を使用するときに、セクションを識別するのに役立ちます 。
- 共焦点イメージング
- 目的に直面して coverslip の段階にスライドを付けることによって走査共焦点顕微鏡倒立レーザ マウント セクション。コントロール スライドに適切な光学系 (目的、レーザー、およびチャネルの設定のようにゲインし、オフセット) を決定し、サンプル 27 間一貫性を保ちます。データの損失を防ぐためにピクセルを飽和を避ける 。
- は、チャネルの設定を使用して選択した二次抗体 fluorophores の Z スタック共焦点画像をキャプチャし、画像ファイル 27 を保存します。各セクションの Z スタックを取得します
。 注意: 次の取得の設定を使用して、 図 2 および 3 に画像を取得するためのパラメーターを複製: モード = XYZ;対物レンズの倍率 = 63 倍油浸漬レンズ;客観的数値絞り = 1.4;Z ステップ = 0.1 μ m;Z 深さ = 16.23 μ m 以下のチャネル設定を使用して: 20% 紫外線範囲レーザー、発光フィルター範囲 DAPI の励起 430-480 nm、光電子増倍管 (PMT) 利得を = = 525 V、および光電子増倍管のオフセット =-1.72%;448 nm 蛍光 (材料の表 を参照してください) 20 %488 nm レーザー、発光フィルター範囲を持つ励起 = 493 573 nm, 光電子増倍管の利得 = 689 V、および光電子増倍管のオフセット =-0.2%;594 nm 蛍光励起 32 %594 nm レーザー、発光フィルター範囲 = 608 706 nm、PMT ゲイン = 768 V、および光電子増倍管のオフセット =-6.8% 。
- のわかりやすい、一意のファイル名で生データ ファイルを保存し、画像解析ソフトウェアで編集用のコピーを作成します 。
- Z-スタック最大予測を表示するためのコンパイル
- パブリック ドメインの 3次元画像解析ソフトウェア (例えば ImageJ) を使用してデータ ファイルのコピーを開きます。各チャンネルは個別のイメージ シーケンス (Z スタック) として表示されることを確認します 。
- メニューの次のシーケンスを使用してイメージのチャンネルに分割: “ チャンネルの画像/色/分割 ”.
- メニューの次のシーケンスを使用して興味のあるチャネルを重ねることによってマージされた画像を作成する: " 画像/色/マージ チャンネル。 " 選択、594 nm、488 nm、偽色が赤、緑、および青それぞれに DAPI チャンネル。チェック " を作成する複合 " 選択 " OK " 28
。 注: 省略より他の 2 つのチャネルの偽色を選択するだけで 図 2 3 のように最大投影で MTs に特有の詳細を伝える DAPI チャネル 。
- は、マージされた Z スタックを調べるし、最初と最後のすべての目に見えるチャネル内部最高 Z 平面の位置をメモしておきます。通常セクションの表面の凹凸のため信号が次善である外側の Z 平面を却下します。詳細については 29 を参照を参照してください 。
- 次の 3次元画像解析メニュー シーケンスを使用して、Z-スタックの最大強度投影を実行して単一の 2 D イメージとしてマージされた Z スタックを視覚化する: " 画像/スタック/Z-プロジェクト " 開始と終了インナー ベストの位置を入力1.11.3 ステップから Z 平面、" スタート スライス " および " 停止スライス " それぞれ。選択 " Max 強度 " 投影タイプとクリック " [ok] "。詳細については、 28 を参照を参照してください 。
- 分類の分析 MT
- 市販の 3次元画像解析ソフトウェアを開きます。選択 " ライブラリの作成 "、画像ライブラリの名前を提供します。クリックして " を作成します。 " ライブラリに共焦点顕微鏡から生成された raw イメージ ファイルをドラッグ。サイズの大きいファイルを転送するためのより多くの時間を必要とします 。
- は、解析するファイルを選択します。選択 " 拡張フォーカス " から、" ビュー " のメイン ウィンドウでチャンネルにマージされた画像を表示するメニュー 。
- は、バック グラウンド信号を減少し、真の信号は堅牢なまで左または右に各チャンネルのスライダー ツールをドラッグして、しきい値を調整します。観察標識分子 (例えば、長方形または分裂核ですが細胞質または agarose からない自家蛍光を示す DAPI チャンネル) の真の信号を各チャンネルに示します 。
- 選択、" フリーハンド関心領域 (ROI) " ツールし、分析する関心領域の概要を説明します。選択、" アクション " タブが続く " 選択範囲で切り抜き " 画像をトリミングします。トリミングされた画像ファイルを新しい名前で保存します。クリックして、" 測定 " 3次元解析に関連する特定のオブジェクトをフィルター処理するためのプロトコルを作成するタブ 。
- ドラッグ " を見つけるオブジェクト " プロトコルのウィンドウに。最初のプロトコルの名前を変更 " DAPI。 " ドロップ ダウン メニューで DAPI チャンネルを選択します。DAPI プロトコルに次の設定をドラッグし、それらを下に置く " を見つけるオブジェクト " (表 1) の次の順序で: " ObjectsŔ で穴を埋める" ObjectsŔ に触れる別" SizeŔ でオブジェクトを除外" ・ ロワに触れていない除外 ".
注: 最初の設定信号を破棄するしきい値分布を 表 1 に設定の目的である、サイズ、分析対象のオブジェクトのサイズと一致しません。たとえば、核となり核をカウントする際に十分に大きくない信号を排除します 。
- Β-チューブリンと他のマーカーは、テーブル 1 の設定を使って残りを組み立てるための順序 (ステップ 1.12.9) フィルターを実行します 。
- 選択 " メジャー " 各プロトコルの下部に。選択 " 強度とボリューム測定 " と " 骨格の長さ " チューブリンのラベル付け、すべてが DAPI 信号元のみ 。
- は、測定する領域の周囲の投資収益率を描画します。下で測定を観察、" 概要 " タブの領域を処理した後ソフトウェア。データをコピーし、表 2 のように、実行可能なスプレッドシートに保存します。後で分析できるようにスプレッドシートのバックアップ コピーを作成します 。
- 選択対象 (たとえば、長さ MT バンドル、MT と明らかにバンドル/核数の測定さまざまなマーカー) スプレッドシートの各グループの平均値を決定するために分析と
。 注: 平均の MT バンドルの長さの合計を =、' β-チューブリンの骨格の長さを意味 ' 各胚の胚の合計数で割った値します。表 2 の 20 行を参照してください。変数群と実験群を簡単にグラフ化できるように、スプレッドシートの書式を設定します 。
2。・ デ ・ ノボMT アセンブリ アッセイ (プロトコル 2)
- 構築とテスト、マルチ フロースルー式装置 ( 図 1) 実験に先立ち 2 日
。 注意: 装置はノコダゾール治療 材料の表 からの装置を使用した実験の複数のグループの同時ウォッシュ アウトできます。シリコーン シーラーは、乾燥の時間それは胚毒性危険をもたらす前に少なくとも 24 時間を必要とします。- 半分の 50 mL 遠心管を分割縦、治具やバンドソーを使用します 。
- カット 7 半円、半径 3 cm、70 μ m のナイロンのメッシュし、トリムに合わせてしっかりと一つに分割の半分はチューブを遠心分離します。水族館の安全なシリコン シーラーを使用して 10 mL グラデーション マーキングに平行の遠心管に半円を接着します。2 日間乾燥し、ビーカーの中 2-3 h. のための水の浸漬による洗浄装置を許可
- トップ (スレッド) 粘土のモデリングの流れを介してデバイスに保持液の高さが 1/4 インチ ( 図 1 2 と 3 のビュー) の深さでカット遠心管末をラインします 。
- は、50 mL シリンジのプランジャーを取り外し、極細管の 12 インチの先端を挿入してウォッシュ アウト装置を準備します。限り、それは行くし、モデリング粘土を用いた関節周りのシール部チューブをプッシュします 。
- は、全体の流れを通してデバイスを介して実行する液体を許可する胚の媒体を使用してメッシュを事前濡れています。角度が、まだすべてのコンパートメントの液体のプール、氷の上のデバイスは、粘土の縁のある前面を空にします。氷 ( 図 1) の流れをデバイス上のウォッシュ アウト装置を中断リング スタンドを使用しています 。
- 氷の上胚中の 200 mL を寒さし、ウォッシュ アウト装置はすべての空気の泡がクリアされる流量が注射器の高さを変更することによって約 7 mL/分の流量の調整をされることを確認するに十分なを注ぐ 。
- Dechorionate 胚酵素によって
- 20 mL 胚中非特定プロテアーゼ株式 10 mg/mL の 1 mL を希釈することによって非特定プロテアーゼの実用的なソリューションを作る 。
- 胚前に彼らが必要な発達段階に到達する予想される場合 timepoint 1 h 化学 dechorionation を実行します。100 mm シャーレから胚媒体を取除くことによってダイジェスト絨毛上演胚および非特定プロテアーゼ作業溶液 20 mL を追加します 。
- 37 ° C で 5 分間加温胚
注: 5 分を超える使用したりしないで非特定プロテアーゼ溶液の高濃度落下胚になるので離れてノコダゾールと一度扱われる 。
- はすぐに非特定プロテアーゼをピペットし、約 25 mL 胚中料理を補充します。もう一度繰り返します 。
- 損傷からそれらを保護するためにガラスに hpf 料理を 1 mL ガラス ピペット、24 歳未満の移植胚を使用しています 。
- 完全な dechorionation 1.1.5 の手順で説明するように、微細鉗子のペアを使用して絨毛を手動で削除します 。
- 希望の発達段階に達するまで、30 分以上 28.5 ° C の定温器で dechorionated 胚を含むガラス シャーレを配置します 。
- Depolymerize 既存の MTs
- 10 mL の氷冷胚中 50 μ L 1 mg/mL 在庫ノコダゾールを組み合わせることにより 5 μ G/ml ノコダゾールの実用的なソリューションを準備します
。 注意: ノコダゾール、皮膚刺激を処理するときに手袋を使用しています 。
- は、10 mL の冷たいノコダゾール実用的なソリューションを持つノコダゾール治療グループの胚培地を交換します。ペトリ皿を氷の発達段階 (4-5 体節胚のたとえば、1 h) の適切な時間の場所します。2.3.4.1 整数型におけるステップのウォッシュ サンプルと一緒に固定する氷の上ペトリ皿での無処理胚をおきます 。
- 移植胚実験グループごとに別々 のコンパートメントを用いたフロースルー装置に火磨かれた 1 mL ガラス ピペットを使用しています。50 mL の注射器の上に注いで氷冷胚媒体によってノコダゾール ウォッシュ アウトを開始します
。 注: は、実験群あたり少なくとも 30 の胚を使用します。実験グループがコントロール胚またはさまざまな morpholino または RNA 注入胚で構成されます。ウォッシュは、氷で MT の成長を阻害しながら追加 8-10 分ごとにウォッシュ アウトする胚中の約 150 mL の合計、ノコダゾールを必要があります。MTs が低温で安定しないと風邪初期胚の開発を遅らせるために、氷の上胚を保つことがこの試金の成功のため不可欠です 。
- を許可する MTs ガラス シャーレに胚を転送することによって RT のウォッシュ アウトの 20 分後に再生するのには暖かい火磨かれた 1 mL ガラス ピペットを使用して (28.5 ° C) 胚中です。胚を転送したとすぐに、タイマーを開始します。
- コントロールとウォッシュ アウトの胚を 1 分で修正、5 分と 1.5 mL に約 10 胚をピペットで 10 分遠心管 1 mL 4 %pfa/MAB 修正 (28.5 ° C) でいっぱいで指示に従うステップ 1.2.3 。
- 10 mL の氷冷胚中 50 μ L 1 mg/mL 在庫ノコダゾールを組み合わせることにより 5 μ G/ml ノコダゾールの実用的なソリューションを準備します
- 反応に従ってセクション 1.3 から 1.5 のためにサンプルを準備します 。
- Immunolabel フローティング セクションととしてイメージ胚一次抗体の仕様に以下の変更と 1.6 1.10 節で説明: マウス抗 β-チューブリンの使用 1: 500 ウサギ抗 γ チューブリンとレバレッジ 。
- プロセスと 1.12 の手順で説明するように 3次元画像解析ソフトウェアを使用して画像を分析します 。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
反応を使用して安定性と動的な MTs の解析
プロトコル 1, Glu チューブリンと Tyr チューブリン マーカーとして使用する安定性と動的な MTs のそれぞれ初期 (神経キール) 中に MT のサブ集団の分布と神経管の開発の後期 (神経ロッド) が明らかに。動的な MTs 一世を風靡菱脳神経キール ステージ (4-5 体節) で (図 2A ~ D)。キールが神経のロッド (11-12 体節)、強化された上皮化の段階に発展する質的少ない MTs は腹側のロッドを中心に反 Tyr チューブリン抗体 (図 2E H) 陽性。対照的に、Glu チューブリンが散在しており神経キール (図 3A ~ D) 全体で点状が MT 管 (図 3E H) に沿って腹側神経ロッドで濃縮されています。矢印は、ラベリングが増加特定の MT バンドルまたは構造体をポイントします。
抗グルタミン酸チューブリンと反 Tyr チューブリン抗体は (二重標識実験の防止) 同一宿主動物種で生産されたが安定的かつ動的な MT のマーカーはほとんどゼブラフィッシュの逃避重複と考えられました。まず、腹側の神経のロッドが動的 (図 2F) MTs よりもより安定した (図 3F) です。背側神経ロッドで傾向を反転、ゼブラフィッシュ モデルと一致して結びつけている神経管までに背側の組織するダイナミックなまま形成20。第二に、分裂が完全に付けニューラル キール (図 2D矢印)、Tyr-チューブリン抗体、中心体と一致するスピンドルのベースだけ中が付いて安定性マーカー Glu チューブリン (図 3D矢印)。Β-チューブリン蛍光抗体法、両方の試金に一般的な通知されますすべて MTs 分布の実験者、非固有のラベルを却下するための基礎を提供しています。
大量の便利な表 (表 2) にまとめることがデータで起因する 3次元画像解析ソフトウェアを使用してオブジェクトを測定します。長さ、数、および面積の測定をするためには、分析に使用できるデータのサブセットのみを用いています。さらに分析することがなくデータのコンポーネントの 1 つは、指定したオブジェクトの数です。この番号は、数する必要がありますいないによって大きく異なるセクション、および MTs に核の比率は単一の治療条件の類似したとどまるべきであるのような内部の品質管理として使用します。外れ値は、調整フィルターを再実行する必要がありますいずれかの分析、またはイメージの分析も不十分なラベルが付いたインジケーターです。したがって、外れ値のすべての画像は、調整した設定を再分析する必要があります。外れ値セクションは、貧しい人々 のラベル付けや異常なオブジェクト カウント可能性があります物理的な損傷の兆候を検討すべき。解析が完了すると、品質管理、有用な情報から回復できる生データはよう合計 MTs の平均の長さと、MTs または合計 MTs (表 3) に安定した mt 比を安定します。これらの測定に加えて (核、中心、等) 他の細胞構造に MTs または彼らの関係に関する推論を描画するために使用することができます 3次元画像解析ソフトウェアを使用して他の多くの統計情報が得られます。
De novoMT アセンブリ アッセイ
ノコダゾール治療 depolymerizes MTs (図 4A、4 D 、 4 G) びまん性ラベリングの結果です。MTs が再生すると、彼らは中心体 (図 4B、4 eと4 H) から延長、ただし、これはそれらの非平面軌道 (図 4C、4 階と4I) による単一の平面でわかりにくい。それにもかかわらず、いくつかの画像解析ソフトウェアはノコダゾール ウォッシュ アウト (表 4) 後 MT 成長の評価を有効にする 3次元の長さを測ることができます。表 4にデータセットから得られる重要な観察、分析神経管のすべての地域でノコダゾール ウォッシュ後の時間の経過とともに増加する MTs の平均の長さが表示されます。前述のように、他のタイプの 3次元画像解析ソフトウェアから得られるメトリックは、mt 法データ (たとえば、比 MTs の核当り) を解釈する携帯電話のコンテキストを提供できます。
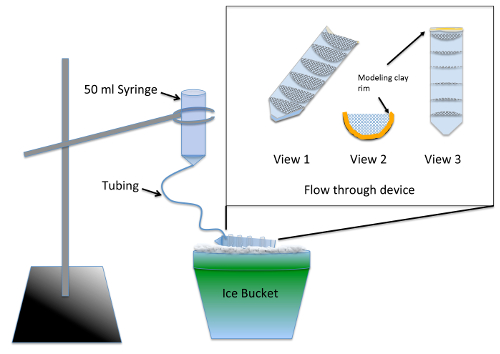
図 1: [ウォッシュ アウト] 装置の図・ デ ・ ノボMT アセンブリ アッセイ。はめ込みカット縦 50 mL 遠心管に接着メッシュから作られた流れを介してデバイスのクローズ アップがあります。メッシュは、複数の実験グループを同時に処理することができますように通過デバイスを個人的します。使用中に胚の中は注射器に追加され、すべての実験グループに一定のリンスを提供するフローを介してデバイスに合わせてチューブをゆっくりと流れます。この図の拡大版を表示するのにはここをクリックしてください。

図 2:動的な山地をイメージさせる反応の利用Dechorionated 胚の適切な段階 ( A ~ Dの 4-5、12-13 体節Eh)、後脳と β-チューブリン ( AとEグリーン) に対する抗体とカルビンディン抗体を介して横断面で固定しました。動的 MT 集団を明らかにするすべての MTs と tyrosinated α-チューブリン ( BとFで赤) をマークします。非常にダイナミックな MTs は、黄色のラベルが表示されます ( D, Hの矢印) の地域として結合画像 (C, G) と (D, H) そのより高い倍率で見ることができます。スケール バー = (A ~ C と E G) 25 μ m と 10 μ m (D ・ H)。この図の拡大版を表示するのにはここをクリックしてください。

Figure 3: 安定した山地をイメージさせる反応の使用Dechorionated 胚は、適切な段階 ( E-Hで-D と 12-13 距離で 4-5 体節) で、菱脳とカルビンディン抗体を切断、修正されました。合計 MTs は一般的な β-チューブリン抗体 ( AおよびEの緑) で視覚化した中安定した MTs は ( BとFで赤) α-チューブリン (Glu-チューブリン) の detyrosinated フォームに対する抗体が付いています。結合画像 (C, G) と (D, H) そのより高い倍率で赤と黄色の信号は、高山の安定度 (D, H の矢印) の領域を表します。スケール バー = (A ~ C と E G) 25 μ m と 10 μ m (D ・ H)。この図の拡大版を表示するのにはここをクリックしてください。

図 4:初期の山地をイメージさせる反応の利用Dechorionated 胚は 4-5 体節に固定され、横菱脳を断面します。セクションだったカルビンディン抗体 β-チューブリン (D、E、およびF) MTs の成長をマークすると γ-チューブリン (A、B、およびC) 核形成ポイント/体をマークします。神経管の背側領域は箱入り (A、D;B、EとC F)、高倍率で表示 (G, H, 私、それぞれ) 核 (DAPI、ブルー) の合計と、(γ-チューブリン ・赤) の由来を明らかにする MTs (β-チューブリン、緑)。白の矢印: MTs と由来。黄色の矢印: セルの第 2 中心小体が表示されます。スケール バー = (A ~ F) 25 μ m と 10 μ m (G-私)。この図の拡大版を表示するのにはここをクリックしてください。

表 1: 既定の 3次元画像解析ソフトウェア内のオブジェクトをフィルター処理するための設定。

表 2: 代表的な raw データ セットを分析するソフトウェアは MTs を安定した 3次元画像解析を用いて。各列では、単一のセクションからの測定値を表します。Min: 最小測定;Max: 最大測定;SD: 標準偏差;SE: 標準誤差です。

テーブル 3: 3次元から得ることができるデータセットの例として画像安定した MTs を定量化する分析ソフトウェア。合計 (β-チューブリン) の平均長さの測定を選択し、MTs (の合計 (Glu-チューブリン) MTs (表 2を参照してください) すべてのサンプルから関連するラベルの平均の骨格の長さの平均と安定の比で計算を安定Glu-tubulin あたり β-チューブリンの縞すじの一手) 平均の Glu チューブリン数で割った平均 β-チューブリン カウントを取得して計算します。

表 4:データセットを分析する 3次元画像解析ソフトウェアから取得可能の例・ デ ・ ノボMT アセンブリ。De novo MT アセンブリからの代表的な結果の実験、ノコダゾール ウォッシュ後に 3 つの回復時間ポイント (1, 5, 10 分) 得られたデータセットを比較します。各時点の核数を由来 (γ-チューブリン涙点)、合計 MTs (β-チューブリン縞) の数の測定が表示されます選択、イメージの領域分析 (開発の神経管の断面)。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
現在多くの方法がありますゼブラフィッシュ胚における MT 動態をイメージングしてタグ付き分子ライブ イメージングからの反応に至るまで固定組織11,12,13,14。単一のセルに MTs は、動的または安定した状態で存在できますが、上皮化は、MTs は徐々 に時間をかけて安定化プロセスです。安定性と動的な MTs のマーカーを使用してこの現象を可視化する方法を提供しています。ここで紹介した方法では 3次元イメージング ソフトウェアの機能を活用して、動的からゼブラフィッシュ胚組織の断面の安定した MT の集団への移行を定量化します。プロトコル 2メソッドを使用して、新生 MTs の明瞭な人口をラベルし、核生成と成長の残業代に従ってください。
MTs が外界にその傾向のため、自然な状態でイメージし悪名高くにくいです。したがって、このメソッドの重要なコンポーネントは、MTs の全体胚全体の急速な固定です。これは、MTs が安定し、胚の浸透性を増加するバッファーを使用して生理的温度で固定を開始によって達成されます。固定時間も過固定は、抗原、抗体の結合と干渉することを隠すことができる間、縮小固定が MTs の逮捕に失敗すると重要です。3-4 時間の推奨固定時間は、半ば原腸陥入で、最大 24 時間後受精胚で動作します。時間スケールの若い終わりに向かって胚修正されますの近い 3 h より古い胚は全体 4 h を必要があります。適切な固定とも切片と反応する必要があります修正から 1 週間以内に発生するので、MTs は時間と外界が。
組織が正しく修正、一度反応を問題が発生することができます。セクションが多すぎますに孵化するで、同じの場合に発生した最も一般的な問題は特に、組織の中心を通って不溶をされています。胚の透過を改善するために洗剤の増加と共に第一次および二次抗体を一次抗体と潜伏期間の濃度を増加させる反応の問題のほとんどを改善します。抗体の分類が固定問題や反応の問題のため失敗した場合、抗体の分類のパターンを調べることによって、原因を特定することが可能です。貧しい固定が膜で強烈なラベリングの結果および細胞質にラベリングを拡散、とき過固定が弱いラベリングする MT アーキテクチャを保持されます。ただし、抗体、不溶と、ラベルなしで、組織の中心エリアとして表示されます。
有意義な方法で MT の画像を分析する能力、高品質の画像に依存します。3 D で MT 長さをキャプチャするには、目的と開口可能な最小の Z ステップ サイズを使用してください。ここで示されている画像は次の生産数値の 1.4 の絞りと油エマーション目的 X 63 で捕獲された: ピクセル = 240 nm、Z ステップ Z スタック サイズ 0.1 μ m の = = 16.252 μ m。シングル MT の幅が 25 nm、約 10 倍以下に顕微鏡、このメトリックの分解能の限界を正確にすることはできませんを用いてこの手法。代わりに、すべての 3 次元で達成可能な最小ピクセル サイズ以上のみの MT 長さを測定できます。行またはフレーム平均 MT 信号定義を強化できます。MT 解析は、高品質のセクションの予約する必要があります。貧しい固定組織をイメージし、分析できない、穏やかな overfixation 慎重にレーザー強度の増加によって相殺することができます、良好なダイナミック レンジを維持しながら弱い信号を検出するを得る。貧しい抗体浸透中最善ではない薄いセクション (5-10 μ m) をイメージングの結果、適切な標識が地域に画像取得を制限することによって修正できます。高いバック グラウンド表示からフィルターの設定を調整することによって補正できます。ただし、これらの調整のいずれかが行われている場合は、ことを確認する必要は Z スタックの各平面上許容できるフィルターのしきい値。
3次元画像解析ソフトでは、MT 長さ、面積、角度、豊富と固定ティッシュ セクションの 3次元空間内の他の基準を定量化する実験者をことができます。ここで説明した方法は、市販のソフトウェアを用いてデータを取得するための方法を説明します。ただし、フィルタ リング モジュールは、関連するプラグインやマクロ、分析のすべてで利用可能と強化されたパブリック ドメイン ソフトウェアに適応させること。分析の前に raw 画像はどちらられる数量化の背景と非特異的シグナルを含むを避けるためにする必要があります。分析を完了すると、実行可能なスプレッドシートにデータを転送、データ セットから多くの推論が可能です。ここで行われる計算の 1 つだった合計 MTs に β-チューブリン縞、または安定した MTs の比率ごとの Glu チューブリン縞 1 が全体の MT 骨格が投資収益率で安定したを表します実験者希望彼らの定量的データを補完する洗練されたタグ付きイメージ ファイルの形式 (TIFF) を生成する場合スケール バー画像が 3次元画像解析ソフトで楽。
このアッセイでは、MT アセンブリ、 in vivoに関与するタンパク質の機能解析をことができます。反応を連続切片を交互に実行する場合は、同じ胚における動的かつ安定的な MTs を勉強するこのプロトコルを使用する可能性があります。今後、増加洗剤または変更された埋め込み角度などの変更は、古い胚のためのこれらのメソッドの使用と解剖学的質問の広い範囲が許可されます。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者が明らかに何もありません。
Acknowledgments
共焦点顕微鏡はから、米国国立科学財団 (NSF)、補助金 #DBI 0722569 資金で購入しました。研究によって、米国国家機関の健康/国立医学研究所の一般的な (NIH/日の出) グラント #GM085290 および米国国防総省 (DOD) グラント #W81XWH-16-1-0466 r. m. ブリュー スターに授与を受けました。E. バイタルは UMBC に前の大学と学部科学教育プログラムを通じてハワード ヒューズ医学研究所からの助成金によって支えられた、#52008090 を付与します。S. p. ブラウンは奨学金教育 GAANN によって米国部のサポートされていた、メイヤーホフ大学院フェローシップ NIH/日の出の補助金によって資金を供給 #GM055036、および資金によって、米国の国防総省グラント #W81XWH-16-1-0466 研究させて。
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
