

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
De geclusterde regelmatig interspaced korte palindromische herhalingen/CRISPR geassocieerde eiwit 9 (CRISPR/Cas9) systeem biedt een veelbelovend instrument voor de gentechnologie, en opent de mogelijkheid van gerichte integratie van transgenen. We beschrijven een homologie-gemedieerde einde deelname aan (HMEJ)-gebaseerd strategie voor efficiënte DNA integratie in vivo gerichte en gerichte gentherapieën met behulp van CRISPR/Cas9.
Abstract
Als een veelbelovende genoom bewerken platform heeft het CRISPR/Cas9-systeem een groot potentieel voor efficiënte genetische manipulatie, met name voor de gerichte integratie van transgenen. Echter, als gevolg van het lage rendement van homologe recombinatie (HR) en verschillende indel mutaties van niet-homologe einde deelname aan (NHEJ)-op basis van strategieën in niet-delende cellen, in vivo genoom bewerken blijft een grote uitdaging. Hier beschrijven we een homologie-gemedieerde einde deelname aan (HMEJ)-CRISPR/Cas9 systeem voor efficiënte in-vivo nauwkeurig gerichte integratie gebaseerd. In dit systeem, de gerichte genoom en de donor vector met homologie wapens (~ 800 bp) geflankeerd door één gids RNA (sgRNA) doel gekloofd sequenties zijn door CRISPR/Cas9. Deze HMEJ gebaseerde strategie realiseert efficiënte transgenic integratie in muis zygoten, evenals in hepatocyten in vivo. Bovendien, een HMEJ gebaseerde strategie biedt een efficiënte aanpak voor correctie van fumarylacetoacetate hydrolase (Fah) Mutatie in de hepatocyten en redt Fah-deficiëntie geïnduceerde lever mislukking muizen. Samen genomen, gericht op integratie gericht, deze HMEJ gebaseerde strategie biedt een veelbelovende instrument voor een verscheidenheid van toepassingen, waaronder generatie transgene diermodellen en gerichte gentherapieën.
Introduction
Nauwkeurige, gerichte genoom bewerken wordt vaak vereist voor de productie van genetisch gemodificeerde diermodellen en klinisch therapieën. Veel inspanning heeft geleverd om verschillende strategieën voor efficiënte gerichte genoom bewerken, zoals zink vinger nuclease (ZFN), transcriptie activator-achtige effector nucleasen (TALENs), en CRISPR/Cas9 systemen. Deze strategieën gerichte DNA dubbele-strand einden (DSB) maken in het genoom, en te profiteren van de intrinsieke DNA repair systemen, zoals homologe recombinatie (HR)1,2, microhomology-gemedieerde einde deelname aan (MMEJ)3 , 4 , 5en niet-homologe einde deelname aan (NHEJ)6,7,8 te induceren gerichte integratie van transgenen1,9. De HR gebaseerde strategie is momenteel de meest gebruikte genoom bewerken benadering, die zeer efficiënt in cellijnen, maar niet gemakkelijk toegankelijk zijn voor niet-delende cellen als gevolg van deze beperkte zich voordoet in de late S/G2-fase is. De HR gebaseerde strategie geldt dus niet voor in vivo genoom bewerken. Onlangs, is de NHEJ gebaseerde strategie ontwikkeld voor efficiënte gene knock in muis weefsels8. Niettemin, introduceert de NHEJ gebaseerde methode meestal microdeleties op de kruispunten, waardoor het moeilijk voor het genereren van precieze genoom bewerken, vooral wanneer het proberen om te bouwen in-frame fusion genen8. MMEJ-gebaseerde gerichte integratie is geschikt voor het bewerken van de precieze genoom. Echter verhoogt het slechts bescheiden de efficiëntie van de gerichte integratie in vorige verslagen5. Verbetering van de efficiëntie van precies gerichte integratie in vivo is daarom dringend nodig voor brede therapeutische toepassingen3.
In een onlangs gepubliceerde werk, we toonden een homologie-gemedieerde einde deelname aan (HMEJ)-op basis van de strategie, waaruit bleek de hoogste efficiëntie gerichte integratie in alle gerapporteerde strategieën beide in vitro en in vivo10. Hier beschrijven we een protocol voor de oprichting van het HMEJ-systeem, en ook de bouw van de vectoren van single-gids RNA (sgRNA) gericht op het gen van belang en de donor vectoren sgRNA target-sites herbergen en ~ 800 bp homologie wapens (Figuur 1) . In dit protocol beschrijven we ook de gedetailleerde stappen voor generatie van DNA knock-in muizen en korte stappen voor gerichte integratie in weefsels in vivo. Bovendien, een studie van de proof-of-concept van de HMEJ gebaseerde strategie aangetoond haar vermogen te corrigeren Fah mutatie en Fah- / - lever mislukking muizen, die verder geopenbaard zijn therapeutisch potentieel te redden.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Alle procedures, met inbegrip van dierlijke onderwerpen zijn goedgekeurd door het biomedische onderzoek ethisch comité bij de Shanghai instituten voor biologische wetenschap (CA's).
1. ontwerp van Donor plasmiden
-
Selectie van sgRNA
- Online CRISPR Ontwerptools gebruiken om te voorspellen sgRNAs op de doelgroep regio11,12,13,14,15. Ontwerpen voor de locus Cdx2 , zes verschillende sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) rond de stop codon met hogere rang en lagere potentieel uit-doelen (Figuur 1A)16.
- Linearize 2 µg Cas9-CMV-EGFP expressievectoren en sgRNA door BbsI spijsvertering (1 µL van BbsI gedurende 2 uur bij 37 ° C op een eindconcentratie van 1 U/µL in een volume van 20 µL). Vervolgens het zuiveren van het product door gel zuivering kit met een 1% agarose gel in 1 × TAE-buffer.
- Mix een paar sgRNA oligonucleotides in 10 µL van 1 × T4 DNA ligase buffer om een eindconcentratie van 50 µM. Incubeer de oligo-oplossing met behulp van de gradiënt van een temperatuur van 95 ° C tot 25 ° C met een temperatuur verandering tarief van 5 ° C/5 min (95 ° C gedurende 5 min dan 90 ° C gedurende 5 min, 85 ° C gedurende 5 min, enz.), die zal de oligos gloeien.
- Mix 4 µL van gegloeid product, 2 µL van de gelineariseerde vector met 1 µL van T4 DNA ligase in 10 µL van 1 × T4 DNA ligase buffer, en vervolgens afbinden bij 22 ° C gedurende 1-2 h (Figuur 1B).
-
Landmeter nuclease kwantitatieve analyse van sgRNA
Opmerking: De targeting efficiëntie van de sgRNA gebruikt voor het experiment knock-in wordt geëvalueerd door landmeter nuclease assay (ook bekend als T7 endonuclease ik (T7EI) assay)17. Selecteer het sgRNA met DNA decollete hoogrenderende en een lage afstand tussen de sgRNA snijden site en de stop codon.- Transfect Cas9-sgRNA-EGFP expressievectoren in N2a cellijnen gekweekte in DMEM aangevuld met foetale runderserum 10%, 1% PSG, en 1% niet-essentiële aminozuur door de transfectie kit (Zie Tabel van materialen). Incubeer bij 37 ° C in 5% CO2transfected cellen.
- Na 48u van incubatie, verzamelen 5.000 transfected cellen (GFP+) door fluorescentie-activated cell sorting (FACS) met behulp van niet-transfected cellen als een besturingselement.
- Verteren de verzamelde cellen in 2-5 µL oflysis buffer (0,1% Triton X-100, 0,1% Tween-20 en 100 µg/mL proteïnase K) op 56 ° C gedurende 30 minuten en vervolgens hitte inactivering van proteïnase K bij 95 ° C gedurende 10 min.
- Versterken van het monster door genestelde PCR (tabel 1) met behulp van de fabrikant-protocol. De grootte van PCR producten is ingesteld op 300-500 bp.
- Voeg 1 µL van lysis product met polymerase van DNA en een paar buitenste inleidingen herkennen de volgorde rond de doelsite sgRNA (0.1 µM, eindconcentratie) (tabel 1), en het uitvoeren van de primaire PCR in een volume van 20 µL.
- Activeren van de polymerase van DNA bij 95 ° C gedurende 5 minuten, en het uitvoeren van de primaire PCR voor 30 cycli bij 95 ° C gedurende 30 s, 60 ° C gedurende 30 s, en 72 ° C gedurende 24 s (1 min/1 kb), met een definitieve verlenging bij 72 ° C gedurende 5 minuten.
- De secundaire PCR met 1 µL van primaire PCR product en een paar genestelde inleidingen van de innerlijke uitvoeren
- Denatureren en opnieuw anneal 300-600 ng van gezuiverde PCR product in 20 µL van 1 × T7EI reactie buffer (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT pH 7,9) met behulp van een verloop van de temperatuur van 95 ° C tot 25 ° C met een tarief van 5 ° C/5 min.
- Voeg 1 µL van T7EI enzym aan de gegloeide PCR producten en digest bij 37 ° C gedurende 2 uur. Voer de spijsvertering product op 2% agarose gel in 1 × TAE-buffer op 120 V voor 40 minuten, totdat de fragmenten worden gescheiden (Zie Tabel van materialen).
- ImageJ gebruiken om te bepalen van de band intensiteiten van knippen en Onbesneden DNA. Bereken de frequentie van de indel met behulp van de methoden zoals eerder gemeld9 (Figuur 1C).
-
Bouw van donor vector
Opmerking: Voor het genereren van HMEJ donor vectoren voor Cdx2 gen, bouwen een donor DNA (800 bp HAL-p2A-mCherry-800 bp HAR) geflankeerd door 23 nt Cdx2-sgRNAs gericht op volgorde aan beide uiteinden (Figuur 1D en Figuur 1E). De PAM van doel sequentie was grenzend aan het einde van de homologe arm. Gibson vergadering wordt aanbevolen voor HMEJ donor klonen.- Versterken van de 800 bp links homologie arm (HAL) met voorwaartse primer-1F (met 15-20 nt overlapping opeenvolging van vector, 23 nt Cdx2-sgRNA gericht op volgorde, en ongeveer 20 nt volgorde van HAL) en reverse primer-1R (met 15-20 bp overlapping sequence p2A-mCherry en ongeveer 20 nt volgorde van HAL) op 0.1 µM eindconcentratie gebruikend muis genomic DNA op 200 ng/µL (Figuur 1D, tabel 1).
- Versterken van het p2A-mCherry invoeging fragment met voorwaartse primer-2F (met 15-20 nt overlapping opeenvolging van HAL en ongeveer 20 nt volgorde van invoeging fragment) en reverse primer-2R (met 15-20 nt overlapping opeenvolging van HAR en ongeveer 20 nt sequence de invoeging fragment) op 0.1 µM-eindconcentratie genomic DNA of de plasmide met verslaggever reeksen op 100 ng/µL of 30 ng/µL (Figuur 1D, tabel 1).
- Versterken van de 800 bp juiste homologie arm (HAR) met voorwaartse primer-3F (met 15-20 nt overlapping opeenvolging van vector, 23 nt Cdx2-sgRNA gericht op volgorde, en ongeveer 20 nt volgorde van HAR) en reverse primer-3R (met 15-20 nt overlapping sequence p2A-mCherry en ongeveer 20 nt volgorde van HAR) op 0.1 µM eindconcentratie gebruikend muis genomic DNA op 200 ng/µL (Figuur 1D, tabel 1).
- Voer alle de PCR producten op 1% agarose gel in 1 × TAE-buffer en zuiveren van de PCR-producten van verwachte grootte door gel extractie kit, volgens de instructies van de fabrikant (tabel 1).
- Het verteren van 50-100 ng van de vector van een constructie met door KpnI en XbaI. Meng 2 µL van de gelineariseerde vector bij 30-40 ng/µL met drie PCR versterkt fragmenten (1 µL voor elke, 100-200 ng/µL) in 2 x Gibson mix. Toevoegen van H2O om het uiteindelijke volume op 10 µL.Incubate de mix bij 50 ° C gedurende 60 minuten.
- Transformeren van bevoegde E. coli cellen met alle geassembleerde product en extract de plasmide met DNA-extractie kit volgens de instructies van de fabrikant construeert. Controleer of de HMEJ donor door DNA sequentiebepaling.
2. genoom bewerken in muis embryo's met behulp van de HMEJ gebaseerde methode
-
Productie van Cas9 mRNA
- Voor Cas9 mRNA voorbereiding, voegt u de T7 promotor volgorde naar de Cas9 regio codering door PCR versterking met behulp van de juiste primer paar vermeld in tabel 1. De primer Cas9 F/R toevoegen om een eindconcentratie van 0.1 µM en 20 ng van Cas9 uiten van vector naar 1 × high-fidelity mix van de polymerase van DNA. Het uiteindelijke volume op 50 µL met H2O.
- Activeren van de polymerase van DNA bij 95 ° C gedurende 5 minuten, en het uitvoeren van PCR voor 36 cycli bij 95 ° C gedurende 30 s, 60 ° C gedurende 30 s, en 68 ° C gedurende 4 min (1 min/1 kb), met de laatste extensie bij 68 ° C gedurende 10 minuten.
- Zuiveren van T7-Cas9 PCR product voor in vitro transcriptie (IVT), en vervolgens het transcriberen van 0,5-1 µg DNA door mRNA transcriptie kit bij 37 ° C gedurende 8 uur in een totaal volume van 20 µL, volgens de instructies van de fabrikant (Zie Tabel van materialen).
- Voeg 1 µL van DNase aan het mengsel te verwijderen van de DNA-sjabloon bij 37 ° C gedurende 15 min. toevoegen een poly-A-staart gedurende 45 minuten bij 37 ° C en herstellen van de Cas9 mRNA door RNA zuivering kit, volgens de instructies van de fabrikant (Zie Tabel van materialen).
-
Productie van sgRNA
- Het genereren van de sjabloon van de sgRNA gedreven door een promotor van de T7 met high-fidelity de polymerase van DNA als hierboven. Kies een sgRNA steiger met vector als de sjabloon. De inleidingen gebruikt zijn vermeld in tabel 1.
- Zuiveren van de T7-sgRNA PCR-product en gebruik van 0,5-1 µg DNA als de sjabloon voor in vitro transcriptie van sgRNA met behulp van een korte RNA transcriptie kit bij 37 ° C gedurende 6 uur in een totaal volume van 20 µL, volgens de instructies van de fabrikant (Zie tabel van materialen < / c11 >).
- Voeg 1 µL van DNase aan het mengsel en blijven de incubatie bij 37 ° C gedurende 15 minuten te verwijderen van de DNA-sjabloon. De sgRNAs zuiveren door RNA zuivering kit, als hierboven (Zie Tabel van materialen).
- Verdun de sgRNA tot 500 ng/µL in RNase-gratis water en opslaan van de monsters bij −80 ° C voor maximaal 3 maanden.
Opmerking: CRISPR ribonucleoproteins (RNPs) zijn een alternatieve substitutie met een beter snijden efficiëntie18,19,20.
-
Embryo collectie, microinjection en in vitro cultuur
- Superovulate vrouwelijke B6D2F1 (C57BL/6 × DBA2J) muizen (7-8 weken oud) door een zwangere merrie serum gonadotrofinen (PMSG), gevolgd door humaan choriongonadotrofine (hCG) 48 uur later. Na de hCG-inspuiting, huis vrouwtjes met B6D2F1 mannetjes overnachting.
- Het offeren van de vrouwtjes door CO2 anesthesie, 24 uur na de hCG-inspuiting. De bevruchte embryo's van hun oviducts (met 30-50 embryo's voor elke vrouwelijke) in M2 medium verzamelen.
- Plaats de bevruchte embryo's (ongeveer 300 eieren voor eendaagse injectie) tot KSOM medium (5.55 g/L NaCl, KCl, 0.05 g/L KH2PO4, 0.05 g/L MgSO4•7H2O, 0,04 g/L Glucose, 1.12 g/L natrium 0,19 g/L lactaat, 2,1 g/L NaHCO3 , 0,02 g/L natrium pyruvaat, 0,25 g/L CaCl2•2H2O 0,004 g/L EDTA, 0.146 g/L L-glutamine en 1 g/L bovien serumalbumine) bij 37 ° C in een broedstoof met 5% CO2.
- Mix Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL), HMEJ donor vector (100 ng/µL) en voeg H2O aan te passen het eindvolume 10 µL. doe het mengsel op het ijs.
- Trek capillaire naalden (buitendiameter 1,0 mm, binnendiameter 0.78 mm met gloeidraad) met een trekker van de micropipet (parameters: warmte, 74; trekken, 60; snelheid, 80, vertraging, 200, druk, 300. Zie tabel van materialen). Commerciële naalden zou een alternatieve vervanging voor de microinjection.
- Injecteren van een waarschijnlijke volume van het mengsel in het cytoplasma van zygoten met welomschreven pronuclei in een druppel van HEPES-CZB medium met 5 µg/mL cytochalasin B met behulp van een microinjector met constante stroom instellingen(Figuur 2)(Zie Tabel van materialen)21.
Opmerking: Elke groep zygoten moet worden geïnjecteerd in 20-30 min. Cytochalasin B kon verhogen de levensvatbaarheid van muis zygote na injectie. Als alternatief, microinjection kan bediend worden met het piëzo-systeem, zoals eerder beschreven22. - Cultuur van de geïnjecteerde zygoten in KSOM medium bij 37 ° C minder dan 5% CO,2 tot het blastocyst stadium na 3,5 dagen voor fluorescentie observatie (cijfers 2B en 2C).
-
Embryotransfer en generatie van muizen
- Metgezel Oestrische ICR vrouwelijke muizen met vasectomized ICR mannelijke muizen op dezelfde dag als injectie.
- De geïnjecteerde zygoten cultuur de fase 2-cel bij 37 ° C onder 5% CO2, en overdracht 25-30 2-cel-embryo's in oviducts van pseudopregnant ICR vrouwtjes op 0,5 dag post coitum (dpc). Ontvangende moeders leveren pups bij 19.5 dpc.
-
Genotypering van Mouse
- Uittreksel muis genomic DNA van teen of staart monsters met behulp van een DNA-extractie kit, volgens de instructies van de fabrikant (Zie Tabel van materialen).
- De 5' en 3' kruising van knock-in evenementen met behulp van 200-400 ng van genomic DNA gemeten door UV/vis-spectrometrie als een sjabloon voor het uitvoeren van de PCR-amplification te identificeren.
- Activeren van de polymerase van DNA bij 95 ° C gedurende 5 minuten, en het uitvoeren van PCR voor 38 cycli bij 95 ° C gedurende 30 s, 60 ° C gedurende 30 s, en 72 ° C gedurende 1 minuut (1 min/1 kb), met de laatste extensie bij 72 ° C gedurende 10 minuten. Voor de 5' kruising, gebruik de voorwaartse primer op de upstream voor de HAL, met de omgekeerde op het fragment knock-in (p2A-mCherry). Over 3' junction, gebruiken de voorwaartse primer op het fragment knock-in (p2A-mCherry), met de omgekeerde op de downstream van de HAR (tabel 1).
- 6 µL van het PCR-product worden uitgevoerd op 1% agarose gel in 1 × TAE-buffer en controleren voor de grootte van de verwachte fragment. Controleer hen door DNA sequencing (Figuur 2D).
3. op HMEJ gebaseerde In Vivo genoom bewerken in hepatocyten
- Plaats van ontvangende C57BL/6J muis (8 weken) in een matigende apparaat en zet de staart door de gleuf.
- Meng HMEJ donor vectoren (30 µg) en spCas9 expressievectoren (30 µg) in 2 mL zuiver zoutoplossing. Voor de controle-experiment, schorten HMEJ donor vectoren (30 µg) in 2 mL zuiver zoutoplossing (Figuur 3A).
- Reinig de muis staart met 70% ethanol. Invoegen de naald in de staart ader en injecteren de plasmide DNA oplossing binnen 5-7 s. Verwijder de naald en laat de muisknop los van de beteugeling apparaat.
- Offeren muizen door CO2 verdoving na 5-9 dagen na de injectie. Perfuse de transcardially van de muizen met zoutoplossing 0,9%, gevolgd door 4% paraformaldehyde gebruik een peristaltische pomp en 's nachts herstellen van de lever bij 4 ° C.
- Uitdrogen van het weefsel met 30% sacharose 's nachts, totdat het zinkt naar de bodem van de buis.
- Sectie het bevroren weefsel bij een dikte van 10 µm voor lever monsters.
- Spoel de secties driemaal in 0,1 M fosfaatgebufferde (PB) en ze uit te broeden met primair antilichaam: anti-mCherry konijn (in 5% verdunde NGS) overnachting bij 4 ° C.
- Wassen van secties driemaal in PB, en ze vervolgens uit te broeden met secundair antilichaam: Cy3-AffiniPure geit anti-konijn IgG gedurende 2 uur bij kamertemperatuur op een roteerschudapparaat.
- Counterstain in de secties met de DAPI voor 20 min en zware montering met glycerine op glas dia's voor verdere fluorescentie observatie (Figuur 3B).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
HMEJ gebaseerde genoom bewerken in muis embryo's: Als u wilt definiëren de efficiëntie knock-in van de HMEJ gebaseerde methode in muis zygoten, verlost we Cas9 mRNA, sgRNA gericht op het Cdx2 -gen en de HMEJ donor in muis zygoten, die werd ontworpen om een gen verslaggever p2A-mCherry aan de laatste codon van de van Cdx2 fuse gen(Figuur 2). De geïnjecteerde zygoten uitgegroeid tot blastocysten in de cultuur. Om te beoordelen van de knock-in efficiëntie, geanalyseerd wij de fluorescentie van mCherry met een fluorescente microscoop, en we vonden dat 12,9% van de ontvangen HMEJ donoren blastocysten waren positief voor mCherry, die strikt in de trophectoderm uitgedrukt was (cijfers 2B 2 C). Door de sequencing van het PCR positieve muizen, vonden wij ook dat alle onderzocht integratie evenementen werden nauwkeurig in-frame integraties bij zowel 5' en 3' kruisingen (Figuur 2D).
HMEJ gebaseerde genoom bewerken in volwassen weefsels en HMEJ-gemedieerde gentherapie: Om te onderzoeken of het bewerken van de HMEJ gebaseerde genoom kan worden toegepast in volwassen weefsels, ingevoegd we de cassette mCherry vlak voor de stop codon van Actb -gen door transducing Actb- HMEJ constructies aan C57/B6J muis levers door staart-veneuze hydrodynamische injectie (Figuur 3A). Na 7 dagen van injecties vonden we dat bijna de helft van de transfected hepatocyten mCherry uitgedrukt als gekleurd op de lever secties (Figuur 3B).
Verkennen van de mogelijkheid van het gebruik van een HMEJ gebaseerde strategie voor gentherapie, wij werkzaam fumarylacetoacetate hydrolase (Fah)-deficiënte muizen. De Fah- / - mouse is een gevestigde erfelijke tyrosinemia typ ik (HTI) muismodel, die havens een invoeging fragment in exon 5 van de Fah -gen, waardoor frameshift mutaties in de volgende volgorde23. Als u wilt behouden Fah- / - muizen, we getrakteerd de Fah- / - muizen met een inhibitor van de omwenteling van de upstream van tyrosine katabole traject, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Hier zetten we uit om te zien of de MMEJ - en HMEJ-gemedieerde gene correctie Fah mutatie in de Fah- / - muis kon redden. We hydrodynamica geïnjecteerd Cas9 construct samen met Fah- MMEJ of Fah- HMEJ constructies, ontworpen voor het invoegen van Fah cDNA van 5 tot en met 14 in intron 4 van Fah gen, exon Fah - / - muis levers () Figuur 3 C). een week na de injectie, NTBC werd ingetrokken voor het opwekken van leverschade (Figuur 3C). Na de intrekking van NTBC, Fah-gecorrigeerde hepatocyten van de Fah- / - muizen ontvangen Fah- HMEJ en Cas9 constructies bleek doeltreffender proliferatie dan MMEJ gebaseerde methode (Figuur 3D ).

Figuur 1 : HMEJ-gemedieerde gerichte integratie in vitro.
De experimentele regeling (A) voor de selectie van sgRNAs: zes verschillende sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) rond de stop codon van het Cdx2 locus met een hoger rang en af-target potentieel werden gekozen op basis van online CRISPR ontwerp gereedschap. De volgorde van de aangrenzende motief (PAM) protospacer is in het rood. (B) experimenteel design: The Cas9-CMV-GFP expressie plasmiden uiting van sgRNA, Cas9 en GFP N2a cellen werden binnengebracht. GFP+ cellen werden gesorteerd op dag 3 voor landmeter assay. (C) Surveyor assay voor Cdx2 targeting: 6 verschillende sgRNAs werden ontworpen voor landmeter assay. Normaal N2a cel genomic DNA dient als controle. *, de sgRNA gebruikt voor Cdx2-2A-mCherry knock-in de experiment. (D) schematisch overzicht van de bouw van donoren van de HMEJ met behulp van Gibson vergadering. (E) schematisch overzicht van HMEJ-gemedieerde gen targeting strategie op Cdx2 locus. HAL/HAR, links/rechts homologie arm; driehoeken, sgRNA target-gebieden; VAN / OR, buitenste vooruit-/ achteruitspoelen primer; Als / IR, innerlijke vooruit-/ achteruitspoelen primer. Figuur aangepast ten opzichte van de vorige verslag-10. Klik hier voor een grotere versie van dit cijfer.

Figuur 2 : Genoom bewerken in muis embryo's via HMEJ-gemedieerde gerichte integratie
(A) experimentele regeling van microinjection: een mengsel van Cas9 mRNA (100 ng/µL), sgRNA (50 ng/µL) en donor plasmiden (100 ng/µL) werden ingespoten in muis zygoten. (B) vertegenwoordiger fluorescentie beelden van muis embryo's bewerkt door de strategie van de HMEJ. Bar, 20 µm. (C) Knock-in efficiëntie aangeduid met percentage van mCherry+ blastocysten. Nummer boven elke bar, totale blastocysten geteld. (D) sequentie analyse van gen-bewerkt muizen op Cdx2 locus. PCR producten versterkt van 5' en 3' junction sites werden gesequentieerd. Homologie bovenarm; paarse, p2A; rood, mCherry; HAR of HAL, rechts of links homologe arm. Onderbroken lijnen markeren de regio weggelaten voor duidelijkheid. Figuur aangepast ten opzichte van de vorige verslag-10. Klik hier voor een grotere versie van dit cijfer.

Figuur 3 : HMEJ-gemedieerde gerichte integratie in vivo.
(A) schematisch overzicht van de hydrodynamische staart veneuze injectie. Een mengsel van plasmiden donor sequentie en sgRNA en plasmiden uiting van spCas9 werden geleverd aan de lever via hydrodynamische staart veneuze injectie. (B) vertegenwoordiger immunofluorescentie beelden van hepatocyten. De lever secties waren verzameld 7 dagen post injectie. Schaal bar, 50 µm. GFP, transfected cellen. (C) plasmiden van beide MMEJ - of HMEJ-gemedieerde gene vervanging strategie die is ontworpen om Fah cDNA van exon 5 tot en met 14 invoegen intron 4 van Fah gen werden geleverd in Fah- / - muis levers door hydrodynamische injectie. NTBC op: Fah- / - muizen werden bijgehouden op NTBC water; NTBC uit: intrekking van NTBC water (de eerste dag van de terugtrekking van de NTBC werd gedefinieerd als dag 0, dat de 7e dag na injectie is). (D) Fah immunohistochemistry kleuring van lever secties van Fah- / - muizen ingespoten met MMEJ of HMEJ plasmiden. Schaal bar, 100 µm. figuur aangepast ten opzichte van eerdere verslagen5,10. Klik hier voor een grotere versie van dit cijfer.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De meest kritische stappen in de bouw van HMEJ donor plasmiden zijn: (1) de selectie van de sgRNA met hoge DNA decollete procesefficiëntie en de lage afstand tussen sgRNA snijden site en stop codon, en (2) goede bouw van HMEJ donor. CRISPR/Cas9-gemedieerde decollete op beide transgenic donor vector (met sgRNA target sites en ~ 800 bp homologie wapens) en gerichte genoom is noodzakelijk voor een efficiënte en nauwkeurige gerichte integratie in vivo. De belangrijkste stappen van de generatie van knock-in muizen met behulp van de HMEJ gebaseerde methode zijn: (1) het opstellen van kwalitatief hoogstaande Cas9 mRNA en sgRNA (geen degeneratie bestaat in Cas9 mRNA en sgRNA), en (2) de opstelling van de hoge kwaliteit HMEJ donor plasmide. Het plasmide bevat geen toxische effecten op de embryonale ontwikkeling.
Onlangs, had ook een NHEJ gebaseerde methode gemeld voor efficiënte in-vivo genoom8bewerken. Verschillende soorten indel mutaties werden echter meestal veroorzaakte op de kruispunten, zoals beschreven in de vorige verslagen8, waardoor het moeilijk om precieze integratie. Hier bleek de HMEJ gebaseerde strategie die we hierboven beschreven nauwkeurig gerichte integratie met nauwelijks indel mutaties. Dus zou een HMEJ gebaseerde strategie een ideaal platform voor het vervangen van een gemuteerde reeks (zoals een punt mutatie) met de juiste is, die is niet van toepassing voor NHEJ gebaseerde methode.
Mozaïcisme is een groot probleem voor het bewerken van het gen in embryo's. Injectie van Cas9 eiwitten in plaats van mRNA in een eerder embryonale stadium kan bereiken transgenic knock-in één cel stadium zonder mozaïcisme. Voor klinische toepassingen is de levering van de CRISPR/Cas9-systemen in volwassen weefsels nog uitdagend.
Er zijn veel toekomstige potentiële toepassingen van genoom HMEJ gebaseerde bewerken. Het kan worden gebruikt voor het genereren van transgene diermodellen. Gezien haar knock-in hoogrenderende in embryo's, deze methode het dierlijke nummer nodig voor het genereren van transgene diermodellen aanzienlijk kan verminderen, en met name de mogelijkheid van het genereren van niet-menselijke primaten genetische modellen opent. HMEJ gebaseerde genoom bewerken kunt lineage trace individuele celtypes in volwassen weefsel, die is vooral handig voor dierlijke modellen, aangezien er een gebrek aan beschikbare dierlijke modellen, zoals niet-menselijke primaten. Het kan worden gebruikt voor gerichte gentherapieën: de meest aantrekkelijke toepassing van een HMEJ gebaseerde strategie is gentherapie voor kliniek gebruikt. In deze studie, we de Fah mutatie van erfelijke tyrosinemia type gecorrigeerd ik muizen door hydrodynamische injectie van de aangegeven vectoren. Levering van het systeem van CRISPR/Cas9 in volwassen weefsels is echter nog steeds de grote technische uitdaging voor klinisch gebruik, zoals hydrodynamische injectie is het onwaarschijnlijk moet worden uitgevoerd bij patiënten. Momenteel, is verbetering van de levering-strategie verder dringend nodig vóór het vertalen van deze HMEJ gebaseerde methode in de kliniek.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
De auteurs hebben niets te onthullen.
Acknowledgments
Dit werk werd gesteund door CAS strategische prioriteit Research Program (XDB02050007, XDA01010409), de nationale Hightech R & D-programma (863 programma; 2015AA020307), de nationale Natural Science Foundation van China (NSFC verleent 31522037, 31500825, 31571509, 31522038), China jeugd duizend talenten programma (HY), Break via project van Chinese Academie van Wetenschappen, project Shanghai City Comité van wetenschap en technologie (16JC1420202 aan HY), het ministerie van wetenschap en technologie van China (de meeste; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Genetica kwestie 133 CRISPR/Cas9 gerichte integratie homologie-gemedieerde einde toetreden In Vivo Embryo gemodificeerde genetisch muizen hydrodynamische injectieErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
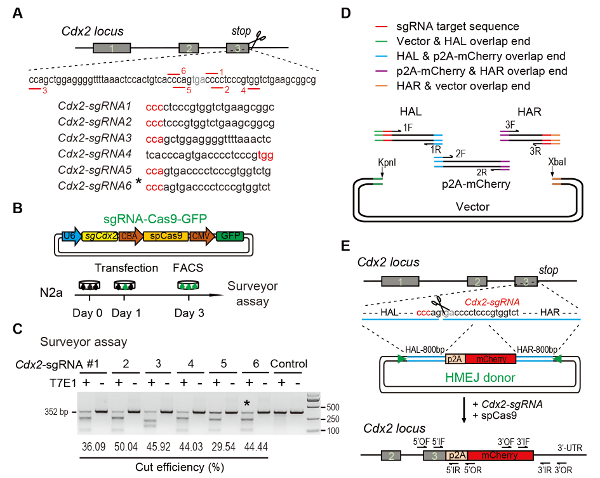
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
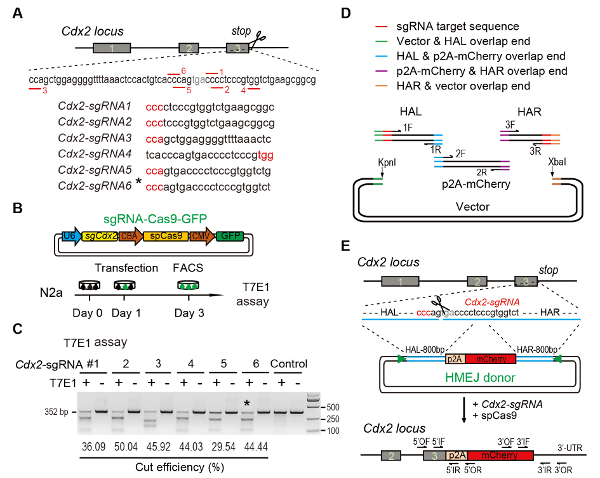
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
