

Summary
Immunolabeling métodos para analisar populações distintas de microtúbulos no cérebro em desenvolvimento zebrafish são descritos aqui, que são amplamente aplicáveis a outros tecidos. O primeiro protocolo descreve um método otimizado para immunolabeling estável e dinâmica de microtúbulos. O segundo protocolo fornece um método para a imagem e quantificar microtúbulos nascentes especificamente.
Abstract
Microtúbulos (MTs) são estruturas dinâmicas e frágeis que são desafiadores para imagem na vivo, particularmente em embriões de vertebrados. Immunolabeling métodos são descritos aqui para analisar populações distintas de MTs em tubo neural em desenvolvimento do embrião do zebrafish. Enquanto o foco estiver em tecido neural, esta metodologia é amplamente aplicável para outros tecidos. Os procedimentos são otimizados para cedo aos embriões de meados-somitogenesis-estágio (1 somite para 12 somitas), no entanto podem ser adaptadas a uma gama de outras fases com relativamente pequenos ajustes. O primeiro protocolo fornece um método para avaliar a distribuição espacial da MTs estáveis e dinâmicos e realizar uma análise quantitativa dessas populações com software de processamento de imagem. Essa abordagem complementa as ferramentas existentes para imagem microtubule dinâmica e distribuição em tempo real, usando linhas transgénicas ou expressão transiente de construções etiquetadas. Com efeito, tais ferramentas são muito úteis, porém eles não facilmente distinguir entre MTs dinâmicos e estáveis. A capacidade de imagem e analisar essas populações distintas do microtubule tem implicações importantes para os mecanismos de compreensão subjacente a polarização celular e morfogênese. O segundo protocolo descreve uma técnica para analisar especificamente o nascentes MTs. Isso é realizado através da captura as propriedades de crescimento de novo do MTs ao longo do tempo, seguindo microtubule despolimerização com a drogas nocodazole e um período de recuperação após o fracasso de drogas. Esta técnica ainda não tiver sido aplicada ao estudo da MTs em embriões de peixe-zebra, mas é um ensaio valioso para investigar a função no vivo das proteínas implicado na montagem de microtúbulos.
Introduction
Microtúbulos (MTs) são polímeros de α - e β-tubulina que reúne numa protofilamentos lineares, vários dos quais se combinam para formar um tubo oco1,2. MTs são estruturas polarizadas, com rápido crescimento além de extremidades e crescimento lento menos extremidades ancorados na centrossoma ou outros microtubule-organização centro (MTOC)3. De novo Formação de MT é iniciada pela nucleação no complexo anel γ-tubulina (γ-TURC), que fornece um modelo para montagem de MT4. Em qualquer determinada célula, duas populações de MTs distinguem-se a curva sobre a taxas diferentes. MTs dinâmicos explorar seu ambiente celular alternando entre fases de crescimento e retração em um processo conhecido como instabilidade dinâmica5. Ao contrário de dinâmicas MTs, MTs estáveis são não-crescimento e tem uma meia-vida mais longa que a dinâmica MTs6.
Décadas de pesquisa em biologia celular tem fornecido um sofisticado conjunto de ferramentas para estudar a função e estrutura de MT e resultou em um grande corpo de conhecimento sobre esses elementos do citoesqueleto. Por exemplo, MTs desempenham um papel central no estabelecimento e na manutenção da polaridade da célula, que é atribuível não só a sua polaridade intrínseca, mas também para a distribuição diferencial subcellular de estábulo versus dinâmico MTs7, 8. em contraste, muito menos é compreendido sobre arquitetura de MT e função em ambientes (3D) tridimensionais mais complexos, tais como os embriões vertebrados, em parte devido ao desafio de citoesqueleto MT em alta resolução de imagem. Apesar desta limitação, a geração recente de transgênico GFP-expressando as linhas esse rótulo MTs ou expressão transiente de marcadores de MT marcados fluorescentemente aumentou nossa compreensão das mudanças dinâmicas que se submetem a MTs e seus celulares e papel do desenvolvimento no embrião zebrafish. Toda a rede de MT pode ser fotografada em linhas transgénicas na qual tubulina ou é diretamente rotulados polímeros9 ou tubulina indiretamente são etiquetados usando proteínas associadas MT Doublecortin-como-quinase (Dclk) ou Ensconsin (EMTB)10, 11. Outras linhas (e construções) foram geradas que permitem avaliação de polaridade intrínseca de MT por rotulagem especificamente MT além termina ou centrossoma ancorada menos termina11,12,13, 14. o poder dessas ferramentas encontra-se na capacidade de estudar a dinâmica de MT em ao vivo, organismos em desenvolvimento. Tais estudos têm revelado, por exemplo, a distribuição espacial e dinâmica de MTs em populações de células específicas, a orientação de mitótica fusos em tecidos submetidos a morfogênese (um indicador do plano de divisão celular), a polaridade do polímero MT no que se refere aos processos, tais como alongamento celular e migração e a taxa de crescimento de MT determinada pelo cometa velocidade9,13,15. A limitação dessas ferramentas é que eles não prontamente qualquer discriminação entre as populações de MT estáveis e dinâmicas.
Desenho da literatura biologia celular rico, immunolabeling métodos de imagem estáveis e dinâmicos MTs no zebrafish embrião são descritos aqui, que são complementares para o uso de linhas transgénicas. O uso generalizado de tais métodos immunolabeling no zebrafish tem sido um pouco dificultado pela dificuldade em preservar a integridade de MT durante o procedimento de fixação. 1 protocolo descreve um método otimizado para immunolabeling total, dinâmico, e MTs estáveis em secções do rombencéfalo zebrafish desenvolvimento transversais. Além disso, um método simples usando comercialmente software disponível é descrita para quantificar estas populações de MT. MTs estáveis são distintos dos MTs dinâmicos com base em várias modificações borne-translational do α-tubulina, tais como a acetilação e detyrosination, que se acumulam na MTs estáveis ao longo do tempo16,17. No embrião zebrafish, acetilação ocorre em ciliares e axonal MTs mas não em interfase estável MTs18, limitando a utilidade deste marcador a um subconjunto de MTs estabilizados. Em contraste, detyrosination parece ocorrer em todos os MTs estáveis em zebrafish embrião18. Esta modificação pós-traducional expõe o ácido glutâmico carboxy-terminal de α-tubulina (detyrosinated tubulina)18 e pode ser detectada usando anti-Glu-tubulina19. Embora detyrosination ocorre preferencialmente na MTs estáveis, evidência experimental indica que esta modificação pós-traducional é um resultado de, ao invés de uma causa de, estabilidade de MT16. Distingue-se a população MT recíproca, composta por MTs dinâmicos, usando um anticorpo, anti-Tyr-tubulina, que reconhece especificamente a forma tyrosinated de α-tubulina19. Após immunolabeling com estes marcadores e imagem latente confocal, análise quantitativa de MTs (comprimento, número e abundância relativa) pode ser realizada em regiões definidas do tubo neural em desenvolvimento. Aqui, é fornecido um método simplificado para realizar esta análise, utilizando o software de processamento de imagem em 3D. Esse método pode ser aplicado para responder a perguntas sobre morfogênese e o estabelecimento ou a maturação de células polaridade20. Com efeito, a elaboração de matrizes polarizadas de estáveis MTs acompanha muitos eventos do desenvolvimento, incluindo fotorreceptoras morfogênese21, epitelização das células no desenvolvimento do tubo neural18 e axônio formação8.
2 protocolo descreve uma adaptação na vivo de um ensaio de biologia de pilha para analisar MTs durante sua montagem fase (nucleação/fixação e crescimento)22,23. MTs nascentes são nucleated na centrossoma e posteriormente ancoradas apêndices subdistal da mãe centriole23. É descrito um método para analisar a nascente MT rebrota após despolimerização. Este protocolo fornece detalhes sobre o tratamento de nocodazole para depolymerize MTs, o processo de esmaecimento de droga e o período de recuperação pós-tratamento. Re-crescimento de MT é monitorado em intervalos regularesesmaecimento de post s por immunolabeling com marcadores para MTs totais (anti β-tubulina) ao lado de marcadores para a centrossoma (anti γ-tubulina) e núcleo (4', 6-diamidino-2-phenylindole (DAPI)), de acordo com os procedimentos gerais descritos no protocolo 1. A etapa de despolimerização MT do presente protocolo é essencial, como permite a avaliação do crescimento de MT de novo , em vez de extensão de MTs preexistentes. Esta técnica é, portanto, distinta de outros procedimentos publicados para medir as taxas de crescimento de MT (na ausência de despolimerização) usando um marcador de ponta mais como fim proteína 3 fundido a proteína verde fluorescente (EB3-GFP), conforme mostrado no Tran et al., 201211. Além disso, este ensaio é particularmente útil para a análise de embriões com defeito na montagem de novo MT, tais como os mutantes NEDD1 relatados anteriormente, em que o recrutamento de γ-tubulina para a centrossoma é prejudicado, resultando em incompleta formação do tubo neural e defeitos neuronal24.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
declaração de ética: os procedimentos descritos abaixo siga a Universidade das orientações de cuidados com animais de Maryland, Baltimore County.
1. análise de estável e dinâmico MTs usando Immunolabeling (protocolo 1)
- dechorionation Manual de embriões antes da fixação
- obter recentemente gerou embriões derramando água em excesso do sistema e Então, coletar embriões restantes em uma placa de Petri de plástico (consulte a Tabela de materiais).
- Remover todos os restos de embriões de água e transferir o sistema para um novo prato cheio de médio de embrião (consulte a Tabela de materiais) para garantir que os embriões se desenvolvem em um ambiente limpo.
- Permitem que os embriões desenvolver a fase desejada em uma incubadora com temperatura controlada em 28,5 ° C.
- Embriões de lugar mais jovens do que 24h pós fertilização (hpf) em um prato de vidro antes da dechorionation.
Nota: Embriões de Dechorionate antes da fixação para maximizar a rápida penetração do fixador e preservar integridade MT. Médio de embrião de uso em vez de água do sistema para fornecer o adicional Ca 2 + necessário durante a dechorionation. - Remover manualmente as chorions de embriões enquanto em placa de Petri, usando a pinça fina sob um microscópio de dissecação.
- Beliscar uma pequena área no córion redondo, transparente que circunda o embrião com um par de fórceps e suavemente puxe fórceps separados para criar uma ruptura na membrana.
- Ampliar a abertura pelo delicadamente curiosos sobre o rompimento córion usando fórceps. Tenha cuidado para não tocar o embrião com o fórceps, como ele poderia romper.
- Fixação de embriões encenados
- transferência encenado, dechorionated embriões para tubos de centrífuga de 1,5 mL. Remover o máximo médio de embrião quanto possível, usando um pipeta Pasteur de vidro.
Nota: Realize tratamentos de fixação e drogas em embriões jovens (meados-somitogenesis), antes da formação dos centros neurais mediando a sensação de dor, que não exigem nenhum procedimento adicional para aliviar a dor durante a eutanásia. Estádios de desenvolvimento são definidos em Kimmel et al., 1995 25. Fase 4-5 e 11-12 somite foram usado para obter imagens para as figuras 2 e 3. - Preparar a montagem /MT 4% paraformaldeído (PFA) fixador de tampão (MAB) (consulte a Tabela de materiais), combinando 1 mL 8% PFA por 1 mL 2 X MAB e adicionar 2 µ l 100% Triton X-100 por 1 mL de volume total.
Cuidado: Use luvas durante o manuseio de soluções contendo PFA e Triton X-100, que são irritantes da pele. - Correção de embriões em fixador PFA/MAB 1 mL 4% por 5 min em 28,5 ° C. Aspire o fixador com uma pipeta, substituí-lo com fixador fresco 1 mL e incubar durante 3 h à temperatura ambiente (RT) sobre um roqueiro.
Nota: Amostras devem ser fixadas rapidamente à temperatura biológica (28,5 ° C para zebrafish) para evitar despolimerização de temperatura-dependente MT.
- transferência encenado, dechorionated embriões para tubos de centrífuga de 1,5 mL. Remover o máximo médio de embrião quanto possível, usando um pipeta Pasteur de vidro.
- Aspire fixador e adicione 1 mL 1 x Tris salino com NP40 tampão (TBS-NP40). Agite suavemente no RT sobre um roqueiro três vezes por 5 min cada. Armazenar os embriões a 4 ° C em 1 mL fresco 1 X TBS-NP40 para não mais de 7 dias.
Cuidado: Use luvas quando manusear soluções contendo NP-40, um irritante da pele.
Conjunto de agarose - set Sectioning embriões para immunolabeling
- calor RT 4% baixo ponto de fusão (LMP), incorporando médio em um recipiente fechado até que a solução torna-se claro usando um prato quente a 50 ° C, posicionado perto de um microscópio de dissecação . Mantenha o recipiente fechado entre amostras e aquecida durante todo o processo de incorporação (passos 1.4.4-1.4.6).
- Transferência de embriões desde a 1,5 mL centrifugar tubos para uma placa de Petri com uma pipeta de vidro e encha-o com 1 X TBS-NP40.
- Remover células de embriões de estágio de somitogenesis (4-5 e 11-12 somitas) grande gema na prato de Petri usando pinça fina sob a ampliação de uma dissecação do microscópio 26. Segure o embrião por bud a cauda com um par de pinças e descascar as células de gema com o outro par para preservar o tecido do rombencéfalo. Transferir os embriões de gema para uma área de Petri livre de detritos de gema.
Nota: Incorporar os embriões no molde cheio de agarose individualmente para evitar endurecimento prematuro de agarose a LMP. - Preenchimento um 12 mm x 5 x 3 mm bem do molde de corte com 200 µ l derreteu LMP agarose utilizando uma micropipeta. Execute as etapas 1.4.5.-1.4.6. rapidamente (dentro de 20 s de enchimento do molde) para incorporar o embrião antes da agarose LMP esfria a RT e solidifica.
- Use pinça fina para transferir um embrião de gema pelo tailbud da caixa de Petri no molde cheio de agarose em direção a sua extremidade pontiaguda sob um microscópio dissecação.
- Uso de fórceps bem orientar o embrião no molde, tal que a vibratome cortes no plano desejado. Crie seções transversais, orientando o embrião tais que o tecido do rombencéfalo corre paralelo ao comprimento do molde com sua superfície dorsal virada para a borda e sua superfície anterior, encarando o fim da região cônico. Repita as etapas 1.4.4-1.4.6 para os restantes embriões.
- Permitir a incorporação de agarose para solidificar durante 5 min à RT.
- Gerar 40 µm seções do eixo mais alto o agarose incorporado embriões (passos 1.4.1-1.4.7) usando um vibratome com o prato de corte cheio de 1 x TBS-NP40. Transferência de seções de interesse para uma placa de 24 em 500 µ l 1 x TBS-NP40 usando pinça fina. Colocar as seções de apenas um embrião por alvéolo.
Nota: Consulte para fazer referência a 18 para mais detalhes. Certifique-se que seções permanecem hidratadas em todos os momentos pelo menos 250 µ l de tampão e rock com velocidade baixa (rpm 10-25) para as etapas restantes evitar a separação da incorporação de agarose. Detergentes presentes no bloqueio e soluções de lavagem devem reduzir a tensão superficial do meio líquido e permitir que a submersão das seções. Verifique as seções permanecem nos poços durante e depois de todas as manipulações. Tenha cuidado para evitar acidentalmente descartando seções durante lavagens.
- Remover o tampão e adicione 500 µ l de solução de bloqueio. Rocha pelo menos 1 h no RT.
Nota: Use uma solução de bloqueio que contém 5% soros das espécies de hospedeiros de cada anticorpo secundário a ser usado (consulte a Tabela de materiais). - Incubar em 300 µ l os anticorpos primários diluídos em tampão de bloqueio para 36-72 h a 4 ° C em uma cadeira de balanço. Lave duas vezes em 600 µ l 1 x TBS-NP40 sobre um roqueiro para 30 min cada, em RT.
Nota: Seções de duplo-rótulo incubando em anticorpos primários contra MTs totais (anti-β-tubulina, ou anti-α-tubulina) e MTs estáveis (anti-Glu-tubulina) ou dinâmicos MTs (tubulina anti-Tyr). Selecione os anticorpos primários que foram levantados em diferentes hospedeiros quando dupla rotulagem para total e post-translationally modificado populações de α-tubulina. Consulte Tabela de materiais para as diluições anticorpo. - Incubar em 300 µ l de anticorpos secundarios conjugados fluoróforo diluído em tampão de bloqueio em um roqueiro para 16-24 h, a 4 ° C, no escuro. Lave duas vezes em 600 µ l 1 x TBS-NP40 sobre um roqueiro para 30 min cada, em RT.
Nota: Passe o prato multi bem contendo anticorpo secundário em folha a partir deste momento e após cada manipulação para evitar a têmpera. Selecione os anticorpos secundários que reagem com a imunoglobulina de anfitrião do anticorpo primário. Escolha o anticorpo secundário fluorophores que têm separados e disjunto de espectros de emissão. Consulte Tabela de materiais para as diluições anticorpo. - Incubar os embriões em 500 µ l de solução DAPI sobre um roqueiro para 30 min, em RT. Wash três vezes em TBS-NP40 balançando no RT por 5 min cada.
Nota: Rotulagem Nuclear fornece contexto celular para a quantificação de MT, realizada na etapa 1.12. - Colocar uma gota de meio de montagem com agente antidesvanece-se no centro de um slide livre de poeira. Use pinça fina a transferência de seções para a gota média de montagem. Coloque uma lamela livre de poeira em cima da amostra. Armazenar slides em um lugar seco, escuro e fresco, envolvido em papel alumínio, até que a imagem é executada.
Nota: Circular as seções na parte de trás do slide usando um marcador permanente ponta fina antes da imagem vai ajudar a identificar seções quando usando o microscópio. - Da imagem latente confocal
- seções de montagem em um laser invertido microscópio confocal apondo o slide para o palco com a lamela enfrentando o objetivo. Determinar a ótica apropriada (objetivo, laser e as configurações de canal como ganho e offset) em um slide de controle e mantê-los consistentes entre amostras 27. Evitar oversaturating os pixels para evitar perda de dados.
- Captura imagens confocal de Z-pilhas usando configurações de canal para o anticorpo secundário selecionados fluorophores e salvar os arquivos de imagem 27. Adquirir pilhas de Z para cada seção.
Nota: Replicar os parâmetros usados para adquirir as imagens nas figuras 2 e 3 usando as seguintes configurações de aquisição: modo = XYZ; ampliação objetiva = 63 X lente de imersão de óleo; Abertura numérica objetiva = 1,4; Z-passo = 0.1 µm; Z-profundidade = 16.23 µm. Use as seguintes configurações de canal: excitação de DAPI com laser UV-intervalo de 20%, intervalo de filtro de emissão = 430-480 nm, fotomultiplicador (PMT) ganho = 525 V e deslocamento de PGTO =-1.72%; 448 nm fluoróforo (consulte a Tabela de materiais) excitação com 20% 488 nm do laser, intervalo de filtro de emissão = 493-573 nm, ganho de PGTO = 689 V e deslocamento de PGTO =-0.2%; excitação de fluoróforo 594 nm com 32% 594 nm do laser, intervalo de filtro de emissão = 608-706 nm, ganho de PGTO = 768 V e deslocamento de PGTO =-6.8%. - Salvar arquivos de dados brutos com nomes de arquivo originais, descritivo e criar uma cópia para edição no software de análise de imagem.
- Compilação de Z-pilhas para exibir projeções máximos
- abra a cópia do arquivo de dados usando o software de análise de imagem 3-d domínio público (por exemplo, ImageJ). Verifique se cada canal é exibido como uma sequência de imagens individuais (Z-stacks).
- Dividir canais de imagem usando a seguinte sequência de menu: “ canais de imagens/cor/Split ”.
- Criar uma imagem mesclada sobrescreve os canais de interesse usando a seguinte sequência de menu: " imagens/cor/mesclar canais. " selecione a 594 nm, 488 nm e canais DAPI ser falsa cor vermelha, verde e azul, respectivamente. Verificar " composto de criar " e selecione " Okey " 28.
Nota: Omitir o canal DAPI para melhor transmitir detalhes específicos para MTs em uma projeção máxima como na Figura 2 e 3, por apenas selecionando cores falsas para os outros dois canais. - Examinar a pilha-Z mesclada e anote o início e término de posições dos melhores Z-suprafísicos para todos os canais visíveis. Descarte os exteriores Z-aviões que normalmente têm sinal de qualidade inferior devido as superfícies irregulares da seção. Consulte a referência de 29 para obter detalhes.
- Visualizar mesclada Z-pilha como uma única imagem 2-D, realizando uma projeção de intensidade máxima da pilha-Z usando a seguinte sequência de menu de análise de imagem 3-d: " imagens/pilha/Z-projeto. " digite o inicial e final posições dos melhores internas Z-aviões da etapa 1.11.3 como o " fatia de início " e " fatia de paragem, " respectivamente. Selecione " intensidade de Max " como o tipo de projeção e clique " Okey ". Consulte a referência de 28 para mais detalhes.
- MT analisando rotulagem
- abrir o software de análise de imagem 3D comercial. Selecione " criar biblioteca " e fornecer um nome descritivo para a biblioteca de imagens. Clique " Create. " arrastar arquivos de imagem raw gerados a partir do microscópio confocal para a biblioteca. Arquivos maiores exigem mais tempo para transferir.
- Selecione um arquivo para analisar. Escolha " foco estendido " do " vista " menu para exibir a imagem do canal-fundiu-se na janela principal.
- Ajustar o limiar, arrastando a ferramenta de controle deslizante para cada canal para a esquerda ou direita até que o sinal de fundo é reduzido e o sinal verdadeiro é robusto. Observar que cada canal mostra um sinal verdadeiro para a molécula chamada (por exemplo, canal DAPI mostrando núcleos oblongos ou mitóticos mas não autofluorescência do citoplasma ou agarose).
- Selecione o " Freehand região de interesse (ROI) " ferramenta e contorne a região de interesse a serem analisados. Selecione o " ações " guia, seguido de " culturas a seleção " para recortar a imagem. Salve o arquivo de imagem recortada sob um novo nome. Clique o " medições " guia para criar o protocolo para filtragem de objetos específicos relevantes para a análise de 3-d.
- Arrastar " encontrar objetos " à janela de protocolo. Renomeie o primeiro protocolo " DAPI. " selecionar o canal DAPI no menu suspenso. Arraste as seguintes configurações para o protocolo DAPI e colocá-los abaixo " encontrar objetos " na seguinte ordem (tabela 1): " preencher buracos em ObjectsŔ " Separado de tocar ObjectsŔ " Excluir objetos por SizeŔ " Excluir não tocar ROIs ".
Nota: O objetivo das configurações na tabela 1 são primeiro definir um limiar que descarta sinais cuja distribuição e tamanho são inconsistentes com o tamanho de objetos sendo analisado. Por exemplo, eliminar um sinal não é grande o suficiente para ser um núcleo quando a contagem de núcleos. - Executar a sequência (etapa 1.12.9) para montar o restante filtros para β-tubulina e outros marcadores usando as configurações na tabela 1.
- Select " medida " na parte inferior de cada protocolo. Escolha " medição de Volume e intensidade " e " comprimento esquelético " para todos os rótulos de tubulina, mas apenas do antigo para o sinal DAPI.
- Desenhar um ROI em torno da região a ser medido. Observar as medições sob a " Resumo " tab após o software processa a região. Copiar os dados e salvá-los em uma planilha praticável, como ilustrado na tabela 2. Criar uma cópia de backup da planilha para análise posterior.
- Selecionar medidas de interesse (por exemplo, comprimento de pacote de MT, número de pacotes de MT/núcleo, como revelado com diferentes marcadores) na planilha e analisar para determinar as médias para cada grupo.
Nota: A MT bundle duração média = soma da ' quer dizer comprimento esquelético para β-tubulina ' para cada embrião dividido pelo número total de embriões. Consulte a linha 20 da tabela 2. Formatar a planilha para que as variáveis e os grupos experimentais são facilmente graficamente.
2. De Novo Ensaio de Assembly de MT (protocolo 2)
- construção e teste o multi bem escoamento aparelho ( Figura 1) 2 dias antes da experiência.
Nota: O aparelho permite lavagem simultânea de múltiplos grupos experimentais nocodazole tratamento usando suprimentos de Materiais da tabela a seguir. Aferidor do silicone requer pelo menos 24 horas de tempo de secagem antes de não coloca nenhum risco de toxicidade para os embriões.- Dividir um tubo de centrífuga de 50 mL ao meio longitudinalmente, usando um gabarito ou serra.
- Semi-círculos cut 7, com um raio de 3 cm, por 70 µm de nylon de malha e redimensioná-las para caber firmemente em uma metade da split centrifugar o tubo. Cola os semi-círculos no tubo de centrífuga paralelo para as marcações de gradação de 10ml usando selante de silicone de aquário-seguro. Deixe-o secar por 2 dias e enxágue por imersão em um copo de água para 2-3 h.
- Linha extremidade superior (roscada) do tubo cortado com argila de modelagem, tal que a altura do líquido retido no dispositivo de passagem tem uma profundidade de ¼ de polegada ( Figura 1, vistas, 2 e 3).
- Preparar o aparato de lavagem, remover o êmbolo de uma seringa de 50 mL e inserindo 12 polegadas de tubo fino na ponta. Empurre o tubo que vai ir e vedação ao redor da articulação utilizando argila de modelagem.
- Pre-molhado da malha usando meio de embrião para permitir que o líquido executar através do dispositivo de passagem inteiro. Ângulo do dispositivo no gelo então aquele líquido piscinas em todos os compartimentos, mas ainda esvazia a frente onde se situa a borda de argila. Usando uma base de anel, suspender o aparato de lavagem acima o dispositivo de passagem no gelo ( Figura 1).
- Chill 200 mL do meio de embrião no gelo e despeje suficiente no aparelho de lavagem para garantir que todas as bolhas de ar estão limpos e que a taxa de fluxo é aproximadamente 7 mL/min. Ajuste a taxa de fluxo, alterando a altura da seringa.
- Enzimaticamente embriões de dechorionate
- fazer uma solução de trabalho de protease específico diluindo-se 1 mL de 10 mg/mL, estoque de protease específico em 20 mL de meio de embrião.
- Dechorionation química de executar em embriões 1 h antes do commit quando eles são esperados para chegar à fase do desenvolvimento desejada. Chorions Digest removendo médio de embrião de 100 mm placas de Petri contendo encenaram embriões e adicionar 20 mL de solução de trabalho específico protease.
- Embriões de incubar a 37 ° C por 5 min.
Nota: Não exceder 5 min ou usar uma maior concentração da solução de protease específico, pois isso resultará em embriões caindo distante uma vez tratados com nocodazole. - Rapidamente Pipetar fora específico protease e reencher pratos com aproximadamente 25 mL de meio de embrião. Repetir uma vez.
- Usando uma pipeta de vidro de 1 mL, transferência de embriões mais jovem do que 24 hpf para vidro pratos para protegê-los contra danos.
- Completa dechorionation removendo manualmente chorions usando um par de pinças bem, conforme descrito na etapa 1.1.5.
- Coloque pratos de Petri de vidro contendo embriões de dechorionated em uma incubadora de 28,5 ° C por um período mínimo de 30 min até atingirem o estágio do desenvolvimento desejado.
- Depolymerize os MTs existentes
- preparar uma solução de trabalho de 5 µ g/mL nocodazole, combinando-se 50 µ l de 1 mg/mL nocodazole estoque com 10 mL de meio de embrião frio gelo.
Atenção: Utilize luvas quando manusear o nocodazole, um irritante da pele. - Trocar o meio de embrião do grupo de tratamento nocodazole com 10 mL de solução de trabalho frio nocodazole. Coloque pratos de Petri no gelo para a hora certa para a fase de desenvolvimento (por exemplo, 1 h para embriões somite de 4-5). Retiradas embriões controle não tratados em uma placa de Petri no gelo a fixar-se ao lado de amostras de esmaecimento na etapa 2.3.4.1.
- Transferência de embriões utilizando uma pipeta de vidro polido de 1 mL de fogo para o aparelho de passagem, usando compartimentos separados para cada grupo experimental. Começar a dispensa de nocodazole por meio de embrião frio gelo derramando na parte superior da seringa de 50 mL.
Nota: Use pelo menos 30 embriões por grupo experimental. Grupos experimentais poderiam consistir de embriões de controle ou uma variedade de Morpholinos ou embriões de RNA-injetado. O esmaecimento exigirá um total de cerca de 150 mL de meio de embrião para ser adicionado a cada 8-10 min. Washout o nocodazole continuando a inibir o crescimento de MT com gelo. Manter os embriões no gelo é essencial para o sucesso deste teste porque MTs são instáveis em baixas temperaturas e fria retarda o desenvolvimento de embriões adiantados. - Permitir MTs de regenerar após 20 min de esmaecimento no RT através da transferência de embriões para pratos de Petri de vidro contendo quente médio de embrião (28,5 ° C) utilizando uma pipeta de vidro polido 1ml fogo. Assim que os embriões são transferidos, inicie um timer.
- Fixar os embriões controle e desbotar a 1 min, 5 min e 10 min pipetando aproximadamente 10 embriões em um 1,5 mL centrifugar o tubo enchido com correção PFA/MAB 1 mL 4% (28,5 ° C) e seguindo as instruções no passo 1.2.3.
- preparar uma solução de trabalho de 5 µ g/mL nocodazole, combinando-se 50 µ l de 1 mg/mL nocodazole estoque com 10 mL de meio de embrião frio gelo.
- Preparar amostras para immunolabeling conforme descrito nas secções 1.3-1.5.
- Immunolabel seções flutuantes e embriões de imagem como descreveram nas seções 1.6-1.10 com as seguintes alterações às especificações do anticorpo primário: uso 1: 500 coelho anti-γ-tubulina e 1: 200 mouse anti-β-tubulina.
- Processo e analisar imagens usando software de análise de imagem 3-d, conforme descrito na etapa 1.12.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Análise de MTs estáveis e dinâmicos usando immunolabeling
No protocolo n º 1, a distribuição das subpopulações de MT durante o início (quilha neural) e os estágios finais (haste neural) do desenvolvimento do tubo neural são revelados, usando Glu-tubulina e Tyr-tubulina como marcadores para MTs estáveis e dinâmicos, respectivamente. MTs dinâmicas predominam o rombencéfalo na fase neural da quilha (4-5 somitas) (Figura 2A-D). Como a quilha desenvolve-se a haste neural (11-12 somitas), uma fase de epitelização reforçada, qualitativamente menos MTs são imunorreativas com o anticorpo anti-Tyr-tubulina (Figura 2E-H), especialmente na haste ventral. Em contraste, Glu-tubulina é dispersada e punctate ao longo da quilha neural (Figura 3A-D), mas é enriquecida da haste neural ventral ao longo de intervalos de MT (Figura 3E-H). Pontas de seta apontam para pacotes específicos de MT ou estruturas onde a rotulagem é aumentada.
Embora tanto anti-Glu-tubulina e Tyr-tubulina anticorpos foram produzidos na mesma espécie hospedeiro (impedindo uma experiência de rotulagem dupla), estes resultados indicam que os marcadores de MT estáveis e dinâmicos raramente se sobreponha o rombencéfalo zebrafish. Em primeiro lugar, a haste neural ventral tem mais estável (Figura 3-F) que dinâmico MTs (Figura 2-F). É inverter a tendência da haste neural dorsal, consistente com um modelo de zebrafish neurulação em que o tecido dorsal permanece dinâmico até que o tubo neural é formado de20. Em segundo lugar, enquanto fusos mitóticos totalmente são rotulados com o anticorpo de Tyr-tubulina na quilha neural (Figura 2D, pontas de seta), apenas a base do eixo, coincidente com a centrossoma, são rotulados com o marcador de estabilidade (Glu-tubulina Figura 3 D, pontas de seta). Imunofluorescência de β-tubulina, comum a ambos os ensaios, informa o experimentador da distribuição de todos os MTs e fornece uma base para julgar não-específicas de rotulagem.
Usando software de análise de imagem em 3D de objetos de medição resulta em uma grande quantidade de dados que podem ser organizados em uma tabela conveniente (tabela 2). Para tornar o comprimento, contagem e medições de área, estamos usando apenas um subconjunto de dados que estão disponíveis para analisar. Um dos componentes dos dados que nós não mais analisar é o número de objetos identificados. Este número é usado como um controle de qualidade interno, como o número não deve variar amplamente entre como seções e a proporção de núcleos para MTs devem ficar semelhante em uma condição única de tratamento. Um outlier é um indicador de que também a análise precisa ser executado novamente com filtros ajustados ou que a imagem é muito mal rotulada para analisar. Assim, todas as imagens de outlier devem ser reanalyzed com configurações ajustadas. A seção de outlier deve ser examinada para sinais de rotulagem pobre ou danos físicos que possam resultar em contagens de objetos incomuns. Uma vez que a análise for concluída e qualidade controlada, informações úteis podem ser recuperadas a partir dos dados brutos tais como comprimento de MTs totais médio e estável MTs ou a relação de estáveis MTs MTS total (tabela 3). Além destas medidas, muitas outras métricas podem ser obtidas usando o software de análise de imagem 3D que pode ser usado para desenhar inferências sobre MTs ou sua relação com outras estruturas celulares (núcleo, centrossoma, etc.).
De novo Ensaio de montagem MT
O tratamento de nocodazole depolymerizes MTs resultando em rotulagem difusa (Figura 4A, 4 D e 4 G). Como os MTs crescerem, estendem-se desde a centrossoma (Figura 4B, 4E e 4 H), no entanto, isto pode não ser óbvio em um único avião devido a suas trajetórias não-plana (Figura 4C, 4F e 4I). No entanto, alguns softwares de análise de imagem são capazes de medir comprimentos em 3-d, permitindo uma avaliação do crescimento de MT após a dispensa de nocodazole (tabela 4). Uma observação importante que pode ser obtida do conjunto de dados na tabela 4 é que o comprimento de MTs parece aumentar ao longo do tempo após a dispensa de nocodazole em todas as regiões do tubo neural analisados. Como mencionado acima, outros tipos de métricas obtidas do software de análise de imagem em 3D podem fornecer contexto celular para interpretar os dados de MT (por exemplo, relação de MTs por núcleo).
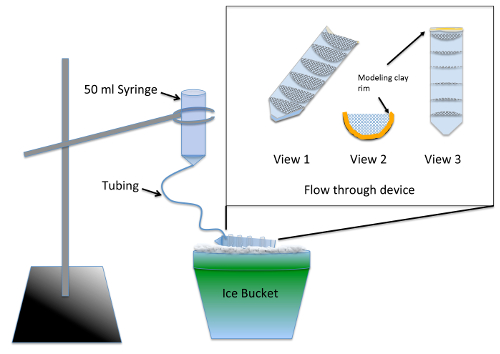
Figura 1 : Ilustração do aparelho de lavagem para de novo Ensaio de montagem MT. O baixo-relevo é um close-up do escoamento dispositivo feito de malha colada em um tubo de centrífuga de 50 mL, corte longitudinal. A malha compartimentaliza o dispositivo de escoamento tal que vários grupos experimentais podem ser processados simultaneamente. Durante o uso, médio do embrião é adicionado para a seringa e lentamente flui através do tubo para encher o dispositivo de passagem, proporcionando uma lavagem constante para todos os grupos experimentais. Clique aqui para ver uma versão maior desta figura.

Figura 2: uso de immunolabeling a imagem dinâmica mts. Dechorionated embriões foram fixados em fases apropriadas (4-5 em A-D - e 12-13 somitas em E-H), seccionados transversalmente através do rombencéfalo e immunolabeled com anticorpos contra a β-tubulina (verde na e E) para marcar todos os MTs e tyrosinated α-tubulina (vermelho em B e F) para revelar a dinâmica das populações de MT. MTs altamente dinâmicos podem ser vistos em imagens mescladas (C, G) e suas ampliações (D, H) como áreas onde a etiqueta amarela é visíveis (pontas de seta em D, H). Escala de barras = 25 µm (A-C e E-G) e 10 µm (D e H). Clique aqui para ver uma versão maior desta figura.

FIkuren 3: uso de immunolabeling a imagem estável mts. Dechorionated embriões foram fixados, seccionados através do rombencéfalo e immunolabeled em fases apropriadas (4-5 somitas em A -somitas D e 12-13 no E-H). MTs estáveis são rotulados com anticorpos contra a detyrosinated forma de α-tubulina (Glu-tubulina) (vermelhos em B e F) enquanto MTs totais foram visualizadas com um anticorpo geral β-tubulina (verde na e E). Sinais vermelhos e amarelos em imagens mescladas (C, G) e suas ampliações (D, H) representam áreas de alta estabilidade de MT (pontas de seta em D, H). Escala de barras = 25 µm (A-C e E-G) e 10 µm (D e H). Clique aqui para ver uma versão maior desta figura.

Figura 4: uso de immunolabeling a imagem nascente mts. Dechorionated embriões foram fixados em 4-5 somitas e seccionados transversalmente através do rombencéfalo. Seções foram immunolabeled com β-tubulina (D, E e F) para marcar crescendo MTs e γ-tubulina (A, Be C) para marcar a ponto de nucleação/centrossoma. Uma região dorsal do tubo neural é encaixotada (A, D; B, E e C-F) e mostrado na maior ampliação (G, H, eu, respectivamente) para revelar o núcleo (DAPI, azul), centríolos (γ-tubulina, vermelho) e total MTs (β-tubulina, verde). Branco de pontas de seta: colocalization de MTs e os centríolos; amarelo com pontas de flechas: o centríolo segundo de uma célula é visível. Escala de barras = 25 µm (A-F) e 10 µm (G-eu). Clique aqui para ver uma versão maior desta figura.

Tabela 1: Configurações padrão para filtragem de objetos em software de análise de imagem em 3D.

Quadro 2: conjunto representativo de dados bruto, obtido usando análise de imagem em 3D, software para analisar estável MTs. Cada coluna representa as medidas de uma única seção. Min: medição menor; Max: medição de maior; SD: desvio-padrão; SE: erro-padrão.

Tabela 3: exemplos de conjuntos de dados que podem ser obtidos em 3-d software de análise para quantificar MTs estáveis de imagem. Selecione as medições do comprimento médio do total (β-tubulina) e estável (Glu-tubulina) MTs calculados tomando-se a média do comprimento médio esquelético para o rótulo relevante de todas as amostras (vide tabela 2) e o rácio de estável para total (MTs Glu-tubulin deixa a marca por listras de β-tubulina) calculado tomando-se a contagem média de β-tubulina dividida pela contagem média de Glu-tubulina.

Tabela 4: Exemplos de conjuntos de dados que podem ser obtidos do software de análise de imagem em 3D para analisar de novo Assembleia de MT. Resultados representativos do conjunto de novo MT experimentar, comparar conjuntos de dados obtidos por três pontos de tempo de recuperação (1, 5 e 10 min) após lavagem nocodazole. Para cada ponto de tempo, obtidas por contagem nuclear, centríolos (γ-tubulina partitura), número de MTs totais (estrias de β-tubulina), estão indicadas as medidas para selecionado regiões da imagem analisaram (seção transversal do tubo neural em desenvolvimento).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Atualmente existem muitos métodos de imagem dinâmica de MT no início do desenvolvimento do zebrafish, variando de imagens ao vivo de moléculas marcou a immunolabeling de fixa tecido11,12,13,14. Embora MTs em uma única célula podem existir nos Estados dinâmicos ou estáveis, epitelização é um processo no qual o MTs são estabilizadas progressivamente ao longo do tempo. Usando marcadores para MTs estáveis e dinâmicos oferece uma maneira de visualizar este fenômeno. O método apresentado aqui aproveita o poder do software de imagens 3-d para quantificar a transição da dinâmica de populações de MT estáveis em uma seção transversal do tecido embrionário zebrafish. No protocolo 2, o método é usado para rotular uma população distinta de nascentes MTs e siga as horas extras nucleação e crescimento.
MTs são notoriamente difíceis de imagem em seu estado nativo devido à sua propensão para depolymerize. Assim, o componente chave deste método é rápida fixação de MTs em todo o embrião inteiro. Isto é conseguido por iniciar a fixação em temperaturas fisiológicas e usando um buffer que estabiliza os MTs e aumenta a permeabilidade do embrião. O tempo de fixação é também importante como fixação cerceada falha para prender MTs enquanto fixação excessiva pode mascarar os resumos, interferindo com a ligação do anticorpo. O tempo de fixação sugerida de 3-4 h trabalha com embriões que estão no meio da gastrulação à pós-fertilização 24 h. Embriões no final da escala de tempo mais jovens devem ser fixados para mais perto de 3 h enquanto embriões mais antigos podem precisar de toda a 4h. Mesmo com fixação adequada, as MTs serão depolymerize com tempo para que seccionamento e immunolabeling devem ocorrer dentro de uma semana de fixação.
Uma vez que o tecido é fixado corretamente, podem surgir problemas com immunolabeling. O problema mais comum encontrado foi pobre penetração através do centro do tecido, particularmente, se muitas seções são incubadas no mesmo poço. Aumentando a concentração do tempo incubação e anticorpo primário para anticorpos primários e secundários, combinada com o aumento dos detergentes para melhorar a permeabilização dos embriões vai amenizar a maioria dos problemas immunolabeling. Se o anticorpo rotulagem falhar devido a problemas de fixação ou immunolabeling problemas, é possível determinar a causa, examinando o anticorpo rotulagem padrão. Fixação pobre resultará na rotulagem intensa na membrana e difusa de rotulagem no citoplasma, enquanto a fixação excessiva resultará em fraco rotulagem que retém arquitetura de MT. Pobre penetração do anticorpo, no entanto, irá aparecer como áreas no centro do tecido sem rotulagem.
A capacidade de analisar imagens de MT em uma maneira significativa é dependente de imagem de alta qualidade. Para capturar o comprimento de MT em 3-d, deve ser usado o tamanho mínimo de Z-passo possível para o objectivo e a abertura numérica. Imagens mostradas aqui foram capturadas com um 63 X objetivo de emersão de óleo com abertura numérica 1.4 produzindo o seguinte: pixel = 240 nm, Z-passo = 0.1 µm, Z-pilha tamanho = 16.252 µm. Porque a largura de um único MT é 25 nm, aproximadamente 10 vezes abaixo do limite de resolução de um microscópio de luz, esta métrica não pode ser precisamente medido usando esta técnica. Em vez disso, apenas comprimentos MT iguais ou maiores que o tamanho do pixel mínimo atingível em todas as três dimensões podem ser medidos. Linha e/ou quadro com média pode melhorar a definição de sinal MT. Análise de MT deve ser reservado para as seções de alta qualidade. Enquanto o tecido com fixação pobre não pode ser fotografado e analisado, suave overfixation pode ser contrabalançada, cuidadosamente, aumentando a intensidade do laser e ganhar para detectar o sinal fraco, mantendo uma boa faixa dinâmica. Penetração de anticorpo pobre, enquanto não ideal, pode ser corrigida por limitar a aquisição de imagens de regiões bem marcadas, resultando em uma seção mais fina (5-10 µm) de imagem. Fundo elevado de rotulagem pode ser compensado ajustando as configurações de filtro. No entanto, se qualquer um destes ajustamentos são feitas, é necessário verificar que o limiar de filtros aceitavelmente em cada avião da Z-pilha.
Software de análise de imagem 3-d permite que o experimentador quantificar MT comprimento, área, ângulo, abundância e outras métricas no espaço 3-d de secções de tecido fixo. O método descrito aqui fornece instruções para a obtenção desses dados usando o software disponível comercialmente. No entanto, os módulos de filtragem podem ser adaptados ao domínio público software melhorado com plugins relevantes e/ou macros, fazendo análise disponível a todos. Antes da análise, imagens raw devem ser thresholded para evitar incluindo plano de fundo e sinais não específicos na quantificação. Uma vez que a análise é concluída e dados são transferidos para uma planilha praticável, muitas inferências podem ser feitas no conjunto de dados. Um dos cálculos feitos aqui foi estrias Glu-tubulina por β-tubulina listras, ou a relação estáveis MTs MTS total onde 1 representa que o citoesqueleto MT inteiro foi estabilizado em um ROI. Se o experimentador deseja complementar seus dados quantitativos, gerando um formato de arquivo de imagem tagged polido (TIFF) imagem com barras de escala é fácil com o software de análise de imagem em 3D.
Este ensaio permite a análise funcional das proteínas implicado na montagem da MT, vivo em. Se immunolabeling é realizada em alternando seções seriais, este protocolo pode ser usado para estudar MTs dinâmicos e estáveis no mesmo embrião. No futuro, modificações como aumento de detergentes ou alterados ângulos incorporação permitirá a utilização desses métodos para embriões mais velhos e uma ampla gama de questões anatômicas.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores não têm nada para divulgar.
Acknowledgments
O microscópio confocal foi comprado com fundos de o US National Science Foundation (NSF), grant #DBI-0722569. A pesquisa foi apoiada por os E.U. institutos de saúde nacional/Instituto Nacional de General Medical Ciências (NIH/NIGMS) concessão #GM085290 e E.U. departamento de defesa (DOD) grant #W81XWH-16-1-0466 atribuído a R.M. Brewster. E. Vital foi apoiada por uma concessão a UMBC do Howard Hughes Medical Institute, através da pre-faculdade e programa de educação científica de graduação, conceder #52008090. S.P. Brown foi apoiado por um departamento E.U. de educação GAANN Fellowship, uma bolsa de pós-graduação Meyerhoff financiado pela concessão de NIH/NIGMS, #GM055036 e um trabalho de assistente de investigação financiado pelos E.U. DOD grant #W81XWH-16-1-0466.
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
