

Summary
I denne artikkelen beskriver vi elektrokjemiske, elektron spinn resonans og UV-synlig og nær infrarød spectroelectrochemical metoder å analysere organiske forbindelser for programmet i organisk elektronikk.
Abstract
Syklisk voltammetry (CV) er en teknikk som brukes i analyse av organiske forbindelser. Denne teknikken er kombinert med elektron spinn resonans (EPR) eller ultrafiolett synlige og nær infrarød (UV-Vis-NIR) spectroscopies, få vi nyttig informasjon om elektron affinitet, ionisering potensial, bandet-gap energier, type belaste bærere og fornedrelse informasjon som kan brukes til å syntetisere stabil organisk elektroniske enheter. I denne studien presenterer vi elektrokjemiske og spectroelectrochemical metoder for å analysere prosessene som oppstår i aktive lag av en organisk enhet som genererte kostnader bærere.
Introduction
Over hele verden, søker forskere stadig etter nye organisk materiale som kan brukes i organisk elektronikk med ønskelig ytelse eller stabilitet, som synker utvidet bruk. Ved økologisk enheter er det viktig å forstå virkemåten til Ladningsbærere å fullt ut kjenne reglene kjøring enheten virkemåten. Analyse av effekten av den molekylære strukturen på generering av kostnader bærer og dynamikk og vedlikehold av saldoen injisert kostnader bærere, både positive (hull) og negative (elektroner), er avgjørende for å forbedre ytelse og stabilitet organisk enhetene. Dette sikrer effektiv rekombinasjon disse individuelle kostnader og dermed forbedrer betraktelig photoluminescence effektiviteten av den økologiske lys emitting diodes (OLED)1,2. For organiske solcellepanel (OPVs)3,4 samt organisk feltet effekt transistorer (OFETs)5,6er det nødvendig å ha materiale med høy kostnad carrier mobilitet. I tillegg til analyse av kostnader bærere, flere viktige parametere av organisk electroactive materialer å forutse hvor materialet kan brukes: ionisering potensial (IP), elektron affinitet (EA) energinivå og bandet-gap mellom dem7 ,8,9,10.
I dette arbeidet presenterer vi en metode for effektiv måling av syklisk voltammetry (CV) som kan brukes i analyser av alle typer electroactive materialer. Denne teknikken gir informasjon om redoks egenskaper, doping/dedoping mekanismen, stabilitet, konvertering og lagring av energi, etc. Det tillater også for estimering elektron affinitet og ioniseringen energi av testen forbindelser i en mye billigere og raskere måte sammenlignet med andre høyt vakuum metoder. Nevnte parameterne samsvarer med energinivået høyeste okkuperte molekylær orbital (HOMO) og laveste ledig molekylær orbital (LUMO).
Metoden som presenteres i denne artikkelen kan brukes til å analysere alle typer konjugert forbindelser som dem med delocalized π-elektroner i deres strukturer. Konjugert forbindelser kan være små molekyler med stor polymere kjeder. Små molekyler kan også være monomerer; under den første reaksjonen kan (fotokjemisk, elektrokjemiske eller kjemiske) monomerer danne polymerer. OLED program er energi nivå verdiene nødvendig å aktivere bruk av riktig vert for emitter i en termisk aktivert forsinket fluorescens (TADF) parkvert systemet eller bestemme som forbindelser exciplex donor-acceptor laget kunne dannet og hva ytterligere lag (elektronet transport layer (ETL), hull transport layer (HTL), elektron blokkerer lag (EBL) og hull blokkerer lag (HBL)) vil være nødvendig å syntetisere stabil effektivt belastet balansert OLED enheter11 , 12 , 13 , 14 , 15 , 16 , 17. ytterligere elektrokjemiske målinger at etterforskningen av mulige siden reaksjoner under av nedbrytning av det aktive laget og dannelsen av lav mobil lade bærere (bipolarons)18,19 ,20,21,22.
Koplingen elektrokjemiske og spectroelectrochemical metoder gir enkel, nøyaktig og pålitelig fastsettelse av graden av oksidasjon eller reduksjon av konjugert forbindelser og deres fornedrelse potensial, som er avgjørende for stabilitet23 , 24 , 25 , 26 , 27 , 28. ultrafiolett synlige og nær infrarød (UV-Vis-NIR) spektroskopi kombinert med elektrokjemi kan beskrive grunnleggende kromatisk egenskapene for alle nye konjugert forbindelser, for eksempel endring av absorpsjon bandet under doping 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30.
I en studie knyttet til doping mekanismen, er det viktig å definere kostnader bærere. I denne prosessen, to klasser av ladet quasiparticles delta, en med uncompensated spinn (polarons) og det andre er diamagnetic (bipolarons); elektron spinn resonans (EPR) spektroskopi gir uvurderlig hjelp, som direkte kan observere og spore endringer i populasjoner av spinn polarons29,30,31,32 . I små molekyler, er det vanskelig å form bipolarons, men disse molekylene kan være ganske likt og har bipolaron-inducing egenskaper; Det er viktig å sjekke om og som polarons og bipolarons er dannet i strukturen. Bipolarons er minst én ordre lavere i mobilitet enn polarons; Derfor, hvis bipolarons er dannet i virksomme innretninger, så det kan føre til en ubalansert forholdet mellom kostnader bærere, som vil resultere i svært høy strøm og overoppheting av OLED enheten eller godt være av nedbrytning33.
Målemetode foreslått i denne studien er billig og raskere og gir mulighet for fastsettelse av mest verdifulle operative parametere for et stort antall electroactive materialer uten behov for spesielle enheter som er basert på nylig syntetisert materialer å sjekke ytelsen. Ved å bruke elektrokjemi og spectroelectrochemistry, er det mulig å velge en materiale som er veldig lovende fra hundrevis av nye materialer. I tillegg er det mulig å få detaljert informasjon om prosessene for doping og deres effekter på den kjemiske strukturen til testen konjugert systemer som bruker elektrokjemiske og spectroelectrochemical metoder, som kan lage mer effektiv organisk elektronikk anordninger.
Protocol
1. forberedelse av eksperimentet
- Forberede 25 mL 0.1 M electrolyte løsning.
Merk: Avhengig av forbindelser under etterforskning, bruke forskjellige elektrolytter som blanding av organisk salt og løsemiddel: den mest felles salter brukes i analyse av organiske molekyler er tetrabutylammonium hexafluorophosphate (Bu4NPF6 ) eller tetrabutylammonium tetrafluoroborate (Bu4NBF4); vanligste løsemiddelet brukes i analysen er diklormetan, acetonitrile eller tetrahydrofuran.- Velg elektrolytt løsemiddelet basert på Løseligheten av testen forbindelser. Det bør være en løsning under etterforskning, men være i en robust begrunne når materialet er brukt til å lage av enheten, som en tynn film avsatt på arbeidsgruppen elektrode overflaten.
- Rengjør elektrodene i eksperimentet29.
- For elektrokjemiske målinger, kan du bruke en 1 mm diameter platina disk elektrode som arbeider elektroden (vi), platina coil eller wire som ekstra elektroden (AE) og en sølv/sølv klorid (Ag/AgCl) elektrode som referanse elektroden (RE).
- For EPR spectroelectrochemical målinger, bruker du platina wire som vi, platina coil eller wire som AE og Ag/AgCl som RE.
- For UV-Vis-NIR spectroelectrochemical mål, kan du bruke en kvarts indium tinn oksid (ITO) eller fluor-dopet tinn oksid (FTO) som vi, en platina wire som AE og en Ag/AgCl elektrode som RE.
- Polsk platina disk elektroden ved å gni ulempen med elektroden (platina elektrode arbeidsområdet) på en polering pad dekket med 1 µm alumina slurry i 3 minutter.
- Skyll elektroden med deionisert vann for å fjerne alle slam fra elektroden og rengjør i ultralydbad (320 W, 37 kHz) med deionisert vann i 15 min.
- Skyll elektroden bruker en 1 mL sprøyte med isopropanol (3 x 1 mL) og deretter med aceton (3 x 1 mL).
- Bruk et papirhåndkle å fjerne alle solid rester og tørr i luften for 3 min.
- Rengjør ITO og FTO elektrodene med deionisert vann. Plass dem i ultralydbad (320 W, 37 kHz) aceton i 15 min og deretter isopropanol i de neste 15 min.
- Brenne platina elektroder, ledninger, og spoler bruker en høy temperatur gass lommelykt (> 1000 ° C) for 1 min. være forsiktig! Bruke pinsett for å holde elektrodene. Vent 5 min etter brenning og før du bruker elektrodene.
- Rengjøre elektrokjemiske og spectroelectrochemical celler (figur 1) med vann og deretter med aceton. Bruk et papirhåndkle for å fjerne alle solid rester. Rengjør alle andre elementer (ca. PTFE deler) med aceton og tørr i luften før bruk.
2. CV analyse
- Slå på potentiostat og datamaskinen.
- Fyll en elektrokjemiske cellen med 1,5 mL elektrolytt-løsning som skal analyseres.
- Putte alle tre elektrodene (vi, AE og RE) i en celle (cellens cap er utstyrt med den PTFE elektrodeholderen, så sette elektroder gjennom hullene i denne holderen) og koble den til en potentiostat. Holde vi og RE som nær hverandre som mulig (figur 1). Følg informasjon om tilkobling av hver ledning av potentiostat til sine respektive elektroden.
- Hvis nødvendig (for eksempel under reduksjon analyse), sette argon (eller nitrogen) røret (gjennom en ekstra hull i elektrodeholderen) og starte boblende løsningen for minst 5 min. Etter dette flytte argon (eller nitrogen) røret over løsningen nivå og beholde gasstrømmen kjører for målingen.
Merk: Cellen ser stengt, men ikke helt lekkasjefri; derfor en metode for å fjerne overflødig mengden gass fra cellen må være tilgjengelig (f.eks., med en ekstra liten hull i elektrodeholderen). Trykket er avhengig av røret brukes; Angi presset til høy som det er nødvendig å sette langsom gasstrømmen. Hvis en svært volatil løsemidler brukes i eksperimentet (f.eks diklormetan) eller eksperimentet tar lang tid (mer enn 30 min), bruk Drechsel flasken for å mette argon (eller nitrogen) gass med løsemiddelet brukes i målingen. - Kjøre potentiostat programvare, velge CV prosedyren og bruk følgende innstillinger: starte potensialet i 0,00 V, øvre toppunktet potensialet i 2.0 V, lavere toppunktet potensialet i 0,00 V (hvis undersøker prosessen med reduksjon og et nedre ytterpunkt potensial på −2.5 V) stoppe potensialet i 0,00 V, nummeret av stopp krysset av 6, og Skann rate på 0,05 V/s.
- I delen eksportere ASCII data, angi et filnavn og velge en mappe å lagre dataene i, og trykk START -knappen (redusere eller øke nedre og øvre toppunktet potensial til å passe vinduet elektrokjemiske eller prøven elektrokjemiske aktivitet)
- Hvis noen topper vises i positiv området potensial, deretter gjenta rengjøring. Hvis det er en topp i fjellkjeden negative potensielle, deretter sette argon (eller nitrogen) røret i løsningen og starte bobler for en ekstra 5 minutter.
- Legge til en dråpe 1 mM ferrocene (i løsemiddelet brukes å forberede elektrolytt) til elektrolytt-løsning med en sprøyte.
- Angi start potensialet til 0,00 V, øvre toppunktet potensielle til 0,85 V, lavere toppunktet potensielle til 0,00 V, stopp potensialet til 0,00 V, antall stopp krysset til 10, og avsøkingshastigheten til 0,05 V/s. I delen eksportere ASCII data, endre filnavn og trykk på START -knappen.
- Etter måling, ferdig ved å gjenta rengjøring fremgangsmåtene som nevnes i trinn 1.2 og 1.3.
- Forberede 4 mL 1 mM test compound i forberedt elektrolytt (trinn 1.1).
- Fyll den elektrokjemiske cellen med test sammensatte løsningen (1,5 mL); sett alle tre elektrodene i en celle og koble til potentiostat (trinn 2.4). Undersøke prosessen med reduksjon, fjerne oksygen (trinn 2.5).
- Angi start potensielle til 0,00 V, øvre toppunktet potensielle 0,50 V (eller 0,00 V ved undersøkelse av reduksjon), lavere toppunktet potensielle til 0,00 V (−0.50 V ved undersøkelse av reduksjon), stopp potensialet til 0,00 V , antall stopp krysset til 10, og avsøkingshastigheten til 0,05 V. I delen eksportere ASCII data, endre filnavn og trykk på START -knappen.
- Gjenta trinn 2.13 ved å øke øvre toppunktet potensialer (eller ved å redusere nedre ytterpunkt potensialet) med 0.1 V til toppen registrering (figur 2a). Hvis etterfølgende skanningen er forskjøvet i potensial (figur 2b), deretter rengjøre RE og la den i elektrolytt-løsning. Deretter gjenta målingen.
- Måle både oksidering og reduksjon prosesser på samme CV for fastsettelse av IP og EA: Angi start potensialet til 0,00 V, øvre toppunktet potensialet til 1,00 V, lavere toppunktet potensial til å −2.70 V og stopp potensialet til 0,00 V. Velg den øvre og nedre vert ex potensial på en måte som registrerer full reduksjon og oksidasjon topper og unngår ytterligere redoks skritt (hvis noen finnes) (figur 2a).
Merk: Beregne IP og EA ved å måle utbruddet potensielle. Det er et par måter å beregne disse parameterne fra CV men nyttigst er å beregne bruker utbruddet potensielle. Noen ganger, er det ikke mulig å observere reversibel redoks par, spesielt for både n og p doping. Blanding av to ulike teknikker er også uønsket som det kan gi feil resultater og konklusjoner.- For å måle utbruddet potensielle, markerer du tangentlinjen til CV peak (Figur 3). Beregne ønskede verdier fra utbruddet potensialet av reduksjon eller oksidasjonsprosessen bruker ligninger (ligninger 1 og 2)10,18,19,36,37 , 38.
 (1)
(1)
hvor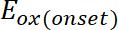 er utbruddet av oksidasjon potensielle kalibrert med ferrocene*
er utbruddet av oksidasjon potensielle kalibrert med ferrocene*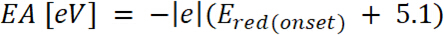 (2)
(2)
hvor er utbruddet av reduksjon potensielle kalibrert med ferrocene*
er utbruddet av reduksjon potensielle kalibrert med ferrocene*
* Potensialet kalibrert med ferrocene betyr at verdien av potensialet fra målingen er redusert oksidasjon potensialet ferrocene.
- For å måle utbruddet potensielle, markerer du tangentlinjen til CV peak (Figur 3). Beregne ønskede verdier fra utbruddet potensialet av reduksjon eller oksidasjonsprosessen bruker ligninger (ligninger 1 og 2)10,18,19,36,37 , 38.
- Trekk ut elektrodene og utøse test løsningen til riktige avfallsbeholderen.
- Gjenta rengjøring (trinn 1.2 og 1.3).
- Gjenta trinn 2.9-2.12 med 0,25 0,10 og 0,20 V/s skanning priser.
- Legg en dråpe 1 mM ferrocene (i løsemiddelet brukes å forberede elektrolytt løsningen) til test løsning bruker en sprøyte. Angi start potensialet til 0,00 V, øvre toppunktet potensielle til 0.60 V, lavere toppunktet potensialet til 0,00, stopp potensialet til 0,00 V, antall stopp Overfarten til 6 og avsøkingshastigheten til 0,05 V. Trykk START -knappen. Når målet er fullført, endre øvre toppunktet potensielle verdien som registrerer ferrocene paret med første toppen av oksidasjon av testen sammensatte.
- Trekk ut elektrodene og utøse test løsningen til riktige avfallsbeholderen.
- Hvis det oppstår electropolymerization under elektrokjemiske oksidasjon, vask vi nøye med elektrolytt løsningen bruker sprøyten (3 ´ 0,5 mL). Rengjør RE, AE, og elektrokjemiske cellen ved hjelp av standard prosedyre (trinn 1.2-1.3).
- Undersøke elektrokjemiske produkter avsatt på vi, går du til trinn 2,23. For å undersøke en løsning, kan du fortsette å gå 1.2.1 og starte på nytt fra trinn 2.3.
- Fyll den elektrokjemiske cellen med en elektrolytt-løsning. Deretter satte alle tre elektrodene i en celle og koble dem til en potentiostat (trinn 2.4). Undersøke reduksjonsprosessen, deoxidate løsningen (trinn 2.5).
- Gjenta trinn 2.10-2.12 og deretter trinn 2,16.
 (1)
(1)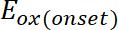 er utbruddet av oksidasjon potensielle kalibrert med ferrocene*
er utbruddet av oksidasjon potensielle kalibrert med ferrocene*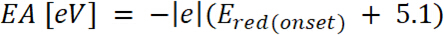 (2)
(2) er utbruddet av reduksjon potensielle kalibrert med ferrocene*
er utbruddet av reduksjon potensielle kalibrert med ferrocene*3. UV-Vis-NIR Spectroelectrochemical analyse
- Måling-forberedelser
- Slå på spectrometer og potentiostat.
- Fyll spectroelectrochemical cellen med 0,5 mL elektrolytt (trinn 1.1).
- Satte PTFE forseglingen på én side av cellen; deretter satt ITO elektroden cellen i denne måten å ha en ledende side av elektroden berøre PTFE segl. Sette resten av PTFE delene som vist i figur 1. Sette RE og AE gjennom den PTFE elektrodeholderen og dekke av ITO elektroden med kobber folie som vist i figur 1 for bedre ledeevne.
- Sett samlet cellen i innehaveren av spectrometer og koble alle elektrodene til en potentiostat. Følg informasjon om tilkobling av hver ledning av potentiostat til sine respektive elektroden.
- Kjøre potentiostat og spectrometer programvaren.
- Spectrometer programvaren, velg fil | Ny | Absorbansen måling.
- Velg ett av de oppførte detektorer.
- Kontroller at knappen på detektorer i "åpen" stilling. Klikk på glødende lyspæren; Lukk detektoren (flytte knappen til lukket posisjon), og klikk deretter på mørke lyspæren.
- Velg det andre merker og gjentar trinn 3.1.7.
- Koble elektroder fra potentiostat og trekk ut elektrodene og andre elementer til spectroelectrochemical-cellen.
- Gjenta rengjøring prosedyrer (trinn 1.2 og 1.3).
- Fyll spectroelectrochemical cellen med en utvannet (1´10−5 M) løsning av testen sammensatte i elektrolytt løsningen hvis tester av lite molekyl. Fyll spectroelectrochemical cellen med elektrolytt hvis tester polymere (oligomeric) laget avsatt på ITO elektroden.
- Montere spectroelectrochemical cellen (trinn 3.1.3).
- Klikk Lagre.
- Velg ett av de oppførte detektorer.
- Definere lagre plassering og navn filen (inkluderer valgte detektoren).
- Velg det andre merker og gjentar trinn 3.1.15.
- Potentiostatic analyse18.
- Bruk en nøytral potensial (dvs. 0,00 V). Dette spekteret blir til Start spectra. Øke potensialet ved 0,1 V og vente ca. 10 s til prosessen stabiliserer og registrerer absorpsjon spectra (trinn 3.1.13–3.1.16). Fortsette målingen til den første endringen i UV-Vis-NIR spectra; vente 10 s og spare til spectra (trinn 3.1.13–3.1.16).
- Øke potensialer ved 0,05 V; vente 10 s og lagre (trinn 3.1.13–3.1.16).
- Gjenta 3.2.2 til å oppnå potensialet i første eller andre oksidasjon potensial (disse potensialene er kjent fra CV målingen); deretter snu potensial og gå tilbake til Start potensial.
- Når målene er fullført, kan du måle CV av test løsning. Legg 1 mM ferrocene (10 μL) og måle CV igjen. Verdien av ferrocene vil hjelpe anslå potensialet i prosessen og forene dataene.
- Denne analysen gir informasjon om optisk band-gap, maksimal bølgelengde undoped og dopet stater og isosbestic poeng av elektrokjemiske prosessen. Beregne de optiske band hullene fra utbruddet verdier π-π* band på UV-Vis spectra og bruke formel 318,19,23,24.
 (3)
(3)
hvor h er Planck's konstant, λutbruddet er sammensatte absorpsjon utbruddet, og c er hastigheten til lys i vakuum. - Tid-løst Potentistatic analyse19,32,33
- Forberede målingen ligner på metoden beskrevet i 3.1.1–3.1.16 og sette argon (eller nitrogen) røret (gjennom en ekstra hull i elektrodeholderen) og boblende løsning minst 5 min før måling. Etter dette flytte argon røret over løsning nivået og beholde gasstrømmen kjører for målingen.
- Kjøre potentiostat programvare, velger chronoamperometry prosedyren med følgende innstillinger: potensial starte potensial til 0,00 V, øvre potensialet 1.0 V, lavere potensial til å −1.00 V, varighet 5 s, intervalltiden som 0.010 s, antall gjentakelser til 10 og forsinkelse tid til 10 s. I delen eksportere ASCII data, definere et filnavn og velger en mappe å lagre dataene.
- Spectrometer programvaren, velg fil | Ny | Høyhastighets oppkjøpet. Et nytt vindu vises. Angi følgende parametere: integrering tid-10 ms, skanner til gjennomsnitt-1, Boxcar Wirth-0, nummeret av skanner-10000. Ikke Klikk knappen gå .
- I potentiostat programvaren, trykk START -knappen. Når nedtellingen drops til 1 s, trykk på Go -knappen i spectrometer programvaren.
- Merk at det ikke er mulig å se absorpsjon resultater til prosessen er ferdig.
- Analysere dataene for å angi parametere. Bruk informasjonen som er lagret i filen slik transmisjon, gjeldende, spenning og nåværende tetthet å gi arbeider parametere:
- Beregne optisk tetthet (ΔOD), absorbansen av testen sammensatte under doping (dedoping) med ligningen 4:
 (4)
(4)
der Tb er transmisjon i dopet form, "blekemiddel" tilstand [%] og Tc er transmisjon i undoped form, en "farget" tilstand [%]. - Beregne Coloration effektivitet (CE, η), mengden elektrokromatiske farge dannet av hvor brukt og som forbindelse mellom injisert/utløst tillegget som en funksjon av elektrode området (Qd) og endring i optisk densitet (ΔOD) verdien på et bestemt bølgelengde (λmax) med ligningen 5. Verdien avhenger på bølgelengde valgt for studien, så verdien måles i λmax optisk absorpsjon bandet av farget.
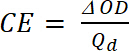 (5)
(5)
hvor ΔOD er optisk densitet [-] og Qd er gratis tetthet [C/cm2]. - Beregne kontrast Ratio (CR), målt intensiteten av fargeendring under elektrokjemiske doping og vanligvis preget av λmax skjemaet farget med ligning 6:
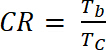 (6)
(6)
der Tb er transmisjon i dopet form, "bleach" tilstand [%] og Tc er transmisjon i undoped form, "farget" tilstand [%].
- Beregne optisk tetthet (ΔOD), absorbansen av testen sammensatte under doping (dedoping) med ligningen 4:
 (3)
(3) (4)
(4)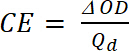 (5)
(5)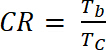 (6)
(6)4. EPR Spectroelectrochemical analyse
- Måling-forberedelser
- Slå på spectrometer og potentiostat.
- Fyll spectroelectrochemical cellen med elektrolytt (trinn 1.1).
- Sett makt til å 1 mW og tune resonator å oppnå et skarpt, sentrert signal i Q-dip (Bruk alternativet autojusterte).
- Angi Mn markøren (mangan standard) til 600, og Lukk vinduet Q-dip. Hvis det ingen er Mn, deretter hoppe over dette trinnet og gå til trinn 4.8.
- Angi "feie bredde ±" til 2.5´ 10 mT, "mod bredde ±" 1.0´0.1 mT, "amplitude" til 5´100, "tidskonstant" 0,03 s og "center feltet" salgsverdi å ca 338 mT og starte målingen.
- Hvis ikke registrerer seks spectral tekstlinjer Mn markør, endre deretter verdien "feltet".
- Når du registrerer seks spectral tekstlinjer Mn markør (Figur 4), setter den "feltet" mellom tredje og fjerde linje og redusere verdien av "feie bredde ±" til en verdi som dekker bare disse to linjene. Starte målingen igjen.
- Gå til alternativet Q-Dip satt Mn markøren til 0, og Lukk vinduet Q-dip.
- Måle ren elektrolytt som bakgrunn og sjekke om det er noen signaler. Hvis signalene vises, kan så signalet være fra forurensning, lav oksidasjon potensielle forbindelser, eller glasset.
- Rengjør spectroelectrochemical cellen (trinn 1.3).
- Undersøkelse av små molekyler (solutions).
- Fyll spectroelectrochemical cellen med en utvannet (1´10−5 M) løsning av testen compound i elektrolytten; sette alle tre elektroder gjennom elektrodeholderen til cellen på denne måten slik at vi og RE er inne spiral AE (figur 1).
- Satt vi nær bunnen av cellen og RE på oversiden av aktivt (metall ikke dekkes av PTFE) del av WE (figur 1). Plasser cellen utstyrt med en elektrode inne i prøven hulrom av spectrometer og koble elektroder med en potentiostat (Følg informasjon om tilkobling av hver ledning av potentiostat til sine respektive elektrode).
- Bruke potensialet tilsvarer utbruddet av første redoks toppen (verdi kjent fra CV måling). Hvis et signal vises, er deretter utstyret satt opp riktig.
- Gå til alternativet Q-Dip, angi Mn markøren til 600, Lukk vinduet Q-dip og starte målingen. Registrering av signal med signalet fra Mn markør gjør beregningen av g-faktor på signalet.
- Gå til alternativet Q-Dip satt Mn markøren til 0, Lukk vinduet Q-dip og vente ca. 5 min.
- Setter den "feltet" i midten av signalet, redusere verdien av "feie bredde ±" til en verdi som dekker signalet, men ikke kuttet ikke av begynnelsen og slutten av signal (når "feie bredde ±" endres, sjekk hele signal), og redusere verdien av "m OD bredde ± "å få godt løst spectra.
- Hvis signalet er liten, så øke "amplitude" verdi og "tid" (oppkjøpet tid) for målingen.
- Du kan redusere signalet til støyforhold, angi oppkjøpet 4, 9 eller 16, avhengig av hvordan støyende signalet er. Hvis signalet er veldig bråkete, Velg 16.
- Fyll spectroelectrochemical cellen med en utvannet (1´10−5 M) løsning av testen compound i elektrolytten; sette alle tre elektroder gjennom elektrodeholderen til cellen på denne måten slik at vi og RE er inne spiral AE (figur 1).
- Undersøkelser av polymere laget avsatt på overflaten av vi.
- Fyll spectroelectrochemical cellen med en elektrolytt-løsning (trinn 1.1).
- Sette elektrodene i cellen. Vær forsiktig med å ødelegge det polymere laget på vi. Koble elektroden med potentiostat (trinn 4.2.1).
- Bruke nøytral potensial (dvs. 0,00 V) å sikre at sammensatt er i en nøytral tilstand; Dette spekteret blir til Start EPR spectra.
- Øke potensialet av 0.10 V. vente ca 10 s til prosessen stabiliserer og registrere EPR spectra.
- Gjenta trinn 4.3.4 når EPR signalet vises.
- Øke potensialet av 0,05 V. vente ca 10 s til prosessen stabiliserer og registrere EPR spectra.
- Gjenta trinn 4.3.6 til først eller andre oksidasjon potensial oppnås og så videre (verdiene for disse potensialene er kjent fra CV måling); deretter snu potensial og gå tilbake til Start potensielle.
- Gjelde potensialet som EPR signal dukket opp i den forrige syklusen (4.3.5), gå til alternativet Q-Dip, angi Mn markøren til 600, Lukk vinduet Q-dip og starte målingen. Registrere signalet i tredje og fjerde spectral linjen av Mn merketråden (4.1.7).
- Når målet er fullført, analysere dataene.
- Noen av analysen som flere spinn kan utføres bare for forbindelsene avsatt (uløselig polymerer) på vi. Hvis du vil beregne antall spill, dobbel-integrere EPR signalet, etterfulgt av en sammenligning av gis verdi med kalibrert Mn interne standard. Beregne antall gjentatte enheter som danner polymer på elektroden bruke ligningen 724,32,33:
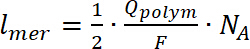 (7)
(7)
der lmer -nummeret av gjentatte enheter i polymer, Qpolym tilkommer polymerisasjon F er Faraday konstant og NA er Avogadros. - Bruk CV av avsatt polymer registrert i en spectroelectrochemical celle før EPR måling, bruker ligningen 8for å beregne doping kostnad:
 (8)
(8)
der Qd er doping kostnad, er gjeldende , E er settet potensial, E0 er første potensialet v er avsøkingshastigheten og e er elementær kostnader. - Bruke beregnede doping kostnad til å beregne doping nivå, ligningen 9:
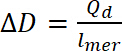 (9)
(9)
hvor ∆D- doping nivå, Qd er doping kostnad og lmer -antall repeterende enheter i polymer - For å beregne hvor mange bipolarons, anta en første doping nivå lik null. Bruke antall polarons per enhet fra konsentrasjonen av spinner, slik at antall bipolarons skal beregnes ved hjelp av ligning 10. I beregningene, anta at den innledende beløpet av bipolarons er null og at faradaic gjeldende er mye høyere enn for ikke-faradaic gjeldende.
 (10)
(10)
der zjeg er gratis transportørens elementær kostnad, njeger antall gratis bærere per polymer enhet (p) er polarons og (b) er bipolarons. - Ekstra g-faktor verdi fra EPR spectra. Fastslå bruker Mn som interne standard (trinn 4.2.3 og 4.3.8), å vite at den fjerde mangan linjen i det har en g-faktor på 2.03324. Beregne linjebredden EPR signal som avstanden i mT mellom minimum og maksimum EPR signalet. Denne verdien er viktig som den kan fortelle hvor (i hvilken atom) radikalene dannes.
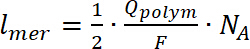 (7)
(7) (8)
(8)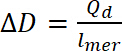 (9)
(9) (10)
(10)Representative Results
Vanligste anvendelse av CV analyse er estimering av IP og EA. Selv om det er et par måter å få CV data, anbefales det å beregne dem basert på utbruddet av redoks topper (Figur 3). Denne tilnærmingen kan forene fremgangsmåten. Ikke alle materialene som testet gjennomgå reversibel oksidasjon/reduksjon prosesser (Figur 3); i slike situasjoner er det ikke mulig å beregne basert gjennomsnittlig potensialer (gjennomsnitt potensialer tilsvarer maksimalt Katodisk-reduksjon og anodic-oksidasjon topper). Det er imidlertid nesten alltid mulig å kjøre tangens til en topp som vist i Figur 3. Med krysset bakgrunn linje og bruk av formler 1 og 2, IP og EA verdiene er beregnet til 5.35 eV og −2.90 eV, henholdsvis. Det finnes også flere forskjellige skalaer brukes til å evaluere IP og EA basert på CV målinger. De vanligst brukte skalaer for organisk materiale er −4.8 og −5.1 eV som tilsvarer 0,00 V versus Normal Hydrogen elektrode (NHE). Men er alle skalaer bare en tilnærming; Husk dette når du sammenligner ulike resultater. Det avgjørende er å oppgi hvilke parametre ble vurdert for beregninger. I dette tilfellet verdien −5.1 eV har valgt som det skal tilsvare formelle potensialet av ferrocene redoks paret; Fermi skalaen er det 0.40 V versus mettet Calomel elektrode (SCE) i acetonitrile, som er i samsvar med forrige måling.
Det er mange artikler publisert om analyse av CV. her, viser vi når prosessen ikke er til å være som det er forventet. Analysen er basert på tiofen derivat: NtVTh (strukturen vist i Figur 4), som gjennomgår fornedrelse ved oksidasjon (figur 5).
NtVTh har to oksidasjon potensialer: først 0.70 V og andre på 0.84 V (figur 5). Under den første syklusen observert reduksjon toppen ikke, som indikerer en. Elektrokjemiske kjennetegner NtVTh viser at polymerisasjon forekommer ikke og etter første oksidasjon potensielle, noen electro inaktive lag reaksjon produkter innskudd på elektroden overflaten, dermed hindre polymerisasjon prosessen. Hva er synlig er reaksjon med den radikale kasjon på vinyl bånd, der molekylet er å miste sin bøyning og form dimers på elektroden.
Mens det er vanskelig å trekke ut informasjon om kostnader bærere fra CV, er det mulig å skille mellom polarons og bipolarons da støttet av en UV-Vis-NIR spectrometer. Den nøytrale poly (OiPrThEE) var preget av to bredt absorpsjon π | π * overganger band med topper maxima på λmax1 = 363 nm og λmax2 = 488 nm, knyttet til aromatiske form av den undoped polythiophene deriverte. Under oksidativt doping genereres nye polaronic og bipolaronic band. UV-Vis spekteret fikk under oksidasjon poly (OjegPrThEE) avslørte det avtagende nøytral polymer absorpsjon bandet (300-550 nm) (figur 6) sammen med dannelsen av en ny absorpsjon band (550-950 nm) av de radikale kasjoner av bithiophene og p- phenylenevinylene med maxima på 692 nm. Isosbestic poenget med oksidasjonsprosessen var på 604 nm. Bipolaronic bandet dukket opp mellom 950 nm og 1700 nm maksimalt ligger på 1438 nm.
Dette inkluderer organisk radikaler39EPR spektroskopi er teknikken som oppdager materialer med en unpaired elektron. Det er flere parametere som kan trekkes ut fra EPR spectra, men en av de mest interessante er å anslå hvor radikalene er lokalisert. Elektroner, ligner på protoner, har spinn. Ved å plassere et elektron i en ekstern magnetfelt, dette kan deles på to måter: parallelle og antiparallel til det magnetiske feltet, gir to energinivå. Dette fenomenet kalles Zeeman effekt40,41. Ved organisk radikaler kommuniserer det unpaired elektronet ikke bare med det eksterne magnetfeltet, men også med magnetiske kjerner (kjerner som har en annen spinn; I≠0). En rekke degenerert energinivå er lik over 2I + 1, hvor jeg er spinn quantum antall kjernen som det unpaired elektronet samhandler42. Samspillet av unpaired elektron med et større antall magnetiske kjerner fører til ytterligere deling av energinivået og hyperfine strukturen i EPR spectra registrering43 (figur 7).
For molekyler hvor unpaired elektron samhandler med et enda større antall kjerner, kan personlige spectral linjen overlappe, som resulterer i Registrering av et enkelt, bred signal44,45,46 (figur 8). Dette er typisk for konjugert polymerer, hvor den genererte radikale ion under en redoks prosess er delocalized47,48.
Kombinasjonen av EPR spektroskopi elektrokjemiske metoder gjør karakterisering av kostnader bærere (radikale ion) generert redoks prosessen samt fastsettelse av mekanismen av disse prosessene49,50 . Hvis godt løst (topper skilles, ikke i form av en bred topp) spectra er registrert, som i tilfellet elektrokjemiske reduksjon av s -tetrazine derivat (figur 7), og analyse av hyperfine spectra fører til konklusjoner om lokalisering av unpaired elektron. En måte å analysere denne typen spectra er simulering med spesiell programvare og å passe simulert spectra med den eksperimentelle en51. Dette er spesielt nyttig når hyperfine er komplisert på grunn av samspillet av unpaired elektron med stort antall protoner. Hvis s- tetrazine derivat vises i figur 7, angir simulering av EPR spectra (rød linje) samspillet av unpaired elektron med fire nitrogen-atomer av s- tetrazine.

Figur 1: elektrokjemiske og spectroelectrochemical celler brukes for målinger. Figuren viser ordningen oppsettet av elektrokjemiske/spectroelectrochemical celler ved hjelp av syklisk voltammetry, UV-synlig og nær-infrarøde (UV-Vis-NIR) og elektron spinn resonans (EPR) spectroelectrochemical målinger. Klikk her for å se en større versjon av dette tallet.

Figur 2: syklisk voltammetry (CV) riktig målt sammensatte COPO1 (a) og CV med ustabile referanse potensielle dibenzothiophene-S, S-karbondioksid med ferrocene (b)52. Figuren viser to syklisk voltammograms. (a) presenterer riktig registrert CV og (b) viser en voltammogram som er registrert ved hjelp av en referanse elektrode med ustabilt potensielle. Klikk her for å se en større versjon av dette tallet.

Figur 3: syklisk voltammetry (CV) av sammensatte COPO1 i en rekke potensialer. Estimering av utbruddet potensial for EA og IP beregninger av COPO1 forbindelser52. EA = −2.90 eV og IP = 5.35 eV. Klikk her for å se en større versjon av dette tallet.

Figur 4. EPR seks spectral linje av mangan standard. Spinn signalet av mangan brukes for kalibrering av signal skiftet. Klikk her for å se en større versjon av dette tallet.

Figur 5: syklisk Voltammetry (CV) av sammensatte NtVTh. Syklisk voltammograms 15 mm NtVTh i 0.1 M Bu4NBF4ugyldig3CN og i ferrocene standard presentere fornedrelse prosessen involvert på arbeider elektroden. Avsøkingshastigheten 0,05 V/s: (en) ble tatt i området −1.4 V til 0,8 V og (b) i området −1.4 V til 0,9 V. Klikk her for å se en større versjon av dette tallet.

Figur 6: ultrafiolett synlige og nær infrarød (UV-Vis-NIR) spectroelectrochemistry av poly (OjegPrThEE) avledede. UV-Vis-NIR spectra presenterer utviklingen av absorpsjon bandet gjennom generasjon kostnad bærere polymer struktur. Klikk her for å se en større versjon av dette tallet.

Figur 7: EPR spectroelectrochemical analyse av tetrazine derivat. (a) strukturen av s- tetrazine avledede; (b) EPR spectra registrert under elektrokjemiske reduksjon av s- tetrazine derivat (svart linje-eksperimentelle og røde linje simulert spektrum). Klikk her for å se en større versjon av dette tallet.

Figur 8: EPR spectroelectrochemical polaron signalet av konjugert polymer. Elektron spinn resonans (EPR) spectra registrert under det første steget av oksidasjon av konjugert polymer (EPR spektra av polaron arter). Klikk her for å se en større versjon av dette tallet.

Figur 9: syklisk voltammetry (CV) ferrocene og decamethylferrocene. Sammenligning av to elektrokjemiske standarder som ren og blanding viser skifte av potensial. Klikk her for å se en større versjon av dette tallet.
Discussion
Elektrokjemiske og spectroelectrochemical har ingen begrensninger; en kan analysere både fast og flytende løsninger i et bredt spekter av temperatur og andre forhold med disse teknikkene. Viktigste i alle disse tilfellene er at forbindelser/materialer er analysert under anvendt potensialet, replikere virkeligheten arbeidsforhold organisk elektronikk anordninger. Den eneste forskjellen er at i elektrokjemi, er dannelsen av kostnader bærere, observert.
Metodene presenteres her viser nytten av analyse av ladet bærere generert i organiske forbindelser som samsvarer med deres anvendelse i organisk elektronikk. Videre de elektrokjemiske og spectroelectrochemical teknikkene er billigere og mindre krevende enn vanlige metoder som brukes i kostnader bærer analyse, men det er noen viktige trinn og modifikasjoner av protokollen som kreves avhengig av hente resultater.
Under elektrokjemiske karakterisering, alltid starte med en bestemt konsentrasjon. Hvis et sett av forbindelser sammenlignes, deretter må alle materialer ha samme molar konsentrasjonen. Best er å starte med 1 mM konsentrasjon og 50 mV/s skanne rate som angitt i protokollen denne studien, men det er godt å vite konsentrasjonen av prøven på observerte elektrokjemiske oppførselen. Prøv alltid å måle minst tre skanninger. De to første skanner er vanligvis forskjellige fordi Start betingelsene (likevekt) er annerledes. Andre og tredje skanner bør være den samme. Hvis den andre og tredje skanner er like, er det trolig ingen siden reaksjoner i dette systemet (figur 2a). I en oksidasjonsprosessen vises en ny topp på en lavere potensial viser at det ledende materialet ble avsatt på vi18,19,24,-25,29,30 , 31 , 32. Hvis høyden på lavere toppen øker i påfølgende skanner, så sannsynligvis den konjugert polymer ble avsatt18,19,24,25,29 , 30 , 31 , 32. Hvis alle strømmene nedgang i påfølgende skanner, så nonconductive produktet av nedbrytning ble avsatt på elektroden. Hvis en liten peak er observert før viktigste oksidasjon eller reduksjon peak (spesielt for polymerer), så er dette trolig kostnad-fangst prosessen19,23,31,34. Hvis en veldig skarp dedoping toppen av oksidasjon eller reduksjon er observert, er dette sannsynligvis forårsaket av nedbryting av krystallinsk strukturer på en elektrode dannet gjennom electrocrystallization prosessen under oksidasjon35.
Kontroller virkemåten til testen sammensatte før, under og etter redoks topper. Det betyr at minst tre CV skanner skal registreres: øvre (i tilfelle oksidasjon) eller nedre ytterpunkt potensialet eller høyere, henholdsvis så potensialet i topp maksimalt, med øvre eller nedre ytterpunkt potensial satt til nøyaktig på toppen maksimal og med toppunktet potensialer høyere (oksidasjon) og lavere (reduksjon) enn potensial peak maksimalt. Observert prosessen kan variere, og noen ganger to prosesser kan observeres under en topp teoretisk. Alltid sammenligne den innsamlede syklisk voltammograms av elektrolytt (trinn 2.6), ferrocene (trinn 2.9), sammensatt (trinn 2.13) og ferrocene med sammensatte (trinn 2.19); Det er flere problemer å bli tatt i betraktning.
Alltid sammenligne CV signalene fra elektrolytten og test sammensatte, hvis noen signaler fra elektrolytt er synlig på de sykliske voltammogram av det målte sammensatt, elektrolytt må endres fordi elektrokjemiske vinduet er for lavt, eller elektrolytt er forurenset. Hvis signalet (redoks par) av ferrocene (trinn 2.9) og ferrocene med sammensatte (trinn 2.19) er i samme posisjon, utføres alt riktig. Hvis toppene er flyttet mellom hverandre, så sjekk RE og gjenta målingen. Hvis signalet (oksidasjon reduksjon og redoks par potensielle) av testen sammensatt med lagt ferrocene (trinn 2.19) på en høyere potensial enn ren sammensatte (trinn 2.13), så vurdere verdiene (oksidasjon reduksjon og redoks par potensielle) fra den sykliske voltammogram av det ren sammensatt. Overgangen skyldes høyere mengden ferrocene i løsningen. Når to oksidasjon prosesser er observert, kan den første prosessen (oksidasjon eller reduksjon) som alltid er på vi påvirke den aktive overflaten; Dette kan forårsake en økning i oksidasjonen potensialet i den andre prosessen (figur 9).
Disclosures
Forfatterne ikke avsløre.
Acknowledgments
Forfatterne takknemlig anerkjenner økonomisk støtte av "Excilight" prosjekt "Donor-Acceptor lys Emitting Exciplexes som materiale for enkel-å-skreddersy supereffektive OLED lyn" (H2020-MSCA-ITN-2015/674990) finansiert av Marie Skłodowska-Curie Handlinger i rammeprogram for forskning og innovasjon "Horizon-2020".
Materials
| Name | Company | Catalog Number | Comments |
| Potentiostat | Metrohm | Autolab PGSTAT100 | |
| EPR | JEOL | JES-FA200 | |
| UV-Vis detector | Oceanoptics | QE6500 | |
| NIR detector | Oceanoptics | NIRQuest | |
| Dichloromethane (DCM) | Sigma-Aldrich | 106048 | |
| Tetrabutylammonium tetrafluoroborate (Bu4NBF4) | Sigma-Aldrich | 86896 | |
| 2-propanol, 99.9% | Sigma-Aldrich | 675431 | |
| Acetone, 99.9% | Sigma-Aldrich | 439126 | |
| Ultrasonic Bath | Elma | S30H | |
| Tetrahydrofuran >99.9% | Sigma-Aldrich | 401757 | |
| ferrocene >98% | Sigma-Aldrich | F408 | |
| decamethylferrocene >97% | Sigma-Aldrich | 378542 |
References
- O'Brien, D. F., Baldo, M. A., Thompson, M. E., Forrest, S. R. Improved energy transfer in electrophosphorescent devices. Applied Physics Letters. 74, 442-444 (1999).
- Chin, B. D., et al. Effects of interlayers on phosphorescent blue organic light-emitting diodes. Applied Physics Letters. 86, 133505-133507 (2005).
- Cardon, C. M., Li, W., Kaifer, A. E., Stockdale, D., Bazan, G. C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bang, A. -M., et al. A novel random terpolymer for high-efficiency bulk-heterojunction polymer solar cells. RSC Advances. 7, 1975-1980 (2017).
- Tybrandt, K., Kollipara, S. B., Berggren, M. Organic electrochemical transistors for signal amplification in fast scan cyclic voltammetry. Sensors and Actuators B: Chemical. 195, 651-656 (2014).
- Schaur, S., Stadler, P., Meana-Esteban, B., Neugebauer, H., Sariciftci, N. S. Electrochemical doping for lowering contact barriers in organic field effect transistors. Organic Electronics. 13, 1296-1301 (2012).
- Mullen, K., Scherf, U. Organic Light Emitting Devices: Synthesis, Properties and Applications. , Wiley-VCH. Weinheim. (2006).
- Monk, P. M. S., Mortimer, R. J., Rosseinsky, D. R. Electrochromism and Electrochromic Devices. , Cambridge University Press. Cambridge. (2007).
- Skotheim, T. A., Elsenbaumer, R. L., Reynolds, J. R. Handbook of Conducting Polymers. , Marcel Dekker. New York. (1998).
- Data, P., et al. Kesterite Inorganic-Organic Heterojunction for Solution Processable Solar Cells. Electrochimica Acta. 201, 78-85 (2016).
- Data, P., Pander, P., Okazaki, M., Takeda, Y., Minakata, S., Monkman, A. P. Dibenzo[a,j]phenazine-Cored Donor-Acceptor-Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters. Angewandte Chemie International Edition. 55 (19), 5739-5744 (2016).
- Jankus, V., et al. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Advanced Functional Materials. 24 (39), 6178-6186 (2014).
- Data, P., et al. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. Journal of Physical Chemistry C. 120 (4), 2070-2078 (2016).
- Dias, F. B., et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science. , (2016).
- Okazaki, M., et al. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo[a,j]phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chemical Science. 8, 2677-2686 (2017).
- Data, P., et al. Efficient p-phenylene based OLEDs with mixed interfacial exciplex emission. Electrochimica Acta. 182, 524-528 (2015).
- Goushi, K., Yoshida, K., Sato, K., Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nature Photonics. 6, 253-258 (2012).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of heteroaryl group on electrochemical and spectroscopic properties of conjugated polymers. Electrochimica Acta. 83, 271-282 (2012).
- Data, P., Motyka, M., Lapkowski, M., Suwinski, J., Monkman, A. Spectroelectrochemical Analysis of Charge Carries as a Way of Improving Poly(p-phenylene) Based Electrochromic Windows. Journal of Physical Chemistry C. 119, 20188-20200 (2015).
- Enengl, C., et al. Explaining the Cyclic Voltammetry of a Poly(1,4-phenylene-ethynylene)-alt-poly(1,4-phenylene-vinylene) Copolymer upon Oxidation by using Spectroscopic Techniques. ChemPhysChem. 18, 93-100 (2017).
- Gudeika, D., et al. Hydrazone containing electron-accepting and electron-donaiting moieties. Dyes Pigments. 91, 13-19 (2011).
- Swist, A., Cabaj, J., Soloducho, J., Data, P., Lapkowski, M. Novel acridone-based branched blocks as highly fluorescent materials. Synthetic Metals. 180, 1-8 (2013).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of alkyl chain on electrochemical and spectroscopic properties of polyselenophenes. Electrochimica Acta. 87, 438-449 (2013).
- Laba, K., et al. Electrochemically induced synthesis of poly(2,6-carbazole). Macromolecular Rapid Communications. 36, 1749-1755 (2015).
- Pluczyk, S., et al. Unusual Electrochemical Properties of the Electropolymerized Thin Layer Based on a s-Tetrazine-Triphenylamine Monomer. Journal of Physical Chemistry C. 120, 4382-4391 (2016).
- Blacha-Grzechnik, A., Turczyn, R., Burek, M., Zak, J. In situ Raman spectroscopic studies on potential-induced structural changes in polyaniline thin films synthesized via surface-initiated electropolymerization on covalently modified gold surface. Vibrational Spectroscopy. 71, 30-36 (2014).
- Laba, K., et al. Diquinoline derivatives as materials for potential optoelectronic applications. Journal of Physical Chemistry C. 119, 13129-13137 (2015).
- Enengl, S., et al. Spectroscopic characterization of charge carriers of the organic semiconductor quinacridone compared with pentacene during redox reactions. Journal of Materials Chemistry C. 4, 10265-10278 (2016).
- Data, P., et al. Electrochemically Induced Synthesis of Triphenylamine-based Polyhydrazones. Electrochimica Acta. 230, 10-21 (2017).
- Brzeczek, A., et al. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigments. 113, 640-648 (2015).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Electrochemistry and spectroelectrochemistry of a novel selenophene-based monomer. Electrochimica Acta. 59, 567-572 (2012).
- Data, P., et al. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochimica Acta. 122, 118-129 (2014).
- Data, P., et al. Evidence for Solid State Electrochemical Degradation Within a Small Molecule OLED. Electrochimica Acta. 184, 86-93 (2015).
- Data, P., et al. Unusual properties of electropolymerized 2,7- and 3,6- carbazole derivatives. Electrochimica Acta. 1282, 430-438 (2014).
- Etherington, M. K., et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nature Communications. 8, 14987 (2017).
- Trasatti, S. The absolute electrode potential: an explanatory note. Pure and Applied Chemistry. 58, 955-966 (1986).
- Cardona, C. M., et al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bredas, J. -L. Mind the gap! Mater Horiz. 1, 17-19 (2014).
- Gerson, F., Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals. , (2003).
- Brustolon, M., Giamello, E. Electron Paramagnetic Resonance. A Practitioner's Toolkit. , John Wiley & Sons Inc. New Jersey. (2009).
- Weil, J., Bolton, J. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. , John Wiley & Sons, Inc. (2007).
- Rieger, P. H. Electron Spin Resonance. Analysis and Interpretation. , The Royal Society of Chemistry. (2007).
- Roznyatovskiy, V. V., Gardner, D. M., Eaton, S. W., Wasielewski, M. R. Radical Anions of Trifluoromethylated Perylene and Naphthalene Imide and Diimide Electron Acceptors. Organic Letters. 16, 16-19 (2013).
- Pluczyk, S., Kuznik, W., Lapkowski, M., Reghu, R. R., Grazulevicius, J. V. The Effect of the Linking Topology on the Electrochemical and Spectroelectrochemical Properties of Carbazolyl Substituted Perylene Bisimides. Electrochimica Acta. 135, 487-494 (2014).
- Ledwon, P., et al. A Novel Donor-acceptor Carbazole and Benzothiadiazole Material for Deep Red and Infrared Emitting Applications. Journal of Materials Chemistry C. 4, 2219-2227 (2016).
- Heinze, J., Frontana-Uribe, B. A., Ludwigs, S. Electrochemistry of Conducting Polymers-Persistent Models and New Concepts. Chemical Reviews. 110, 4724-4771 (2010).
- Kurowska, A., Kostyuchenko, A. S., Zassowski, P., Skorka, L., Yurpalov, V. L., Fisyuk, A. S., Pron, A., Domagala, W. Symmetrically Disubstituted Bithiophene Derivatives of Properties. Journal of Physical Chemistry C. 118, 25176-25189 (2014).
- Zotti, G., Schiavon, G., Zecchin, S., Morin, J. F., Leclerc, M. Electrochemical, Conductive, and Magnetic Properties of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules. 35, 2122-2128 (2002).
- Kaim, W., Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chemical Society Reviews. 38, 3373-3382 (2009).
- Duling, D. R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. Journal of Magnetic Resonance, Series B. 104, 105-110 (1994).
- Benson, C. R., et al. Multiplying the Electron Storage Capacity of a Bis-Tetrazine Pincer Ligand. Dalton Transactions. 43, 6513-6524 (2014).
- Nobuyasu, R. S., et al. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Effi ciency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Advanced Optical Materials. 4, 597-607 (2016).
