

Summary
En este artículo, describimos electroquímico, resonancia paramagnética electrónica y electroquímicas de ultravioleta-visible e infrarrojo cercano métodos para analizar compuestos orgánicos para su aplicación en electrónica orgánica.
Abstract
Voltametría cíclica (CV) es una técnica utilizada en el análisis de compuestos orgánicos. Cuando esta técnica se combina con resonancia paramagnética electrónica (EPR) o espectroscopias de ultravioleta-visible e infrarrojo cercano (UV-Vis-NIR), obtenemos información útil como la afinidad electrónica, potencial de ionización, energías de banda prohibida, el tipo de portadores de la carga y la información de degradación que puede utilizarse para sintetizar dispositivos electrónicos orgánicos estables. En este estudio, presentamos electroquímico y electroquímicas métodos para analizar los procesos que ocurren en las capas activas de un dispositivo orgánico, así como los portadores de carga generados.
Introduction
En todo el mundo, los investigadores continuamente están buscando nuevos materiales orgánicos que pueden ser utilizados en electrónica orgánica con rendimiento deseable o estabilidad, que cae debido a un uso prolongado. En el caso de dispositivos orgánicos, es importante entender el comportamiento del portador del cargo conocer completamente las reglas del comportamiento del dispositivo. Análisis del efecto del molecular de la estructura en la generación del portador de la carga y la dinámica y el mantenimiento del equilibrio de portadores de la carga inyectada, tanto positivas (huecos) y negativas (electrones), es crucial mejorar la eficiencia y la estabilidad de los dispositivos orgánicos. Esto asegura la recombinación efectiva de estos cargos individuales y, en consecuencia, mejora significativamente la eficiencia de la fotoluminiscencia de la luz orgánica emisión de diodos (OLED)1,2. Fotovoltaica orgánica (OPV)3,4 así campo orgánico efecto transistores (OFETs)5,6, es necesario tener materiales con movilidad de portador de alta carga. Además del análisis de portadores de la carga, varios parámetros importantes de los materiales electroactivos orgánica ayudan en la predicción de que el material podría ser utilizado: potencial de ionización (PI), niveles de energía de afinidad (EA) de electrón y boquete de la venda entre ellos7 ,8,9,10.
En este trabajo presentamos un método para la medición eficiente de voltametría cíclica (CV) que puede ser utilizado en el análisis de todo tipo de materiales electroactivos. Esta técnica proporciona información sobre las propiedades redox, el mecanismo de dopaje/dedoping, la estabilidad, la conversión y almacenamiento de energía, etcetera. También permite la estimación de la energía de afinidad e ionización de electrones de los compuestos de prueba una manera mucho más barato y más rápido en comparación con otros métodos de vacío alta. Los parámetros antes mencionados se correlacionan con los niveles de energía del orbital molecular ocupado más alto (HOMO) y el orbital molecular más bajo desocupado (LUMO).
El método presentado en este artículo puede utilizarse para analizar todos los tipos de compuestos conjugados como los π-electrones deslocalizadas en sus estructuras. Compuestos conjugados pueden ser moléculas pequeñas con grandes cadenas poliméricas. Moléculas pequeñas también pueden ser monómeros; durante la reacción inicial (fotoquímicos, electroquímicos o químicos) monómeros pueden formar polímeros. En aplicación de OLED, los valores de nivel de energía son necesarios permitir el uso de host correcto para el emisor en un termal activados retrasado sistema de guest-host de fluorescencia (TADF) o decidir con que compuestos de la capa de donantes-aceptador exciplex podría ser formado y qué capas adicionales (transporte de capa (ETL), transporte de capa (HTL), capa de bloqueo electrónico (EBL) y capa de bloqueo de agujero (HBL) orificio de electrón) será necesarias sintetizar eficientemente cargada estable equilibrado dispositivos OLED de11 , 12 , 13 , 14 , 15 , 16 , 17. medidas electroquímicas adicionales permiten la investigación de reacciones secundarias posibles durante el proceso de degradación de la capa activa y la formación de bajos móviles portadores de carga (bipolarons)18,19 ,20,21,22.
Acoplamiento de electroquímica y electroquímicas métodos permite una determinación fácil, precisa y fiable del grado de oxidación o reducción de compuestos conjugados y su degradación potencial, que es crucial para la estabilidad23 , 24 , 25 , 26 , 27 , 28. espectroscopia del ultravioleta-visible e infrarrojo cercano (UV-Vis-NIR) juntada con electroquímica puede caracterizar las propiedades fundamentales de la cromáticas de los nuevos compuestos conjugados, como el cambio de la banda de absorción durante el dopaje 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30.
En un estudio relacionado con el mecanismo de dopaje, es importante definir el tipo de portadores de la carga. En este proceso participan dos clases de quasiparticles cargadas, uno con spin no compensada (polarons) y el segundo ser diamagnético (bipolarons); Espectroscopia de resonancia paramagnética (EPR) del electrón proporciona valiosa ayuda, que directamente nos permite observar y seguir los cambios en las poblaciones de polarons paramagnético29,30,31,32 . En moléculas pequeñas, es difícil de forma bipolarons, pero estas moléculas pueden ser conjugadas bastante y tienen propiedades de inducción de bipolaron; es importante comprobar si y en la que potenciales polarons y los bipolarons se forman en la estructura. Bipolarons son al menos un orden en la movilidad más baja que la de polarons; por lo tanto, si se forman bipolarons en dispositivos de trabajo, entonces podría llevar a una relación desequilibrada de los portadores de carga, que resultarían de gran intensidad y sobrecalentamiento del dispositivo OLED o bien pueden ser los centros de degradación33.
El método de medición propuesto en este estudio es barato y más rápido y permite la determinación de los parámetros operativos más valiosos para un gran número de materiales electroactivos sin necesidad de dispositivos especiales que se basan en recién sintetizado materiales para comprobar su funcionamiento. Mediante la aplicación de la electroquímica y spectroelectrochemistry, es posible seleccionar un material que es realmente prometedor de cientos de nuevos materiales. Además, es posible obtener información detallada sobre los procesos de dopaje y sus efectos sobre la estructura química de la prueba conjugan sistemas electroquímicos y métodos electroquímicas, que permite construir más dispositivos de electrónica orgánica eficiente.
Protocol
1. preparación del experimento
- Preparar 25 mL de la solución de 0,1 M de electrolito.
Nota: Dependiendo de los compuestos objeto de investigación, utilizar electrolitos diferentes como la mezcla de sal orgánica y solvente: el más común las sales utilizadas en el análisis de moléculas orgánicas son hexafluorofosfato de Tetrabutilamonio (Bu4NPF6 ) o tetrafluoroborato de Tetrabutilamonio (Bu4NBF4); el solvente más común usado en el análisis son el diclorometano, el acetonitrilo o el tetrahidrofurano.- Elija el disolvente electrolitos basado en la solubilidad de los compuestos de prueba. Debe ser una solución durante la investigación pero estar en estado sólido cuando el material se utiliza en la fabricación del dispositivo, como una película delgada que se deposita en la superficie del electrodo de trabajo.
- Limpie los electrodos a utilizar en el experimento29.
- Para medidas electroquímicas, use un electrodo de platino disco de diámetro de 1 mm como el electrodo de trabajo (WE), platino bobina o alambre como electrodo auxiliar (AE) y un electrodo de plata/cloruro de plata (Ag/AgCl) como el electrodo de referencia (RE).
- Para mediciones electroquímicas EPR, use alambre de platino como la WE, una bobina de platino o un alambre como la AE y Ag/AgCl como el RE.
- Para mediciones electroquímicas de UV-Vis-NIR, utilice un indio-óxido de cuarzo de estaño (ITO) óxido de estaño dopado de flúor (OFT) como nosotros, un alambre de platino como la AE y un electrodo de Ag/AgCl como el RE.
- Polaco el electrodo de platino disco frotando el lado negativo del electrodo (el área de trabajo de electrodo de platino) sobre una almohadilla de pulido cubierta con mezcla de alúmina de 1 μm por 3 min.
- Enjuague el electrodo con agua desionizada para quitar todos las mezclas de los electrodos y limpiar en un baño ultrasónico (320 W, 37 kHz) con agua desionizada durante 15 min.
- Enjuague el electrodo con una jeringa de 1 mL con isopropanol (3 x 1 mL) y luego con acetona (3 x 1 mL).
- Use una toalla de papel para eliminar todos los residuos sólidos y secar al aire durante 3 minutos.
- Limpie los electrodos con agua desionizada ITO y FTO. Colocarlos en un baño de ultrasonido (320 W, 37 kHz) en acetona durante 15 minutos y luego en isopropanol para los próximos 15 minutos.
- Quemar los electrodos de platino, cables y bobinas usando un soplete de gas de alta temperatura (> 1000 ° C) por 1 minuto tenga cuidado! Utilice pinzas para sujetar los electrodos. Espere 5 minutos después de la quema y antes de usar los electrodos.
- Limpie las células electroquímicas y electroquímicas (figura 1) con agua y luego con acetona. Utilice una toalla de papel para eliminar todos los residuos sólidos. Limpie todos los demás elementos (ca. piezas PTFE) con acetona y secar al aire antes de usar.
2. CV análisis
- Encienda la computadora y del potenciostato.
- Llene una celda electroquímica con 1,5 mL de la solución electrolítica a analizar.
- Poner los tres electrodos (nosotros, AE y RE) en una celda (tapa de la celda está equipada con el portaelectrodo PTFE, así que Ponte los electrodos a través de los orificios en el soporte) y conectarlo a un potenciostato. Mantener la WE y como cerca uno del otro como sea posible (figura 1). Siga la información sobre la conexión de cada cable del potenciostato a su respectivo electrodo.
- Si es necesario (por ejemplo durante el análisis de la reducción), colocar el tubo de argón (o nitrógeno) (a través de un agujero adicional en el soporte del electrodo) y empezar a burbujear la solución durante al menos 5 minutos. Después, mueva el tubo de argón (o nitrógeno) sobre el nivel de la solución y mantener el flujo de gas durante la medición.
Nota: La celda parece cerrada pero no puede ser completamente a prueba de fugas; por lo tanto, debe estar disponible un método para eliminar la cantidad redundante de gas de la celda (por ej., por un orificio adicional en el soporte del electrodo). La presión depende de la pipa usada; Ajuste la presión a alta como es necesaria para establecer el flujo de gas lento. Si se utiliza un solvente altamente volátil en el experimento (por ejemplo, diclorometano) o el experimento está tomando mucho tiempo (más de 30 minutos), luego use la botella de Drechsel para saturar el gas argón (o nitrógeno) con el solvente utilizado en la medición. - Ejecute el software de potenciostato, escoger el método de la CV y utilice la siguiente configuración: Inicio potencial de 0.00 V, potencial del vértice superior de 2.0 V, menor potencial de vértice de 0.00 V (si se investiga el proceso de reducción, luego un menor potencial de vértice de −2.5 V), deje de potencial de 0.00 V, número de la travesía de la parada del 6 y tasa de 0.05 V/s de lectura.
- En la sección datos de la exportación ASCII, definir un nombre de archivo y elija una carpeta para guardar los datos en y presione el botón START (disminuir o aumentar el potencial de vértice inferior y superior para adaptarse a la ventana electroquímica o la muestra actividad electroquímica)
- Si los picos son visibles en el rango positivo de potencial, repita el procedimiento de limpieza. Si hay un pico en el rango negativo potencial, entonces ponga el tubo de argón (o nitrógeno) en la solución y empezar a burbujear un adicional 5 min.
- Añada una gota de 1 mM de ferroceno (en el disolvente utilizado para preparar el electrolito) a la solución electrolítica usando una jeringa.
- Establecer el inicio potencial de 0.00 V, el vértice superior potencial de 0,85 V, el potencial de 0.00 V de vértice inferior, el potencial de parada 0.00 V, el número de parada cruce a 10 y la frecuencia de barrido a 0.05 V/s. En la sección datos de la exportación ASCII, cambiar el nombre de archivo y presione el botón START .
- Después de la medida, acabado repitiendo los procedimientos de limpieza, como se mencionó en los pasos 1.2 y 1.3.
- Preparar 4 mL de prueba de 1 mM en el electrolito preparado (paso 1.1).
- Llenar la celda electroquímica con la solución compuesta (1,5 mL); poner todos los tres electrodos en una celda y conectarse el potenciostato (paso 2.4). Para investigar el proceso de reducción, eliminar el oxígeno (paso 2.5).
- Establezca el inicio potencial en 0.00 V, el vértice superior potencial 0.50 V (o V 0.00 en el caso de investigación del proceso de reducción), el vértice más bajo potencial de 0.00 V (−0.50 V en el caso de investigación del proceso de reducción), el potencial de parada 0.00 V , el número de parada cruce a 10 y la frecuencia de barrido a 0,05 V. En la sección datos de la exportación ASCII, cambiar el nombre de archivo y presione el botón START .
- Repita el paso 2.13 aumentando potenciales de vértice superior (o disminuir el potencial de vértice inferior) por 0.1 V hasta registro de pico (Figura 2a). Si el análisis sucesivos se cambia de puesto en el potencial (figura 2b), luego limpiar el RE y dejar en la solución electrolítica. A continuación, repita la medición.
- Medir procesos de oxidación y reducción en el mismo CV para la determinación del IP y EA: establece el potencial de inicio 0.00 V, el potencial de vértice superior 1.00 V, el menor potencial de vértice a −2.70 V y el potencial de parada en 0.00 V. elige el vert superior e inferior ex potencial en una forma que registra completa reducción y oxidación picos y evita pasos redox (si existe alguno) (Figura 2a).
Nota: Calcular IP y EA midiendo la aparición potencial. Hay un par de formas de calcular estos parámetros de CV pero es más útil calcular utilizando el potencial de inicio. A veces, no es posible observar el par redox reversible, especialmente de n y p dopaje. Mezcla de dos diferentes técnicas también es deseable que pueda proporcionar conclusiones y resultados equivocados.- Para medir el potencial de inicio, marque la línea tangente a la cima de la CV (figura 3). Calcular los valores deseados de la posibilidad del inicio de la reducción o el proceso de oxidación mediante las ecuaciones (ecuaciones 1 y 2)10,18,19,36,37 , 38.
 (1)
(1)
donde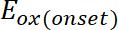 es el inicio de la oxidación potencial calibrado con ferroceno*
es el inicio de la oxidación potencial calibrado con ferroceno*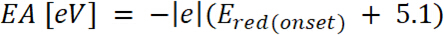 (2)
(2)
donde es el inicio de la reducción potencial de calibrado con ferroceno*
es el inicio de la reducción potencial de calibrado con ferroceno*
* Calibrado con ferroceno potencial significa que se reduce el valor del potencial de la medida por el potencial de oxidación de ferroceno.
- Para medir el potencial de inicio, marque la línea tangente a la cima de la CV (figura 3). Calcular los valores deseados de la posibilidad del inicio de la reducción o el proceso de oxidación mediante las ecuaciones (ecuaciones 1 y 2)10,18,19,36,37 , 38.
- Retirar los electrodos y derramar la solución en el recipiente de desechos adecuado.
- Repita el procedimiento de limpieza (pasos 1.2 y 1.3).
- Repita los pasos 2.9, 2.12 con 0.25, 0.10 y 0,20 V/s tarifas de exploración.
- Añada una gota de 1 mM de ferroceno (en el disolvente utilizado para preparar la solución electrolítica) a la solución de prueba utilizando una jeringa. Establecer el potencial inicio de 0.00 V, el vértice superior potencial a 0,60 V, el potencial inferior vértice de 0.00, el potencial de parada 0.00 V, el número de parada cruce a 6 y la frecuencia de barrido a 0,05 V. Pulse el botón iniciar . Una vez finalizada la medición, cambia el vértice superior potencial a un valor que registra la pareja ferroceno con el primer pico de oxidación de la sustancia de ensayo.
- Retirar los electrodos y derramar la solución en el recipiente de desechos adecuado.
- Si el electropolymerization se produce durante la oxidación electroquímica, lavar cuidadosamente la WE con la solución electrolítica usando la jeringa (3 ´ 0,5 mL). Limpie la celda electroquímica mediante el procedimiento estándar (pasos 1.2-1.3), RE y AE.
- Para investigar productos electroquímicos que deposita en nosotros, vaya al paso 2.23. Para investigar una solución, proceder a paso 1.2.1 y empezar de nuevo desde el paso 2.3.
- Llenar la celda electroquímica con una solución electrolítica. Luego, poner los tres electrodos en una celda y conectarlos a un potenciostato (paso 2.4). Investigar el proceso de reducción, deoxidate la solución (paso 2.5).
- Repita los pasos 2.10, 2.12 y luego paso 2.16.
 (1)
(1)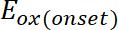 es el inicio de la oxidación potencial calibrado con ferroceno*
es el inicio de la oxidación potencial calibrado con ferroceno*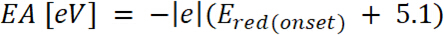 (2)
(2) es el inicio de la reducción potencial de calibrado con ferroceno*
es el inicio de la reducción potencial de calibrado con ferroceno*3. UV-Vis-NIR electroquímicas análisis
- Preparación de la medición
- Encienda el espectrómetro y el potenciostato.
- Llenar la celda electroquímicas con 0,5 mL de solución electrolítica (paso 1.1).
- Ponga el sello PTFE en un lado de la célula; luego poner el electrodo ITO a la celda de esta manera tener un lado conductor del electrodo de la tocar el sello PTFE. Poner el resto de las piezas PTFE como se muestra en la figura 1. Coloco el RE y el AE a través del soporte de electrodo PTFE y cubrir la parte superior del electrodo ITO con hoja de cobre como se muestra en la figura 1 para mejor conductividad.
- Poner la célula montada en el soporte del espectrómetro y conectar los electrodos a un potenciostato. Siga la información sobre la conexión de cada cable del potenciostato a su respectivo electrodo.
- Ejecute el software potenciostato y espectrómetro.
- En el software del espectrómetro, seleccionar archivo | Nuevo | Absorbancia medida.
- Elija uno de los detectores mencionados.
- Asegúrese que el botón en los detectores en la posición "abierta". A continuación, haga clic en la bombilla que brilla intensamente; cierre el detector (mueva el botón a la posición cerrada) y a continuación, haga clic en la bombilla de luz oscura.
- Seleccione el segundo detector y repita el paso 3.1.7.
- Vuelva a conectar los electrodos desde el potenciostato y retire los electrodos y otros elementos de la celda electroquímicas.
- Repita los procedimientos de limpieza (pasos 1.2 y 1.3).
- Llenar la celda electroquímicas con un diluido (1´10−5 M) solución de la prueba en la solución de electrolito si pruebas la molécula pequeña. Llenar la celda electroquímicas con el electrolito si pruebas la capa (oligomérica) polimérica depositada sobre el electrodo ITO.
- Montar la celda electroquímicas (paso 3.1.3).
- Haga clic en Guardar.
- Seleccione uno de los detectores mencionados.
- Definir el guardar ubicación y el nombre del archivo (incluya el detector seleccionado).
- Seleccione el segundo detector y repita el paso 3.1.15.
- Análisis de Potentiostatic18.
- Se aplica un potencial neutro (es decir, 0,00 V). Este espectro serán los espectros a partir. Aumentar la posibilidad de 0.1 V y esperar aprox. 10 s hasta que el proceso se estabiliza y registra el espectro de absorción (medidas 3.1.13–3.1.16). Continuar la medición para el primer cambio en espectros UV-Vis-NIR; Espere de 10 s y luego guardar los espectros (medidas 3.1.13–3.1.16).
- Potenciales de aumento por 0.05 V; Espere de 10 s y guardar (medidas 3.1.13–3.1.16).
- Repetir 3.2.2 hasta alcanzar el potencial el potencial de oxidación primera o segunda (estos potenciales son conocidos de la medición de la CV); a continuación, invertir el potencial y volver al potencial de partida.
- Una vez finalizadas las medidas, medida de la CV de la solución de ensayo. Añadir 1 mM ferroceno (10 μL) y medir otra vez el CV. El valor de ferroceno ayudará a estimar el potencial del proceso y unificar los datos.
- Este análisis proporciona información sobre la óptica boquete de la venda, longitud de onda máxima de lámpara y dopado y isosbestic puntos del proceso electroquímico. Calcular los ópticos banda-vacíos de valores inicio de bandas π-π* en los espectros UV-Vis y utilizando la ecuación 318,19,23,24.
 (3)
(3)
donde h es constante de la de Planck, λInicio inicio de absorción compuesto y c es la velocidad de la luz en el vacío. - Análisis tiempo-Resolved de Potentistatic19,32,33
- Preparar la medición similar al método descrito en 3.1.1–3.1.16 y colocar el tubo de argón (o nitrógeno) (a través de un agujero adicional en el soporte del electrodo) y burbujear la solución por lo menos 5 min antes de la medición. Después, mueva el tubo de argon por encima del nivel de la solución y mantener el flujo de gas durante la medición.
- Ejecutar el software de potenciostato, elegir el procedimiento de chronoamperometry con los siguientes valores: potencial Inicio potencial de 0.00 V, potencial superior a 1.0 V, menor potencial de −1.00 V, duración 5 s, intervalo de tiempo como 0.010 s, número de repeticiones a 10 y el retardo tiempo de 10 s. En la sección datos de la exportación ASCII, definir un nombre de archivo y elija una carpeta para guardar los datos.
- En el software del espectrómetro, seleccionar archivo | Nuevo | Adquisición de alta velocidad. Aparecerá una nueva ventana; configurar los siguientes parámetros: tiempo de integración, 10 ms, exploraciones a media — 1, Boxcar Wirth — 0, número de exploraciones, 10000. Hace clic en el botón ir .
- En el software del potenciostato, pulse el botón iniciar . Cuando la cuenta regresiva desciende a 1 s, pulse el botón Go en el software del espectrómetro.
- Tenga en cuenta que no es posible ver los resultados de absorción hasta que termine el proceso.
- Analizar los datos para proporcionar parámetros. Utilizar la información guardada en el archivo de dicha transmisión, corriente, voltaje y densidad de corriente para proporcionar parámetros de trabajo:
- Calcular la densidad óptica (ΔOD), la absorbancia de la prueba del compuesto durante el doping (dedoping) con la ecuación 4:
 (4)
(4)
donde Tb transmitancia en forma dopada, un estado de "bleach" [%] y Tc es transmitancia en forma de lámpara, un estado de "color" [%]. - Calcular la eficiencia de coloración (CE, η), la cantidad de electrocrómico color formado por la cantidad usada de carga y como la conexión entre la carga inyectada/expulsado como una función de área de electrodo (Qd) y el cambio en la densidad óptica (ΔOD) valor en una determinada longitud de onda (λmáx.) con la ecuación 5. Su valor depende de la longitud de onda elegida para el estudio, por lo que el valor se mide a λmax de la banda de absorción óptica del estado color.
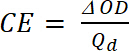 (5)
(5)
donde ΔOD es la densidad óptica [-] y Qd es la densidad de la carga [C/cm2]. - Calcular la relación de contraste (CR), la intensidad medida del cambio de color durante el dopaje electroquímico y caracterizada generalmente por λmáx de la forma coloreada con ecuación 6:
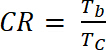 (6)
(6)
donde Tb transmitancia en forma dopado, "bleach" estado [%] y Tc es transmitancia en forma de lámpara, "color" estado [%].
- Calcular la densidad óptica (ΔOD), la absorbancia de la prueba del compuesto durante el doping (dedoping) con la ecuación 4:
 (3)
(3) (4)
(4)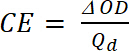 (5)
(5)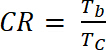 (6)
(6)4. EPR electroquímicas análisis
- Preparación de la medición
- Encienda el espectrómetro y el potenciostato.
- Llenar la celda electroquímicas con electrolito (paso 1.1).
- Conjunto el poder de 1 mW y tune el resonador para conseguir una señal fuerte, centrada en Q-dip (utilice la opción autotuned).
- Establece el marcador de Mn (manganeso estándar) en 600 y cerrar la ventana de Q-dip. Si no hay ningún marcador de Mn, luego saltear este paso y vaya al paso 4.8.
- Establecer "barrido anchura ±" en 2.5´ 10 mT, "mod ancho ±" 1.0´0.1 mT, "amplitud" para 5´100, la "constante de tiempo «a 0,03 s y"campo"valor a ca. 338 mT y comenzar la medición.
- Si no registrando seis líneas espectrales de marcador Mn, luego cambiar el valor "center field".
- Al registrar seis líneas espectrales del marcador de la Mn (figura 4), establezca el valor "center field" entre tercera y cuarta línea y disminuir el valor de "barrido anchura ±" por un valor que cubre sólo estas dos líneas. Iniciar la medición otra vez.
- Ir en la opción de Q-Dip, el marcador de Mn se establece en 0 y cerrar la ventana de Q-dip.
- Medir el electrolito puro como telón de fondo y comprobar si hay cualquier señal. Si las señales son visibles, la señal puede ser la contaminación, compuestos potenciales de oxidación bajo, o el vidrio.
- Limpie la celda electroquímicas (paso 1.3).
- Investigación de moléculas pequeñas (soluciones).
- Llenar la celda electroquímicas con un diluido (1´10−5 M) solución de la prueba en el electrolito; poner todos los tres electrodos a través del soporte del electrodo a la celda de esta manera para que la WE y RE están dentro de la espiral AE (figura 1).
- Puesto que cerca de la parte inferior de la celda y la en el lado superior del activo (metal no recubierto de PTFE) parte de la WE (figura 1). Coloque la celda equipada con un electrodo dentro de la cavidad de la muestra del espectrómetro y conectar los electrodos con un potenciostato (seguir la información sobre la conexión de cada cable del potenciostato a su respectivo electrodo).
- Aplicar el potencial correspondiente al inicio del primer pico de redox (valor de medición de la CV). Si aparece una señal, entonces el equipo está correctamente configurado.
- Ir a la opción Q-Dip, definir el marcador Mn a 600, cerrar la ventana del baño Q y comenzar la medición. El registro de la señal con la señal del marcador de la Mn permite calcular g-factor de la señal recibida.
- Ir a la opción Q-Dip, establezca el marcador Mn 0, cerrar la ventana del baño Q y esperar ca. 5 min.
- Establezca el valor "center field" en el centro de la señal, disminuir el valor de "barrido anchura ±" por un valor que cubre la señal, pero no interrumpe el inicio y final de la señal (cuando se cambia el "± ancho de barrido", toda señal de control) y la disminución el valor de "m OD ancho ± "para obtener espectros bien resueltos.
- Si la señal es pequeña, entonces aumentar el valor de "amplitud" y "tiempo" (adquisición) de la medida.
- Para disminuir la relación señal a ruido, establece la adquisición en 4, 9 o 16 dependiendo cómo ruidoso de la señal. Si la señal es muy ruidosa, seleccione 16.
- Llenar la celda electroquímicas con un diluido (1´10−5 M) solución de la prueba en el electrolito; poner todos los tres electrodos a través del soporte del electrodo a la celda de esta manera para que la WE y RE están dentro de la espiral AE (figura 1).
- Investigaciones de la capa polimérica depositado en la superficie de la WE.
- Llenar la celda electroquímicas con una solución electrolítica (paso 1.1).
- Colocar electrodos en la celda. Tenga cuidado de no destruir la capa polimérica en la WE. Conectar el electrodo con el potenciostato (paso 4.2.1).
- Aplicar el potencial neutro (es decir 0.00 V) para que el compuesto esté en un Estado neutral; este espectro será los partida espectros EPR.
- Aumentar potencial 0.10 V. esperar aprox. 10 s hasta que se estabilice el proceso y registrar los espectros EPR.
- Repita el paso 4.3.4 cuando aparece la señal EPR.
- Aumentar el potencial de 0.05 V. esperar aprox. 10 s hasta que se estabilice el proceso y registrar los espectros EPR.
- Repetir paso 4.3.6 hasta el primer o segundo potencial de oxidación se consigue y así sucesivamente (los valores de estos potenciales son conocidos de medición de la CV); a continuación, invertir el potencial y volver a partir de potenciales.
- Aplicar el potencial que el EPR señal apareció en el ciclo anterior (4.3.5), ir a la opción Q-Dip, definir el marcador Mn a 600, cerrar la ventana del baño Q y comenzar la medición. Registrar la señal con la tercera y cuarta línea espectral del marcador de Mn (4.1.7).
- Cuando se haya completado la medición, análisis de datos.
- Algunos de los análisis como un número de vueltas se puede realizar solamente para los compuestos depositan (polímeros insolubles) en la WE. Para calcular el número de giros, doble de integrar la señal EPR, seguida de una comparación del valor obtenido con el calibrado estándar interno de Mn. Calcular el número de unidades que forman el polímero sobre el electrodo mediante la ecuación 724,32,33:
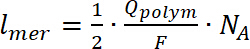 (7)
(7)
donde lmer – número de unidades de repitentes en el polímero, polímeros de Q es la carga de la polimerización, F es la constante de Faraday y NA es el número de Avogadro. - Para calcular la carga de dopaje, utilizar el CV de polímero depositado en una celda electroquímicas antes de la medición de APE, utilizando la ecuación 8:
 (8)
(8)
Qd Dónde está la acusación de dopaje, i es la corriente, E es el conjunto potencial, E0 el potencial inicial, v es la frecuencia de barrido, y e es la carga elemental. - Utilizar carga dopaje calculado para calcular el nivel de dopaje, utilizando la ecuación 9:
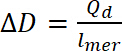 (9)
(9)
donde ∆D- nivel, Qd el dopaje es la carga de dopaje y lmer – número de unidades en el polímero - Para calcular el número de bipolarons, asumir a un dopaje inicial nivel igual a cero. Utilice el número de polarons por unidad de la concentración de tiradas, por lo que el número de bipolarons debe calcularse usando ecuación 10. En los cálculos, suponga que la cantidad inicial de bipolarons es cero y que corriente faradaic es mucho mayor que la de corriente no faradaic.
 (10)
(10)
donde z es la carga elemental del portador de la carga, nies el número de portadores de la carga por unidad de polímero (p) es el polarons y (b) es el bipolarons. - Extracto de la g-factor de valor de los espectros EPR. Determinar utilizando Mn como estándar interno (pasos 4.2.3 y 4.3.8), sabiendo que la cuarta línea de manganeso en él tiene un g-factor de 2.03324. Calcular el ancho de línea de señal de EPR como la distancia en metros entre el mínimo y máximo de la señal EPR. Este valor es importante ya que le diga donde (en que átomo) se forman los radicales.
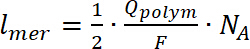 (7)
(7) (8)
(8)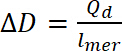 (9)
(9) (10)
(10)Representative Results
La aplicación más común de análisis de CV es la estimación de IP y EA. A pesar de que hay dos formas para obtener datos de CV, se recomienda calcular basado en la aparición de picos de redox (figura 3). Este enfoque permite unificar el procedimiento de cálculo. No todos los materiales probados se someten a procesos de oxidación/reducción reversible (figura 3); en estos casos, no es posible calcular basado en promedio (promedio de potenciales corresponden a un máximo de reducción catódica y anódica oxidación picos) los potenciales. Sin embargo, casi siempre es posible ejecutar la tangente a un pico como se muestra en la figura 3. Con la intersección con la línea de fondo y el uso de las ecuaciones 1 y 2, los valores de IP y EA se estimaron como 5,35 eV y −2.90 eV, respectivamente. También hay varias escalas diferentes utilizados para evaluar la IP y EA basado en las mediciones de la CV. Los más utilizados escalas de materiales orgánicos son −4.8 y −5.1 eV como equivalente a 0.00 V electrodo Normal versus el hidrógeno (NHE). Sin embargo, todas las escalas son sólo una aproximación; Recuerde esto cuando se comparan diferentes resultados. Lo crucial es qué parámetros se consideraron para los cálculos del estado. En este caso, el valor −5.1 eV ha sido elegido como debe corresponder al potencial formal del par ferroceno redox; en la escala de Fermi, es 0.40 V versus electrodo de Calomel saturado (SCE) en acetonitrilo, que está de acuerdo con la medición anterior.
Hay muchos artículos publicados sobre el análisis del CV adjunto, os mostramos cuando el proceso no va a ser como se espera. El análisis se basa en derivados de tiofeno: NtVTh (estructura que se muestra en la figura 4), que sufre una degradación por oxidación (figura 5).
NtVTh tiene dos potenciales de oxidación: la primera en 0.70 V y la segunda en 0.84 V (figura 5). Durante el primer ciclo, el pico de reducción no se observó, indicando un proceso irreversible. Características electroquímicas de NtVTh muestran que la polimerización no ocurre después de la primera oxidación potencial, algunos electro-inactivo capa de productos de la reacción se deposita en la superficie del electrodo, por lo tanto, lo que dificulta el proceso de polimerización. Lo que es visible es la reacción con el catión radical sobre el vínculo de vinilo, donde la molécula pierde sus Conjugación y forma dímeros en el electrodo.
Si bien es difícil extraer información de portadores de la carga de la CV, es posible distinguir entre polarons y los bipolarons cuando apoyado por un espectrómetro UV-Vis-NIR. El poli neutral (OiPrThEE) se caracterizó por dos amplia absorción π | π * transición de bandas con los máximos picos en λmax1 = 363 nm y λmax2 = 488 nm, relacionados con la forma aromática del derivado polythiophene lámpara. Durante el dopaje oxidativo, se generan nuevas bandas polaronic y bipolaronic. El espectro UV-Vis obtenido durante la oxidación de poly (OiPrThEE) reveló la disminución de la banda de absorción del polímero neutro (300-550 nm) (figura 6) junto con la formación de una nueva banda de absorción (550-950 nm) de los cationes radicales de bithiophene y p- phenylenevinylene con maxima en 692 nm. El punto isosbestic del proceso de oxidación se encuentra en 604 nm. La banda de bipolaronic apareció entre los 950 nm y 1700 nm con un máximo situado en 1438 nm.
Espectroscopia EPR es la técnica que detecta materiales con un electrón desapareado, lo que incluye los radicales orgánicos39. Existen varios parámetros que se podrían extraer de espectros EPR, pero uno de los más interesantes es estimar donde se localizan los radicales. Electrones, similares a los protones, poseen spin. Al colocar un electrón en un campo magnético externo, este giro puede dividirse dos maneras: paralela y antiparalela al campo magnético, dando dos niveles de energía. Este fenómeno se conoce como el efecto de Zeeman40,41. En el caso de los radicales orgánicos, el electrón desapareado interactúa no sólo con el campo magnético externo, sino también con los núcleos magnéticos (núcleos que tienen un spin distinto de cero; I≠0). Un número de niveles de energía degenerados es igual a 2I + 1, donde I es el número del quántum de la vuelta del núcleo que el electrón desapareado interactúa42. La interacción del electrón desapareado con un mayor número de núcleos magnéticos conduce a partir adicional de los niveles de energía y estructura hiperfina del EPR espectros Registro43 (figura 7).
Para las moléculas donde el electrón desapareado interactúa con un mayor número de núcleos, la línea espectral individual podía se superponen, que resulta en el registro de una señal única, amplio44,45,46 (figura 8). esto es típico de los polímeros conjugados, donde el ion radical generado durante un proceso redox es deslocalizadas47,48.
La combinación de espectroscopia EPR con métodos electroquímicos permite la caracterización de los portadores de carga (ion radical) generados durante el proceso de redox así como la determinación del mecanismo de estos procesos49,50 . Si bien resuelto (picos están separados; no en la forma de un amplio pico) se registran los espectros, como en el caso de la reducción electroquímica de s -derivado tetrazina (figura 7), a continuación, el análisis de la estructura hiperfina de los espectros conduce a conclusiones acerca de la localización del electrón desapareado. Una forma de analizar este tipo de espectros es para llevar a cabo la simulación con software especial y espectros simulados con los experimentales un51. Esto es especialmente útil cuando la estructura hiperfina es compleja debido a la interacción de los electrones no apareados con un gran número de protones. Simulación de espectros EPR (línea roja) en el caso del s- tetrazina derivado que se muestra en la figura 7, indica que la interacción del electrón desapareado con cuatro átomos de nitrógeno de s- tetrazina.

Figura 1: electroquímica y células electroquímicas utilizadas para medidas de. La figura presenta la configuración del esquema de las células electroquímicas/electroquímicas mediante voltametría cíclica, ultravioleta-visible e infrarrojo cercano (UV-Vis-NIR) y electrón resonancia paramagnética (EPR) electroquímicas mediciones. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: voltametría cíclica (CV) de COPO1 compuesto correctamente medido (a) y el CV con una inestable de referencia potencial dibenzothiophene-S, S-dióxido con ferroceno (b)52. La figura muestra dos voltagramas cíclicos. (a) presenta correctamente registrada CV y (b) muestra un voltagrama registrado con un electrodo de referencia no estables posibles. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: voltametría cíclica (CV) de COPO1 compuesto en una amplia gama de potenciales. Estimación de los potenciales de inicio para los cálculos de IP y EA de la COPO1 compuestos de52. EA = −2.90 eV y IP = 5,35 eV. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4. EPR seis línea espectral de manganeso standard. La señal paramagnética de manganeso utilizado para la calibración de la cambio de la señal. Haga clic aquí para ver una versión más grande de esta figura.

Figura 5: voltametría cíclica (CV) del compuesto NtVTh. Voltamperogramas cíclicos de 15 mM NtVTh en 0.1 M Bu4NBF4/CH3CN y estándar de ferroceno que presenta el proceso de degradación en el electrodo de trabajo. Tarifa de exploración 0.05 V/s: (una) fue tomada en la gama −1.4 V a 0.8 V y (b) en la gama −1.4 V a 0,9 V. haga clic aquí para ver una versión más grande de esta figura.

Figura 6: ultravioleta-visible e infrarrojo cercano (UV-Vis-NIR) spectroelectrochemistry de derivados de polietileno (OiPrThEE). Espectros UV-Vis-NIR que presenta la evolución de la banda de absorción a través de la generación de portadores de la carga sobre la estructura del polímero. Haga clic aquí para ver una versión más grande de esta figura.

Figura 7: análisis EPR electroquímicas de derivados tetrazina. (a) estructura del s- tetrazina derivado; (b) espectros EPR registran durante la reducción electroquímica de s- tetrazina derivado (espectro simulado de línea negra de la línea experimental y rojo). Haga clic aquí para ver una versión más grande de esta figura.

Figura 8: señal de polaron EPR electroquímicas del polímero conjugado. Espectros de resonancia paramagnética (EPR) electrónica registran durante el primer paso de la oxidación del polímero conjugado (espectros EPR de polaron especies). Haga clic aquí para ver una versión más grande de esta figura.

Figura 9: voltametría cíclica (CV) de ferroceno y decamethylferrocene. Comparación de dos normas electroquímicas como puro y mezcla que muestra el cambio del potencial. Haga clic aquí para ver una versión más grande de esta figura.
Discussion
Técnicas electroquímicas y electroquímicas no tienen limitaciones; se pueden analizar el estado sólido y líquidas soluciones en una amplia gama de temperatura y otras condiciones con estas técnicas. Lo importante en todos estos casos es que los compuestos y materiales se analizan bajo el potencial aplicado, replicar las condiciones reales de trabajo dispositivos electrónicos orgánicos. La única diferencia es que en electroquímica, se observa la formación de portadores de carga.
Los métodos presentados aquí muestran la utilidad del análisis de portadores cargados en compuestos orgánicos que se correlacionan con su aplicabilidad en electrónica orgánica. Por otra parte, las técnicas electroquímicas y electroquímicas son más baratos y menos exigentes que la de los métodos típicos usados en análisis de portador de carga, pero hay algunos pasos críticos y modificaciones al Protocolo de que se necesitan dependiendo de la resultados obtenidos.
Durante la caracterización electroquímica, comience siempre con una concentración particular. Si se compara con un conjunto de los compuestos, todos los materiales deben tener la misma concentración molar. Lo mejor es empezar con concentración de 1 mM y 50 mV/s tasa de lectura como se indica en el protocolo de este estudio, pero es bueno saber la concentración de la muestra en el comportamiento electroquímico observado. Trate siempre de medir al menos tres análisis. Las dos primeras exploraciones son generalmente diferentes porque son diferentes las condiciones iniciales (equilibrio). El segundo y el tercer análisis deben ser el mismo. Si la segunda y tercera son la misma, no hay probablemente ningún lado las reacciones observadas en este sistema (Figura 2a). En un proceso de oxidación, un nuevo pico en un menor potencial aparece mostrando que el material conductor se depositó en el18,19,24,25,29,30 , 31 , 32. Si aumenta la altura del pico más bajo en las exploraciones sucesivas, entonces probablemente el polímero conjugado fue depositado18,19,24,25,29 , 30 , 31 , 32. Si todas las corrientes disminuyen en las exploraciones sucesivas, entonces el producto no conductora de degradación fue depositado en el electrodo. Si se observa un pico muy pequeño antes de la oxidación principal o pico de reducción (especialmente para los polímeros), entonces esto es probablemente carga captura proceso19,23,31,34. Si se observa un pico muy agudo dedoping de oxidación o reducción, entonces esto es probablemente causado por la descomposición de estructuras cristalinas en un electrodo formado a través del proceso de electrocristalitzación durante la oxidación35.
Siempre verifique el comportamiento de la prueba compuesta antes, durante y después de picos de redox. Esto significa que se deben registrar al menos tres análisis de CV: con la parte superior (en el caso oxidación) o menor potencial de vértice superior o inferior, respectivamente, entonces el potencial máximo de pico, con potencial de vértice superior o inferior establecido en exactamente en el pico máximo y con vértice los potenciales más altos (oxidación) y menor (reducción) que el potencial del máximo pico. El proceso observado puede variar y algunas veces dos procesos pueden observarse bajo un pico teóricamente. Comparar siempre los voltamperogramas cíclicos recogidos del electrólito (paso 2.6), el ferroceno (paso 2.9), compuesto (paso 2.13) y el ferroceno con compuesto (paso 2,19); Hay varias cuestiones a tener en cuenta.
Siempre comparar las señales de la CV del electrolito y la prueba de compuesto, si las señales desde el electrolito es visible en el voltagrama cíclico del compuesto medido, entonces el electrolito debe cambiarse porque su ventana electroquímico es demasiado baja, o la electrolito está contaminado. Si la señal (par redox) de ferroceno (paso 2.9) y el ferroceno con compuesto (paso 2.19) están en la misma posición, entonces todo se realiza correctamente. Si los picos se desplazan entre sí, entonces Compruebe la RE y repita la medición. Si la señal (oxidación, reducción o par redox potencial) de la prueba de compuesto con agregado ferroceno (paso 2.19) estará a un potencial más alto que el de la pura compuesto (paso 2.13), entonces considera los valores (oxidación, reducción o redox pareja potencial) de el voltagrama cíclico del compuesto puro. El cambio es causado por la mayor cantidad de ferroceno en la solución. Cuando se observan dos procesos de oxidación, el primer proceso (oxidación o reducción) que está siempre en el que puede afectar la superficie activa; Esto puede provocar un aumento en el potencial de oxidación del segundo proceso (figura 9).
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
Los autores reconocen con agradecimiento el apoyo financiero del proyecto "Excilight" "Donador aceptor luz emisor Exciplexes como materiales para fácil-a-sastre relámpago OLED ultra eficiente" (H2020-MSCA-ITN-2015/674990) financiado por Marie Skłodowska-Curie Acciones dentro del programa marco de investigación y las innovaciones "Horizonte 2020".
Materials
| Name | Company | Catalog Number | Comments |
| Potentiostat | Metrohm | Autolab PGSTAT100 | |
| EPR | JEOL | JES-FA200 | |
| UV-Vis detector | Oceanoptics | QE6500 | |
| NIR detector | Oceanoptics | NIRQuest | |
| Dichloromethane (DCM) | Sigma-Aldrich | 106048 | |
| Tetrabutylammonium tetrafluoroborate (Bu4NBF4) | Sigma-Aldrich | 86896 | |
| 2-propanol, 99.9% | Sigma-Aldrich | 675431 | |
| Acetone, 99.9% | Sigma-Aldrich | 439126 | |
| Ultrasonic Bath | Elma | S30H | |
| Tetrahydrofuran >99.9% | Sigma-Aldrich | 401757 | |
| ferrocene >98% | Sigma-Aldrich | F408 | |
| decamethylferrocene >97% | Sigma-Aldrich | 378542 |
References
- O'Brien, D. F., Baldo, M. A., Thompson, M. E., Forrest, S. R. Improved energy transfer in electrophosphorescent devices. Applied Physics Letters. 74, 442-444 (1999).
- Chin, B. D., et al. Effects of interlayers on phosphorescent blue organic light-emitting diodes. Applied Physics Letters. 86, 133505-133507 (2005).
- Cardon, C. M., Li, W., Kaifer, A. E., Stockdale, D., Bazan, G. C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bang, A. -M., et al. A novel random terpolymer for high-efficiency bulk-heterojunction polymer solar cells. RSC Advances. 7, 1975-1980 (2017).
- Tybrandt, K., Kollipara, S. B., Berggren, M. Organic electrochemical transistors for signal amplification in fast scan cyclic voltammetry. Sensors and Actuators B: Chemical. 195, 651-656 (2014).
- Schaur, S., Stadler, P., Meana-Esteban, B., Neugebauer, H., Sariciftci, N. S. Electrochemical doping for lowering contact barriers in organic field effect transistors. Organic Electronics. 13, 1296-1301 (2012).
- Mullen, K., Scherf, U. Organic Light Emitting Devices: Synthesis, Properties and Applications. , Wiley-VCH. Weinheim. (2006).
- Monk, P. M. S., Mortimer, R. J., Rosseinsky, D. R. Electrochromism and Electrochromic Devices. , Cambridge University Press. Cambridge. (2007).
- Skotheim, T. A., Elsenbaumer, R. L., Reynolds, J. R. Handbook of Conducting Polymers. , Marcel Dekker. New York. (1998).
- Data, P., et al. Kesterite Inorganic-Organic Heterojunction for Solution Processable Solar Cells. Electrochimica Acta. 201, 78-85 (2016).
- Data, P., Pander, P., Okazaki, M., Takeda, Y., Minakata, S., Monkman, A. P. Dibenzo[a,j]phenazine-Cored Donor-Acceptor-Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters. Angewandte Chemie International Edition. 55 (19), 5739-5744 (2016).
- Jankus, V., et al. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Advanced Functional Materials. 24 (39), 6178-6186 (2014).
- Data, P., et al. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. Journal of Physical Chemistry C. 120 (4), 2070-2078 (2016).
- Dias, F. B., et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science. , (2016).
- Okazaki, M., et al. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo[a,j]phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chemical Science. 8, 2677-2686 (2017).
- Data, P., et al. Efficient p-phenylene based OLEDs with mixed interfacial exciplex emission. Electrochimica Acta. 182, 524-528 (2015).
- Goushi, K., Yoshida, K., Sato, K., Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nature Photonics. 6, 253-258 (2012).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of heteroaryl group on electrochemical and spectroscopic properties of conjugated polymers. Electrochimica Acta. 83, 271-282 (2012).
- Data, P., Motyka, M., Lapkowski, M., Suwinski, J., Monkman, A. Spectroelectrochemical Analysis of Charge Carries as a Way of Improving Poly(p-phenylene) Based Electrochromic Windows. Journal of Physical Chemistry C. 119, 20188-20200 (2015).
- Enengl, C., et al. Explaining the Cyclic Voltammetry of a Poly(1,4-phenylene-ethynylene)-alt-poly(1,4-phenylene-vinylene) Copolymer upon Oxidation by using Spectroscopic Techniques. ChemPhysChem. 18, 93-100 (2017).
- Gudeika, D., et al. Hydrazone containing electron-accepting and electron-donaiting moieties. Dyes Pigments. 91, 13-19 (2011).
- Swist, A., Cabaj, J., Soloducho, J., Data, P., Lapkowski, M. Novel acridone-based branched blocks as highly fluorescent materials. Synthetic Metals. 180, 1-8 (2013).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of alkyl chain on electrochemical and spectroscopic properties of polyselenophenes. Electrochimica Acta. 87, 438-449 (2013).
- Laba, K., et al. Electrochemically induced synthesis of poly(2,6-carbazole). Macromolecular Rapid Communications. 36, 1749-1755 (2015).
- Pluczyk, S., et al. Unusual Electrochemical Properties of the Electropolymerized Thin Layer Based on a s-Tetrazine-Triphenylamine Monomer. Journal of Physical Chemistry C. 120, 4382-4391 (2016).
- Blacha-Grzechnik, A., Turczyn, R., Burek, M., Zak, J. In situ Raman spectroscopic studies on potential-induced structural changes in polyaniline thin films synthesized via surface-initiated electropolymerization on covalently modified gold surface. Vibrational Spectroscopy. 71, 30-36 (2014).
- Laba, K., et al. Diquinoline derivatives as materials for potential optoelectronic applications. Journal of Physical Chemistry C. 119, 13129-13137 (2015).
- Enengl, S., et al. Spectroscopic characterization of charge carriers of the organic semiconductor quinacridone compared with pentacene during redox reactions. Journal of Materials Chemistry C. 4, 10265-10278 (2016).
- Data, P., et al. Electrochemically Induced Synthesis of Triphenylamine-based Polyhydrazones. Electrochimica Acta. 230, 10-21 (2017).
- Brzeczek, A., et al. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigments. 113, 640-648 (2015).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Electrochemistry and spectroelectrochemistry of a novel selenophene-based monomer. Electrochimica Acta. 59, 567-572 (2012).
- Data, P., et al. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochimica Acta. 122, 118-129 (2014).
- Data, P., et al. Evidence for Solid State Electrochemical Degradation Within a Small Molecule OLED. Electrochimica Acta. 184, 86-93 (2015).
- Data, P., et al. Unusual properties of electropolymerized 2,7- and 3,6- carbazole derivatives. Electrochimica Acta. 1282, 430-438 (2014).
- Etherington, M. K., et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nature Communications. 8, 14987 (2017).
- Trasatti, S. The absolute electrode potential: an explanatory note. Pure and Applied Chemistry. 58, 955-966 (1986).
- Cardona, C. M., et al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bredas, J. -L. Mind the gap! Mater Horiz. 1, 17-19 (2014).
- Gerson, F., Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals. , (2003).
- Brustolon, M., Giamello, E. Electron Paramagnetic Resonance. A Practitioner's Toolkit. , John Wiley & Sons Inc. New Jersey. (2009).
- Weil, J., Bolton, J. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. , John Wiley & Sons, Inc. (2007).
- Rieger, P. H. Electron Spin Resonance. Analysis and Interpretation. , The Royal Society of Chemistry. (2007).
- Roznyatovskiy, V. V., Gardner, D. M., Eaton, S. W., Wasielewski, M. R. Radical Anions of Trifluoromethylated Perylene and Naphthalene Imide and Diimide Electron Acceptors. Organic Letters. 16, 16-19 (2013).
- Pluczyk, S., Kuznik, W., Lapkowski, M., Reghu, R. R., Grazulevicius, J. V. The Effect of the Linking Topology on the Electrochemical and Spectroelectrochemical Properties of Carbazolyl Substituted Perylene Bisimides. Electrochimica Acta. 135, 487-494 (2014).
- Ledwon, P., et al. A Novel Donor-acceptor Carbazole and Benzothiadiazole Material for Deep Red and Infrared Emitting Applications. Journal of Materials Chemistry C. 4, 2219-2227 (2016).
- Heinze, J., Frontana-Uribe, B. A., Ludwigs, S. Electrochemistry of Conducting Polymers-Persistent Models and New Concepts. Chemical Reviews. 110, 4724-4771 (2010).
- Kurowska, A., Kostyuchenko, A. S., Zassowski, P., Skorka, L., Yurpalov, V. L., Fisyuk, A. S., Pron, A., Domagala, W. Symmetrically Disubstituted Bithiophene Derivatives of Properties. Journal of Physical Chemistry C. 118, 25176-25189 (2014).
- Zotti, G., Schiavon, G., Zecchin, S., Morin, J. F., Leclerc, M. Electrochemical, Conductive, and Magnetic Properties of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules. 35, 2122-2128 (2002).
- Kaim, W., Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chemical Society Reviews. 38, 3373-3382 (2009).
- Duling, D. R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. Journal of Magnetic Resonance, Series B. 104, 105-110 (1994).
- Benson, C. R., et al. Multiplying the Electron Storage Capacity of a Bis-Tetrazine Pincer Ligand. Dalton Transactions. 43, 6513-6524 (2014).
- Nobuyasu, R. S., et al. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Effi ciency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Advanced Optical Materials. 4, 597-607 (2016).
