

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Le cluster régulièrement entrecoupées de protéine de courtes répétitions/CRISPR palindromique associé 9 (CRISPR/Cas9) système fournit un outil prometteur pour le génie génétique et ouvre la possibilité d’une intégration ciblée des transgènes. Nous décrivons une fin médiée par homologie rejoindre (HMEJ)-basé de stratégie pour ADN efficace visant l’intégration en vivo et ciblé des thérapies géniques en utilisant CRISPR/Cas9.
Abstract
Comme une plateforme de montage prometteurs du génome, le système CRISPR/Cas9 a un grand potentiel pour des manipulations génétiques efficace, surtout pour l’intégration ciblée des transgènes. Cependant, en raison de la faible efficacité de la recombinaison (HR) et différentes mutations indel de fin non homologue rejoindre (NHEJ)-base de stratégies dans les cellules non-divisant, in vivo du génome édition reste un grand défi. Nous décrivons ici une fin médiée par homologie rejoindre (HMEJ)-CRISPR/Cas9 système d’intégration ciblées précis efficace in vivo . Dans ce système, le génome ciblé et le donateur vecteur contenant armes d’homologie (~ 800 bp) flanqué de cible de RNA (sgRNA) guide unique séquences sont clivés par CRISPR/Cas9. Cette stratégie axée sur les HMEJ réalise intégration du transgène efficace zygotes de souris, ainsi que dans les hépatocytes in vivo. En outre, une stratégie HMEJ propose une approche efficace pour la correction de la fumarylacétoacétate hydrolase (Fah) mutation dans les hépatocytes et sauve Fah-carence induite par souris d’insuffisance hépatique. Pris ensemble, en se concentrant sur ciblé intégration, cette stratégie HMEJ fournit un outil prometteur pour une variété d’applications, y compris la génération de modèles animaux génétiquement modifiés et les thérapies géniques ciblées.
Introduction
Montage précis et ciblé du génome est souvent requis pour produire des modèles animaux génétiquement modifiés et thérapies cliniques. Beaucoup s’est efforcé de développer diverses stratégies pour génome ciblé efficace, édition, comme la nucléase de doigt de zinc (ZFN), nucléases effecteur comme activateur de transcription (TAPS) et les systèmes CRISPR/Cas9. Ces stratégies de créer des cassures double brin de l’ADN (DSB) dans le génome et tirent parti des systèmes de réparation de l’ADN intrinsèques, tels que la recombinaison (HR)1,2, microhomology-mediated fin rejoindre (MMEJ)3 , 4 , 5et à la fin non homologue rejoindre (NHEJ)6,7,8 pour provoquer l’intégration ciblée des transgènes1,9. La stratégie axée sur les ressources humaines est actuellement le plus utilisé du génome édition approche, qui est très efficace dans les lignées cellulaires, mais pas facilement accessible aux non-diviser des cellules en raison de son occurrence limitée à la fin de la phase S/G2. Ainsi, la stratégie axée sur les ressources humaines n’est pas applicable pour l’édition de génome en vivo . Récemment, la stratégie axée sur la NHEJ a été développée pour le gène efficace knock-in dans la souris tissus8. Néanmoins, la méthode de NHEJ introduit généralement indels au niveau des jonctions, rendant difficile générer l’édition génome précis, surtout lorsque vous essayez de construire dans le cadre de fusion de gènes8. MMEJ l’intégration ciblée est capable de génome précis édition. Toutefois, elle n'augmente que modestement l’efficacité de l’intégration ciblée dans les précédents rapports5. Par conséquent, l’amélioration de l’intégration ciblée précise en vivo est absolument nécessaire pour des applications thérapeutiques large3.
Dans un ouvrage récemment publié, nous avons démontré une fin médiée par homologie rejoindre (HMEJ)-basé de stratégie, qui a montré la plus grande efficacité de l’intégration ciblée dans des stratégies signalés tous les deux in vitro et in vivo10. Ici, les auteurs décrivent un protocole portant création du système HMEJ, et aussi la construction des vecteurs RNA (sgRNA) single-guide ciblant le gène d’intérêt et le donateur vecteurs hébergeant sites de cibles sgRNA et ~ 800 bp d’armes homologie (Figure 1) . Dans ce protocole, nous décrivons également les étapes détaillées pour la génération de souris knock-in DNA et brèves étapes pour une intégration ciblée dans les tissus en vivo. Par ailleurs, une étude de validation de la stratégie axée sur les HMEJ a démontré sa capacité à corriger la mutation Fah et sauvetage Fah- / - foie échec souris, qui de plus a révélé son potentiel thérapeutique.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Toutes les procédures, y compris les sujets animaux ont été approuvés par le Comité d’éthique de recherche biomédicale dans les instituts de Shanghai pour les sciences biologiques (CAS).
1. conception des plasmides de donateurs
-
Sélection de sgRNA
- Utiliser des outils de conception en ligne CRISPR pour prédire les sgRNAs sur la cible région11,12,13,14,15. Design pour le locus Cdx2 , six différents sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) autour de l’arrêt codon avec potentiel plus élevé de rang inférieur et le hors-cibles (Figure 1A)16.
- Linéariser 2 µg de vecteurs d’expression Cas9-CMV-EGFP et sgRNA par digestion BbsI (1 µL de BbsI pendant 2 h à 37 ° C, à une concentration finale de 1 U/µL dans un volume de 20 µL). Alors purifier le produit en kit de purification du gel avec un gel d’agarose de 1 % dans un tampon TAE × 1.
- Mélanger une paire des oligonucléotides sgRNA dans 10 µL de tampon de ligase de 1 × T4 ADN à une concentration finale de 50 µM. Incuber la solution oligo en utilisant un gradient de température de 95 ° C à 25 ° C avec un taux de variation de température de 5 ° C/5 min (95 ° C pendant 5 min puis à 90 ° C pendant 5 min, 85 ° C pendant 5 min, etc..), qui va recuire les oligos.
- Mix 4 µL de produit, 2 µL du vecteur linéarisé avec 1 µL de T4 DNA ligase en 10 µL de tampon de 1 × T4 DNA ligase, de recuit et ligaturer puis à 22 ° C pendant 1 à 2 h (Figure 1B).
-
Dosage de nucléase arpenteur de sgRNA
Remarque : L’efficacité de ciblage de le sgRNA utilisé pour l’expérience de knock-in est évaluée par dosage nucléase arpenteur (également connu sous le nom T7 endonucléase j’ai (T7EI) dosage)17. Sélectionnez le sgRNA avec la haute efficacité de clivage de l’ADN et une faible distance entre le site de découpe de sgRNA et le codon stop.- Transfecter vecteurs d’expression Cas9-sgRNA-EGFP dans les lignées cellulaires N2a cultivées en DMEM additionné de sérum de veau fœtal 10 %, 1 % PSG et acide aminés non-essentiels à 1 % par la transfection nécessaire (voir la Table des matières). Incuber les cellules transfectées à 37 ° C à 5 % de CO2.
- Après 48 h d’incubation, recueillir 5 000 cellules transfectées (GFP+) par cellule activée par fluorescence triant (FACS) à l’aide de cellules non transfectées comme un contrôle.
- Digérer les cellules recueillies dans 2 à 5 µL oflysis un tampon (0,1 % X-100 Triton, 0,1 % Tween 20 et 100 µg/mL protéinase K) à 56 ° C pendant 30 min, et puis la chaleur inactiver protéinase K à 95 ° C pendant 10 min.
- Amplifier l’échantillon par PCR nichée (tableau 1) à l’aide du protocole par le fabricant. La taille des produits PCR est définie à 300-500 PB.
- Mélanger 1 µL de produit de lyse avec ADN polymérase et une paire d’amorces externes reconnaissant la séquence autour du site de cible sgRNA (0,1 µM, concentration finale) (tableau 1) et réaliser la PCR primaire dans un volume de 20 µL.
- Activer l’ADN polymérase à 95 ° C pendant 5 min et effectuer le PCR primaire pour cycles de 30 à 95 ° C pendant 30 s, 60 ° C pendant 30 s et 72 ° C pendant 24 s (1 min/1KO), avec une extension finale à 72 ° C pendant 5 min.
- Effectuer la PCR secondaire à l’aide de 1 µL de produit PCR primaire et une paire d’amorces internes imbriqués.
- Dénaturer et re-recuire 300-600 ng du produit PCR purifié dans 20 µL de 1 tampon de réaction T7EI × (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT pH 7,9) utilisant un gradient de température de 95 ° C à 25 ° C avec un taux de 5 ° C/5 min.
- Ajouter 1 µL d’enzyme T7EI aux produits PCR recuits et digérer à 37 ° C pendant 2 h. Avant d’exécuter le produit de digestion sur gel d’agarose 2 % dans un tampon TAE 1 × à 120 V pendant 40 min jusqu'à ce que les fragments sont séparés (voir Table des matières).
- ImageJ permet de déterminer les intensités de bande de coupe et d’ADN non circonci. Calculer la fréquence indel utilisant les méthodes tel que déjà signalé9 (Figure 1C).
-
Construction du vecteur de donateurs
NOTE : Pour générer des vecteurs donneur HMEJ Cdx2 gène, construire un donneur de l’ADN (bp HAL-p2A-mCherry-800 800 bp HAR) flanqué de 23 nt Cdx2-sgRNAs ciblage séquence aux deux extrémités (Figure 1D etEde la Figure 1). La PAM de la séquence cible était adjacente à l’extrémité du bras homologue. Assemblée de Gibson est recommandée pour les donateurs HMEJ clonage.- Amplifier le bras gauche une homologie de 800 bp (HAL) avec apprêt avant-1F (contenant 15-20 nt chevauchement des séquences du vecteur, 23 nt Cdx2-sgRNA séquence de ciblage et environ 20 séquence nt de HAL) et reverse primer-1R (contenant la séquence de chevauchement de Pb de 15-20 de p2A-mCherry et environ 20 séquence nt de HAL) à la concentration finale de 0,1 µM à l’aide de l’ADN génomique de souris à 200 ng/µL (Figure 1D, tableau 1).
- Amplifier le fragment d’insertion p2A-mCherry avec avant apprêt-2F (contenant la séquence de chevauchement de nt de 15-20 de HAL et environ 20 séquence nt de fragment d’insertion) et reverse primer-2R (contenant la séquence de chevauchement de nt de 15-20 de HAR et environ 20 séquence nt de le fragment d’insertion) à la concentration finale de 0,1 µM en utilisant l’ADN génomique ou plasmide avec des séquences de journaliste à 100 ng/µL ou 30 ng/µL (Figure 1D, tableau 1).
- Amplifier le bras de droite homologie bp 800 (HAR) avec apprêt avant-3F (contenant 15-20 nt chevauchement des séquences du vecteur, 23 nt Cdx2-sgRNA séquence de ciblage et environ 20 séquence nt de HAR) et reverse primer-3R (contenant la séquence de chevauchement de nt de 15-20 de p2A-mCherry et environ 20 séquence nt de HAR) à la concentration finale de 0,1 µM à l’aide de l’ADN génomique de souris à 200 ng/µL (Figure 1D, tableau 1).
- Exécutez tous les produits PCR sur gel d’agarose à 1 % dans 1 × un tampon TAE et purifier les produits PCR de taille attendue par gel kit d’extraction, conformément aux instructions du fabricant (tableau 1).
- Digérer les 50-100 ng d’un vecteur de construction avec KpnI et XbaI. Mix 2 µL du vecteur linéarisé à 30-40 ng/µL avec trois PCR amplification fragments (1 µL de chaque, 100-200 ng/µL) dans 2 x mix Gibson. Ajouter H2O pour ajuster le volume final à 10 µL.Incubate le mélange à 50 ° C pendant 60 min.
- Compétentes d' Escherichia coli de transformer les cellules avec tous les produits assemblés et extrait le plasmide construit par kit d’extraction d’ADN selon les instructions du fabricant. Vérifiez le donateur HMEJ par séquençage de l’ADN.
2. génome édition dans des embryons de souris à l’aide de la méthode axée sur les HMEJ
-
Production d’ARNm Cas9
- Pour la préparation de l’ARNm de Cas9, ajouter la séquence du promoteur T7 à la Cas9 région codante de l’amplification par PCR à l’aide de la paire d’amorces appropriés énumérée au tableau 1. Ajouter l’apprêt Cas9 F/R à une concentration finale de 0,1 µM et 20 ng de Cas9 exprimant vector à 1 × haute fidélité mélange de l’ADN polymérase. Ajuster le volume final à 50 µL avec H2O.
- Activer l’ADN polymérase à 95 ° C pendant 5 min et effectuer le PCR pour 36 cycles à 95 ° C pendant 30 s, 60 ° C pendant 30 s et 68 ° C pendant 4 min (1 min/1KO), avec une extension finale à 68 ° C pendant 10 min.
- Purifier le produit PCR de T7-Cas9 pour la transcription in vitro (IVT) et ensuite transcrire 0,5 à 1 µg ADN par kit de transcription ARNm à 37 ° C pendant 8 h dans un volume total de 20 µL, selon les instructions du fabricant (voir Table des matières).
- Ajouter 1 µL de DNase dans le mélange pour supprimer la matrice d’ADN à 37 ° C pendant 15 min. Ajouter une queue poly-A 45 min à 37 ° C et récupérer l’ARNm Cas9 par kit de purification d’ARN, selon les instructions du fabricant (voir Table des matières).
-
Production de sgRNA
- Générer le modèle de sgRNA moteur par un promoteur T7 avec haute fidélité ADN polymérase comme ci-dessus. Choisissez un échafaudage sgRNA contenant le vecteur comme modèle. Les amorces utilisées sont énumérés au tableau 1.
- Purifier le produit PCR de T7-sgRNA et l’utilisation de 0,5 à 1 µg ADN comme modèle pour la transcription in vitro de sgRNA à l’aide d’un kit de transcription d’ARN court à 37 ° C pendant 6 h dans un volume total de 20 µL, selon les instructions du fabricant (voir Table des matières < / c11 >).
- Ajouter 1 µL de DNase dans le mélange et continuer de l’incubation à 37 ° C pendant 15 min enlever la matrice d’ADN. Purifier le sgRNAs en kit de purification d’ARN, comme indiqué ci-dessus (voir Table des matières).
- Diluer le sgRNA à 500 ng/µL dans de l’eau exempte de RNase et stocker les échantillons à −80 ° C pendant 3 mois.
NOTE : Ribonucléoprotéines CRISPR (RNP) sont une alternative substitution avec un découpage meilleure efficacité18,19,20.
-
Culture collection, microinjection et in vitro d’embryons
- Superovulate B6D2F1 (C57BL/6 × DBA2J) des souris femelles (7-8 semaines) de gonadotrophine sérique de jument gravide (PMSG), suivies par la gonadotrophine chorionique humaine (hCG) 48 h plus tard. Après l’injection d’hCG, maison les femelles avec les mâles B6D2F1 pendant la nuit.
- Sacrifier les femelles en CO2 anesthésie, 24 h après l’injection d’hCG. Recueillir les embryons fécondés de leurs oviductes (avec des embryons de 30 à 50 pour chaque femelle) dans un milieu M2.
- Place les embryons fécondés (environ 300 oeufs pour une journée injection) dans un milieu HLGHW (5,55 g/L de NaCl, 0,19 g/L de KCl, 0,05 g/L KH2PO4, 0,05 g/L MgSO4•7H2O, 0,04 g/L de Glucose, 1,12 g/L Sodium lactate, 2,1 g/L NaHCO3 , pyruvate de Sodium 0,02 g/L, 0,25 g/L CaCl2•2H2O, 0,004 g/L d’EDTA, 0,146 g/L, L-glutamine et 1 g/L d’albumine sérique Bovine) à 37 ° C dans un incubateur avec 5 % de CO2.
- Mix Cas9 ARNm (100 ng/µL), sgRNA (50 ng/µL) et donateurs HMEJ vector (100 ng/µL) et ajouter H2O pour ajuster le volume final à 10 µL. mettre le mélange sur la glace.
- Tirer les aiguilles capillaires (diamètre extérieur 1,0 mm, diamètre intérieur 0,78 mm avec filament) à l’aide d’une Micropipette extracteur (paramètres : chaleur, pression, 300 ; vitesse, 80, 74 ; temporisation/200 ; traction, 60. Voir Table des matières). Les aiguilles commerciales serait une substitution alternative pour la microinjection.
- Injecter un volume probable du mélange dans le cytoplasme des zygotes pronucléus bien définies dans une goutte de milieu HEPES-CZB contenant 5 µg/mL cytochalasine B à l’aide d’un microinjector avec la constante de débit de paramètres (Figure 2A) (voir Table des matières)21.
Remarque : Chaque groupe de zygotes doit être injecté dans les 20-30 min. cytochalasine B pourrait augmenter la viabilité du zygote de souris après injection. Par ailleurs, microinjection peut fonctionner avec le système piezo, comme décrit plus haut22. - Culture les zygotes injectés dans un milieu HLGHW à 37 ° C, moins de 5 % de CO2 jusqu'à ce que le blastocyste étape après 3,5 jours pour observation de fluorescence (Figures 2 b et 2C).
-
Transfert d’embryons et de génération de souris
- S’accouplent œstrus souris femelles ICR avec vasectomisés souris mâles ICR sur le même jour que l’injection.
- Culture les zygotes injectées dans l’étape 2 cellules à 37 ° C sous 5 % CO2et des embryons de 2 cellules de transfert 25-30 dans les oviductes des femelles pseudo-gravides de l’IC à 0,5 jour post coitum (dpc). Mères bénéficiaires fournissent des chiots à 19,5 dpc.
-
Génotypage de souris
- Extraire l’ADN génomique de souris d’orteil ou queue des échantillons à l’aide d’un kit d’extraction d’ADN, selon les instructions du fabricant (voir Table des matières).
- Identifier la jonction 5' et 3' de knock-in les événements à l’aide de 200 à 400 ng d’ADN génomique mesurée par spectrométrie UV/vis en tant que modèle pour effectuer l’amplification PCR.
- Activer l’ADN polymérase à 95 ° C pendant 5 min et effectuer le PCR pour 38 cycles à 95 ° C pendant 30 s, 60 ° C pendant 30 s et 72 ° C pendant 1 minute (1 min/1KO), avec une extension finale à 72 ° C pendant 10 min. Pour la jonction 5', utiliser l’apprêt avant à l’amont de la HAL, avec la marche arrière sur le fragment de knock-in (p2A-mCherry). Quant à 3' junction, utiliser l’apprêt avant sur le fragment de knock-in (p2A-mCherry), avec celui inverse sur l’aval de l’HAR (tableau 1).
- Courir 6 µL du produit PCR sur gel d’agarose de 1 % à 1 tampon TAE × et vérifier la taille des fragments attendus. Puis vérifier leur ADN ordonnançant (Figure 2D).
3. base HMEJ In Vivo génome d’édition dans les hépatocytes
- Placer la souris C57BL/6J bénéficiaire (8 semaines) dans un dispositif de retenue et de mettre la queue dans la fente.
- Mélanger HMEJ donneur vecteurs (30 µg) et des vecteurs d’expression spCas9 (30 µg) dans 2 mL de solution saline. Pour l’expérience de contrôle, suspendre les vecteurs de donateurs HMEJ (30 µg) dans 2 mL de solution saline (Figure 3A).
- Nettoyer la queue de la souris avec l’éthanol à 70 %. Insérer l’aiguille dans la queue de la veine et injecter la solution d’ADN de plasmide visé par l’article 5-7, retirer l’aiguille et relâchez le bouton de la souris depuis le dispositif de retenue.
- Sacrifier les souris en CO2 anesthésie après 5 à 9 jours après l’injection. Perfuse le transcardially de la souris avec la solution saline à 0,9 %, suivie de paraformaldéhyde à 4 % à l’aide d’une pompe péristaltique et fixer le foie pendant la nuit à 4 ° C.
- Déshydrater le tissu à l’aide de 30 % de saccharose du jour au lendemain, jusqu'à ce qu’il coule au fond du tube.
- Section du tissu congelé sur une épaisseur de 10 µm pour les échantillons de foie.
- Rincez les sections trois fois en 0,1 M tamponnée au phosphate (PB) et leur incubation avec l’anticorps primaire : anti-mCherry de lapin (dilué à 5 % NGS) durant la nuit à 4 ° C.
- Laver les sections trois fois dans PB et puis les incuber avec anticorps secondaire : Cy3-AffiniPure chèvre anti-lapin IgG pendant 2 h à température ambiante dans un agitateur orbital.
- Contre-coloration les sections au DAPI pendant 20 min et monter avec de la glycérine sur lames de verre pour une observation de fluorescence (Figure 3B) supplémentaires.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Base HMEJ génome d’édition dans des embryons de souris : Pour définir la knock-in l’efficacité de la méthode axée sur les HMEJ zygotes de souris, nous avons livré Cas9 ARNm, sgRNA ciblant le gène Cdx2 et le donateur HMEJ en zygotes de souris, qui a été conçu pour fusionner un gène rapporteur de p2A-mCherry pour le dernier codon de le Cdx2 gène (Figure 2A). Les zygotes injectés est devenue blastocystes dans la culture. Pour évaluer l’efficacité de knock-in, nous avons analysé la fluorescence mCherry avec un microscope à fluorescence, et nous avons trouvé que 12,9 % des blastocystes recevant des donateurs HMEJ étaient positifs pour mCherry, qui a été strictement exprimé dans le trophectoderme (Figures 2 b 2C). En séquençant souris PCR positifs, nous avons aussi constaté que tous examinés événements d’intégration ont été précis dans le cadre intégrations à la jonction 5' et 3' (Figure 2D).
Base HMEJ génome montage en tissus adultes et thérapie génique médiée par HMEJ : Afin de vérifier si le génome basée sur HMEJ édition pourrait s’appliquer dans les tissus adultes, nous avons inséré la cassette de mCherry juste avant le codon du gène Actb par transduction constructions - HMEJ Actbaux foies de souris C57/B6J de queue veineuse injection hydrodynamique (Figure 3A). Après 7 jours d’injections, nous avons constaté que près de la moitié des hépatocytes transfectés exprimé mCherry comme colorées sur les sections du foie (Figure 3B).
Pour explorer la possibilité d’utiliser une stratégie axée sur les HMEJ pour la thérapie génique, nous avons utilisé la fumarylacétoacétate hydrolase (Fah)-souris déficientes. La souris de- / - Fahest une bien établie tyrosinémie héréditaire de type I modèle murin (HTI), qui abrite un fragment d’insertion dans l’exon 5 du gène Fah , causant des mutations de déphasage dans le suivant de la séquence23. Pour maintenir la Fah- / - souris, nous avons traité les Fah- / - souris avec un inhibiteur de l’amont de la voie catabolique de tyrosine, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Ici, nous partons pour voir si MMEJ et HMEJ-médiée par la correction de gène pourrait sauver Fah mutation chez la souris- / - de Fah. Nous avons injecté hydrodynamique Cas9 construct avec Fah- MMEJ ou des constructions HMEJ - Fah, conçues pour insérer Fah cDNA de l’exon 5 à 14 dans l’intron 4 du gène de la Fah , Fah - / - souris (foies Figure 3 C). une semaine après l’injection, NTBC a été retiré pour induire des dommages au foie (Figure 3C). Après le retrait du NTBC, Fah-corrigé les hépatocytes des souris Fah- / - réception Fah- HMEJ et constructions de Cas9 a montré une prolifération plus efficace que la méthode axée sur les MMEJ (Figure 3D ).

Figure 1 : HMEJ médiation visé integration in vitro.
Le régime expérimental (A) pour la sélection de sgRNAs : Six différents sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) autour du codon stop du locus Cdx2 avec un plus haut rang hors cible potentiel choisi reposaient sur la ligne design CRISPR outil. La séquence de motif adjacent (PAM) de protospacer est en rouge. (B) plan expérimental : plasmides d’expression le Cas9-CMV-GFP exprimant sgRNA, Cas9 et GFP ont été introduits dans les cellules N2a. Cellules GFP+ étaient triés à jour 3 pour l’analyse de l’arpenteur. (C) arpenteur dosage pour le ciblage Cdx2 : 6 sgRNAs différents ont été conçus pour l’analyse de l’arpenteur. ADN génomique de la cellule normale N2a sert de contrôle. *, le sgRNA utilisé pour Cdx2-2 a-expérience de knock-in mCherry. (D) aperçu schématique de la construction des donateurs HMEJ avec Gibson assembly. (E) aperçu schématique des gènes HMEJ ciblant la stratégie Cdx2 locus. Bras d’homologie HAL/HAR, gauche/droite ; triangles, sites de sgRNA de cibles ; DE / OR, amorce de marche avant/marche arrière extérieure ; IF / IR, amorce de marche avant/marche arrière intérieure. Figure modifiée du précédent rapport10. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 2 : Génome édition dans des embryons de souris via HMEJ médiation visé intégration
(A) schéma expérimental de microinjection : un mélange d’ADN messagère Cas9 (100 ng/µL), sgRNA (50 ng/µL) et les donateurs plasmides (100 ng/µL) ont été injectés à des zygotes de souris. (B) les images de fluorescence représentatif des embryons de souris sous la direction de la stratégie de HMEJ. Bar, 20 µm. (C) Knock-in efficacité indiquée par le pourcentage de mCherry+ blastocystes. Nombre au-dessus de chaque barre, des blastocystes totales comptés. (D) analyse de gène-édité souris Cdx2 locus d’ordre. Les produits PCR amplifiés à partir des sites de jonction 5' et 3' ont été séquencés. Bras d’homologie supérieur, "Purple", p2A ; rouge, mCherry ; HAR ou HAL, droite ou gauche bras homologues. Pointillés marquent la région omise pour plus de clarté. Figure modifiée du précédent rapport10. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 3 : HMEJ médiation visé integration in vivo.
(A) aperçu schématique de l’injection dans la veine queue hydrodynamique. Un mélange de plasmides exprimant sgRNA et séquence de donateur et plasmides exprimant les spCas9 ont été livrés à la foie par injection dans la veine queue hydrodynamique. (B) les images d’immunofluorescence représentatif des hépatocytes. Les sections du foie ont été collectées 7 jours post injection. Echelle, 50 µm. GFP, transfected des cellules. (C) plasmides de chaque stratégie de remplacement de gène MMEJ HMEJ-médiée par ou conçu pour insérer Fah cDNA de l’exon 5 à 14 dans intron 4 du gène de la Fah ont été livrés dans le foie de souris Fah- / - par injection hydrodynamique. NTBC sur : souris Fah- / - ont été maintenus sur l’eau NTBC ; NTBC off : prélèvement d’eau NTBC (le premier jour du retrait NTBC a été défini comme jour 0, qui est le 7e jour après l’injection). (D) Fah immunohistochemistry souillant des sections du foie de Fah- / - souris injectées avec des plasmides MMEJ ou HMEJ. De précédents rapports5,10, Echelle, 100 µm. Figure modifiée. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Les étapes plus critiques dans la construction des plasmides de donateurs HMEJ sont : (1) sélection de la sgRNA avec la haute efficacité de clivage de l’ADN et de la faible distance entre site de coupe sgRNA et codon stop (2) bonne interprétation du donneur HMEJ. CRISPR/Cas9-mediated clivage sur les deux vecteurs de donneur transgène (contenant des sites cibles sgRNA et armes de l’homologie ~ 800 bp) et le génome ciblée est nécessaire pour l’intégration ciblée efficace et précise in vivo. Les étapes plus critiques de la génération de souris knock-in à l’aide de la méthode axée sur les HMEJ sont : (1) la préparation de haute qualité de Cas9 ARNm et sgRNA (aucune dégénérescence n’existe en ARNm Cas9 et sgRNA) et (2) la préparation du plasmide donneur HMEJ haute qualité. Le plasmide ne montre aucun effet toxique sur le développement embryonnaire.
Récemment, une méthode axée sur le NHEJ avait également signalée pour efficace in vivo du génome édition8. Néanmoins, différents types de mutations indel sont généralement induites au niveau des jonctions, tel que décrit dans les précédents rapports8, rendant difficile de réaliser l’intégration précise. Ici, la stratégie axée sur les HMEJ que nous décrit ci-dessus a montré précise intégration ciblée avec pratiquement aucun des mutations indel. Ainsi, une stratégie axée sur les HMEJ pourrait être une plate-forme idéale pour le remplacement d’une séquence mutée (par exemple une mutation ponctuelle) avec le bon, qui n’est pas applicable pour la méthode NHEJ.
Un mosaïcisme est un problème majeur pour l’édition de gène chez les embryons. Injection de la protéine Cas9 au lieu de l’ARNm à un plus haut stade embryonnaire peut atteindre transgène knock-in à un stade de cellules sans un mosaïcisme. Pour des applications cliniques, la livraison des systèmes CRISPR/Cas9 dans les tissus adultes est toujours difficile.
Il existe de nombreuses utilisations potentielles futures du génome HMEJ base montage. Il peut être utilisé pour générer des modèles animaux génétiquement modifiés. Compte tenu de sa grande knock-in efficacité chez les embryons, cette méthode pourrait réduire sensiblement le nombre d’animaux nécessaire pour générer des modèles animaux génétiquement modifiés et en particulier ouvre la possibilité de générer des modèles génétiques primate non humain. Axée sur les HMEJ génome édition peut types de cellules individuelles de trace de lignée dans les tissus adultes, qui est particulièrement utile pour des modèles animaux, puisqu’il y a un manque de modèles animaux disponibles, tels que les primates non humains. Il peut être utilisé pour les thérapies géniques ciblées : l’application plus attrayante d’une stratégie axée sur les HMEJ est la thérapie génique à usage clinique. Dans cette étude, nous avons corrigé la mutation Fah de tyrosinémie héréditaire de type j’ai souris par injection hydrodynamique des vecteurs indiqués. Toutefois, la livraison du système CRISPR/Cas9 dans les tissus adultes est encore l’important défi technique pour l’usage clinique, comme injection hydrodynamique est peu susceptible d’être effectuée chez les patients. Actuellement, outre l’amélioration de la stratégie de distribution est absolument nécessaire avant de traduire cette méthode axée sur les HMEJ dans la clinique.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs n’ont rien à divulguer.
Acknowledgments
Ce travail a été soutenu par CAS priorité stratégique Research Programme (XDB02050007, XDA01010409), le National Hightech R & D Program (programme 863 ; 2015AA020307), la Fondation nationale des sciences naturelles de Chine (NSFC accorde 31522037, 31500825, 31571509, 31522038), Chine programme jeunes – mille Talents (HY), Break par le biais de projet de l’Académie des Sciences de Chine, Shanghai ville Comité des sciences et techniques du projet (16JC1420202 à HY), le ministère des sciences et technologies de Chine (la plupart ; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Génétique numéro 133 CRISPR/Cas9 intégration ciblée fin médiée par homologie rejoindre In Vivo embryon souris Injection hydrodynamique génétiquement modifiéesErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
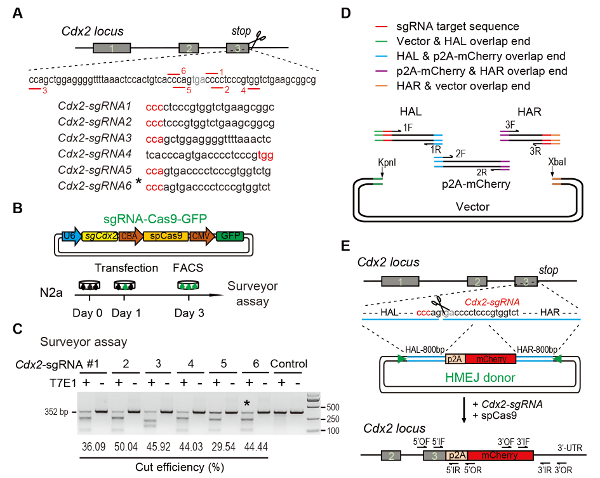
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
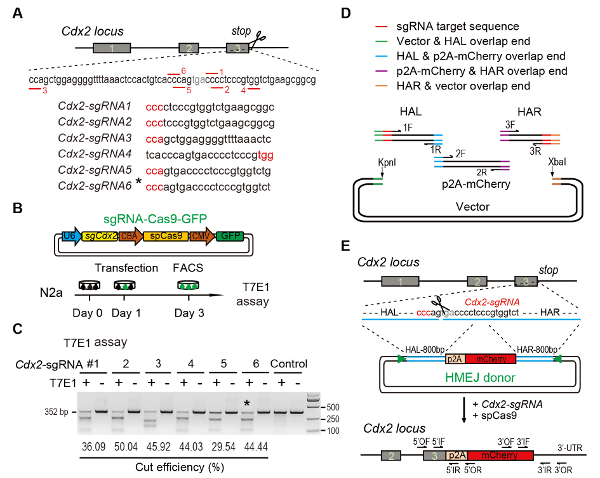
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
