

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Die gruppierten regelmäßig dazwischen kurze palindromische Wiederholungen/CRISPR verbundenen Protein 9 (CRISPR/Cas9) System bietet ein viel versprechendes Instrument für Gentechnik und eröffnet die Möglichkeit, gezielte Integration der transgene. Wir beschreiben eine Homologie-vermittelte Ende verbinden (HMEJ)-basierte Strategie für effiziente DNA Integration in Vivo gezielte und gezielte Gentherapien verwenden CRISPR/Cas9.
Abstract
Als eine viel versprechende Genom-editing-Plattform hat das CRISPR/Cas9 System großes Potential für effiziente Genmanipulation, vor allem für die gezielte Integration von transgenen. Aufgrund der geringen Effizienz der homologen Rekombination (HR) und verschiedenen Indel Mutationen von nicht-homologe Ende verbinden (NHEJ)-basierte Strategien in nicht-teilenden Zellen, in Vivo Genom Bearbeitung bleibt eine große Herausforderung. Hier beschreiben wir eine Homologie-vermittelte Ende verbinden (HMEJ)-basiertes CRISPR/Cas9 System zur effizienten in Vivo präzise gezielte Integration. In diesem System, das gezielte Genom und der Spender Vektor mit Homologie Arme (~ 800 bp) flankiert von einzelnen Guide RNA (SgRNA) Ziel Sequenzen sind durch CRISPR/Cas9 gespalten. Diese HMEJ-basierte Strategie erzielt effiziente Transgen Integration in Maus Zygoten sowie in Hepatozyten in Vivo. Darüber hinaus eine HMEJ-basierte Strategie bietet einen effizienten Ansatz zur Korrektur von Fumarylacetoacetate Hydrolase (Fah) Mutation in den Hepatozyten und rettet Fah-Mangel induziert Leberversagen Mäuse. Zusammen genommen, mit Schwerpunkt auf Integration ausgerichtet, diese HMEJ-basierte Strategie bietet ein viel versprechendes Instrument für eine Vielzahl von Anwendungen, einschließlich der Erzeugung von gentechnisch veränderten Tiermodelle und gezielte Gentherapien.
Introduction
Präzise, gezielte Genom-Bearbeitung ist oft für die Herstellung gentechnisch veränderter Tiermodellen und klinischen Therapien erforderlich. Viel Anstrengungen unternommen worden, um verschiedene Strategien für effiziente zielgerichtete Genom bearbeiten, wie z. B. Zink Nuklease (ZFN), Transkription Aktivator-ähnliche Effektor Nukleasen (TALENs) und CRISPR/Cas9 Systeme. Diese Strategien gezielte DNA-Doppel-Strangbrüchen (DSB) im Genom zu erstellen und profitieren Sie von inneren DNA-Reparatursysteme, wie homologe Rekombination (HR)1,2, Microhomology-vermittelte Ende verbinden (MMEJ)3 , 4 , 5und nicht-homologe Ende verbinden (NHEJ)6,7,8 , gezielte Integration der transgene1,9zu induzieren. Die HR-Strategie ist derzeit die am häufigsten verwendeten Genom Bearbeitung Ansatz, der in Zell-Linien sehr effizient, aber nicht teilenden Zellen aufgrund seines beschränkten Auftretens in der Spätphase der S/G2 nicht ohne weiteres zugänglich ist. Der HR-Strategie gilt somit nicht für in Vivo Genom-Bearbeitung. Vor kurzem wurde die Grundstrategie des NHEJ für effiziente gen-Knock-in Maus Gewebe8entwickelt. Dennoch stellt die NHEJ-basierte Methode in der Regel Indels an den Verbindungsstellen, macht es schwierig, präzise Genom-Bearbeitung, vor allem wenn Sie versuchen, im Rahmen Fusion Gene8konstruieren zu generieren. MMEJ-basierte gezielte Integration ist in der Lage, präzise Genom-Bearbeitung. Es erhöht jedoch nur in bescheidenem Maße die gezielte Integration Effizienz in vorherigen Berichten5. Verbesserung der Effizienz der präzise gezielte Integration in Vivo ist daher für breite therapeutische Anwendungen3dringend erforderlich.
In einer kürzlich veröffentlichten Arbeit zeigten wir eine Homologie-vermittelte Ende verbinden (HMEJ)-basierte Strategie, die die höchste Effizienz der gezielte Integration in alle gemeldeten Strategien sowohl in Vitro und in Vivo10zeigte. Hier beschreiben wir ein Protokoll für die Einrichtung des HMEJ Systems, und auch der Bau der Einzel-Guide RNA (SgRNA) Vektoren gezielt das gen des Interesses und des Spenders Vektoren beherbergen SgRNA Ziel-Sites und ~ 800 bp Homologie Wappen (Abbildung 1) . In diesem Protokoll erläutern wir im folgenden die einzelnen Schritte zur Erzeugung von DNA-Knock-in Mäusen und kurze Schritte für die gezielte Integration in Geweben in Vivo. Darüber hinaus zeigte eine Proof-of-Concept-Studie über die HMEJ-basierte Strategie seine Fähigkeit, Fah Mutation zu korrigieren und Fah- / - Leber Versagen Mäuse, die weiter seine therapeutische Potenzial offenbart zu retten.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Alle Verfahren einschließlich tierische Themen wurden von der biomedizinischen Forschung-Ethik-Kommission der Shanghai Institute für biologische Wissenschaften (CAS) genehmigt.
1. Aufbau des Spenders Plasmide
-
Auswahl des sgRNA
- Verwenden Sie Online-CRISPR-Design-Tools SgRNAs am Ziel Region11,12,13,14,15vorherzusagen. Design für die Cdx2 Locus, sechs verschiedene SgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) um die Haltestelle Codon mit höheren Rang und niedrigeren Potential aus-Ziele (Abb. 1A)16.
- Linearisieren Sie 2 µg Cas9-CMV-EGFP Expressionsvektoren und SgRNA durch BbsI Verdauung (1 µL der BbsI für 2 h bei 37 ° C auf eine Endkonzentration von 1 U/µL in einem Volumen von 20 µL). Dann reinigen Sie das Produkt durch Gel-Reinigung-Kit mit einem 1 % Agarose-Gel in 1 x TAE-Puffer.
- Die Oligo-Lösung mit einem Temperaturgradienten von 95 ° C bis 25 ° C mit einer Änderungsrate der Temperatur von 5 ° C/5 min (95 ° C für 5 min inkubieren Sie Mix ein paar SgRNA Oligonukleotide in 10 µL 1 × T4 DNA-Ligase Puffer, eine Endkonzentration von 50 µM. dann 90 ° C für 5 min, 85 ° C für 5 min, etc..), die wird die Oligos Tempern.
- Mix 4 µL Produkt, 2 µL der linearisierten Vektor mit 1 µL T4 DNA-Ligase in 10 µL 1 × T4 DNA-Ligase Puffer, geglüht und dann bei 22 ° C für 1-2 h (Abbildung 1B) verbinden.
-
Landvermesser Nuklease-Test von sgRNA
Hinweis: Die targeting Effizienz der SgRNA für die Knock-in-Experiment verwendet wird ausgewertet, indem Surveyor Nuclease Assay (auch bekannt als T7 Endonuklease ich (T7EI) assay)17. Wählen Sie die SgRNA mit hoher DNA Spaltung Effizienz und einen geringen Abstand zwischen Ortsbild SgRNA schneiden und das Stopp-Codon.- Cas9-SgRNA-EGFP Expressionsvektoren in N2a Zelllinien transfizieren kultiviert in DMEM mit 10 % fötalen Rinderserum ergänzt, 1 % PSG und 1 % nicht-essentielle Aminosäure durch die Transfektion kit (siehe Tabelle der Materialien). Inkubieren Sie die transfizierten Zellen bei 37 ° C in 5 % CO2.
- Nach 48 h Inkubation, sammeln Sie 5.000 transfizierte Zellen (GFP+) durch Fluoreszenz-aktivierte Zelle sortieren (FACS) Verwendung nicht transfizierten Zellen als ein Steuerelement.
- Die gesammelten Zellen in 2 bis 5 µL Oflysis-Puffer (0,1 % Triton x-100, 0,1 % Tween 20 und 100 µg/mL Proteinase K) bei 56 ° C für 30 min zu verdauen, und dann Hitze inaktivieren Proteinase K bei 95 ° C für 10 Minuten.
- Die Probe von verschachtelten PCR (Tabelle 1) mit der Hersteller-Protokoll zu verstärken. Die Größe der PCR-Produkte wird auf 300-500 bp festgelegt.
- Mix 1 µL Lyse Produkt mit DNA-Polymerase und ein paar von äußeren Primern erkennen die Sequenz auf dem SgRNA-Ziel-Gelände (0,1 µM, Endkonzentration) (Tabelle 1), und führen Sie die primäre PCR in einem Volumen von 20 µL.
- DNA-Polymerase bei 95 ° C für 5 min zu aktivieren, und führen Sie die primäre PCR für 30 Zyklen bei 95 ° C für 30 s, 60 ° C für 30 s und 72 ° C für 24 s (1 min/1 kb), mit der letzten Erweiterung bei 72 ° C für 5 Minuten.
- Führen Sie die sekundäre PCR mit 1 µL des primären PCR-Produkt und ein paar verschachtelte innere Primer.
- Denaturieren und Tempern wieder 300-600 ng des gereinigten PCR-Produkt in 20 µL 1 × T7EI Reaktion Puffer (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT pH 7,9) mit einem Gefälle von Temperatur von 95 ° C bis 25 ° C mit einer Rate von 5 ° C/5 min.
- Die geglühten PCR-Produkte und Digest bei 37 ° C für 2 h 1 µL T7EI Enzym hinzufügen. Führen Sie dann die Verdauung Produkt auf 2 % Agarose-Gel in 1 × TAE Puffer bei 120 V 40 min, bis die Fragmente getrennt sind (siehe Tabelle der Materialien).
- Verwenden Sie ImageJ um die Band-Intensitäten von Schnitt und ungeschnitten DNA zu bestimmen. Berechnen Sie die Indel Frequenz mit den Methoden wie zuvor9 (Abbildung 1C) berichtet.
-
Bau des Spenders Vektor
Hinweis: Um HMEJ Spender Vektoren für Cdx2 gen zu erzeugen, konstruieren einen Spender DNA (800 bp HAL-p2A-mCherry-800 bp HAR) flankiert mit 23 nt Cdx2-SgRNAs targeting-Sequenz an beiden Enden (Abbildung 1D und Abbildung 1E). Die PAM Zielsequenz grenzte an das Ende der homologen Arm. Gibson Montage empfiehlt sich für HMEJ Spender Klonen.- Verstärken Sie die 800 bp linken Homologie Arm (HAL) mit forward Primer-1F (mit 15-20 nt Überlappung Sequenz von Vector, 23 nt Cdx2-SgRNA-targeting-Sequenz und etwa 20 nt-Sequenz von HAL) und reverse Primer-1R (mit 15-20 bp Überlappung Sequenz von p2A-mCherry und etwa 20 nt-Sequenz von HAL) bei 0,1 µM Endkonzentration mit Maus genomischer DNA bei 200 ng/µL (Abbildung 1D, Tabelle 1).
- Verstärken die p2A-mCherry Einfügung Fragment mit forward Primer-2F (mit 15-20 nt Überlappung Sequenz von HAL und etwa 20 nt-Sequenz aus Einfügung Fragment) und reverse Primer-2R (mit 15-20 nt Überlappung Sequenz von HAR und etwa 20 nt-Sequenz aus Das Fragment einfügen) bei 0,1 µM Endkonzentration mit genomische DNA oder Plasmid mit Reporter Sequenzen bei 100 ng/µL oder 30 ng/µL (Abbildung 1D, Tabelle 1).
- Verstärken der 800 bp richtige Homologie Arm (HAR) mit forward Primer-3F (mit 15-20 nt Überlappung Sequenz von Vector, 23 nt Cdx2-SgRNA-targeting-Sequenz und etwa 20 nt-Sequenz von HAR) und reverse Primer-3R (mit 15-20 nt Überlappung Sequenz von p2A-mCherry und etwa 20 nt-Sequenz von HAR) bei 0,1 µM Endkonzentration mit Maus genomischer DNA bei 200 ng/µL (Abbildung 1D, Tabelle 1).
- Laufen Sie die PCR-Produkte auf 1 % Agarosegel auf 1 x TAE-Puffer, und reinigen Sie die PCR-Produkte der erwarteten Größe Gel Extraction Kit, gemäß den Anweisungen des Herstellers (Tabelle 1).
- 50-100 ng eines Vektors Konstrukt mit KpnI und XbaI zu verdauen. Mix 2 µL der linearisierten Vektor auf 30-40 ng/µL mit drei PCR verstärkt Fragmente (1 µL für jedes, 100-200 ng/µL) 2 x Gibson Mischung. Fügen Sie H2O um die letzte Lautstärke auf 10 µL.Incubate die Mischung bei 50 ° C für 60 min hinzu.
- Verwandeln Sie kompetente E. Coli Zellen mit dem montierten Produkt und Extrakt das Plasmid DNA Extraktion Kit gemäß den Anweisungen des Herstellers baut. Überprüfen des HMEJ Spenders durch DNA-Sequenzierung.
(2) Genom-Bearbeitung in Mausembryonen mithilfe der HMEJ-basierte Methode
-
Produktion von Cas9 mRNA
- Vorbereitung der Cas9 mRNA fügen Sie T7 Promotorsequenz, der Cas9 Region durch PCR-Amplifikation mit der entsprechenden Primerpaar in Tabelle 1aufgeführten Codierung hinzu. Fügen Sie den Primer Cas9 F/R auf eine Endkonzentration von 0,1 µM und 20 ng des Cas9 mit dem Ausdruck Vektor 1 × High Fidelity DNA-Polymerase Mischung. Die letzte Lautstärke zu 50 µL mit H2O.
- DNA-Polymerase bei 95 ° C für 5 min zu aktivieren, und führen Sie die PCR für 36 Zyklen bei 95 ° C für 30 s, 60 ° C für 30 s und 68 ° C für 4 min (1 min/1 kb), mit der letzten Erweiterung bei 68 ° C für 10 Minuten.
- T7-Cas9 PCR-Produkt für in-vitro- Transkription (IVT) zu reinigen, und dann transkribieren 0,5-1 µg DNA mRNA-Transkription-Kit bei 37 ° C für 8 h in einem Gesamtvolumen von 20 µL, gemäß den Anweisungen des Herstellers (siehe Tabelle der Materialien).
- Die Mischung der DNA Schablone bei 37 ° C für 15 min. Add ein Poly-A-Endstück für 45 min bei 37 ° C zu entfernen und Wiederherstellen der Cas9 mRNA durch RNA-Reinigung-Kit, gemäß den Anweisungen des Herstellers 1 µL DNase hinzufügen (siehe Tabelle der Materialien).
-
Herstellung von sgRNA
- Angetrieben von einem T7 Promotor mit High-Fidelity-DNA-Polymerase als oben SgRNA-Vorlage generieren. Wählen Sie ein SgRNA Gerüst mit Vektor als Vorlage. Die Primer verwendet, sind in Tabelle 1aufgeführt.
- Reinigen der T7-SgRNA PCR-Produkt und verwenden Sie 0,5-1 µg DNA als Vorlage für in-vitro- Transkription von SgRNA mit einem kurzen RNA Transkription-Kit bei 37 ° C für 6 Stunden in einem Gesamtvolumen von 20 µL, gemäß den Anweisungen des Herstellers (siehe Tabelle der Materialien < / c11 >).
- 1 µL DNase zur Mischung hinzugeben und weiter die Inkubation bei 37 ° C für 15 min, die DNA-Vorlage zu entfernen. Reinigen Sie die SgRNAs durch RNA-Reinigung-Kit, wie oben (siehe Tabelle der Materialien).
- Die SgRNA um 500 ng/µL RNase-freies Wasser zu verdünnen und die Proben bei −80 ° C bis zu 3 Monate lang zu speichern.
Hinweis: CRISPR Ribonucleoproteins (RNPs) sind eine alternative Substitution mit besser schneiden Effizienz18,19,20.
-
Embryo-Sammlung, Mikroinjektion und in-vitro- Kultur
- Superovulate weibliche B6D2F1 (C57BL/6 × DBA2J) Mäusen (ca. 7-8 Wochen alt) von trächtige Stute Serum Gonadotropin (PMSG), gefolgt von humanes Choriongonadotropin (hCG) 48 h später. Nach der hCG-Injektion Übernachtung Haus Frauen mit B6D2F1 Männchen.
- Die Weibchen durch CO2 Narkose, 24 Stunden nach der hCG-Injektion zu opfern. Sammeln Sie die befruchteten Embryonen aus ihrer Eileiter (mit 30-50 Embryonen für jedes Weibchen) in M2 Medium.
- Ort der befruchteten Embryonen (ca. 300 Eiern für Tages-Injektion) in KSOM Medium (5,55 g/L NaCl, 0,19 g/L KCl, 0,05 g/L KH2PO4, 0,05 g/L MgSO4•7H2O, 0,04 g/L Glukose, 1,12 g/L Natrium Laktat, 2,1 g/L Nahco33 , 0,02 g/L Natrium Pyruvat, 0,25 g/L CaCl2•2H2O 0,004 g/L EDTA, 0,146 g/L L-Glutamin und 1 g/L Rinderserumalbumin) bei 37 ° C in einem Inkubator mit 5 % CO2.
- Mix Cas9 mRNA (100 ng/µL), SgRNA (50 ng/µL), HMEJ Spender Vektor (100 ng/µL) und H2O die letzte Lautstärke auf 10 µL. setzen Sie die Mischung auf dem Eis hinzufügen.
- Kapillare Nadeln (Außendurchmesser 1,0 mm, Innendurchmesser 0,78 mm mit Filament) ziehen mit einer Mikropipette Abzieher (Parameter: Wärme, 74, ziehen, 60, Geschwindigkeit, 80; Zeitverzögerung/200, Druck, 300. Siehe Tabelle der Materialien). Kommerzielle Nadeln wäre eine alternative Ersatz für die Mikroinjektion.
- Injizieren eine wahrscheinliche Volumen der Mischung in das Zytoplasma der Zygoten mit klar definierten Vorkerne in ein Tröpfchen HEPES-CZB Medium mit 5 µg/mL Cytochalasin B mit Hilfe eines Microinjector mit konstanten flow-Einstellungen (Abb. 2A) (siehe Tabelle der Materialien)21.
Hinweis: Jede Gruppe von Zygoten injiziert werden sollten innerhalb von 20-30 min. Cytochalasin B könnte die Lebensfähigkeit der Maus Zygote nach der Injektion zu erhöhen. Mikroinjektion kann alternativ mit dem Piezo-System betrieben werden, wie zuvor beschrieben,22. - Kultur der injizierten Zygoten in KSOM-Medium bei 37 ° C weniger als 5 % CO2 bis Blastozyste Bühne nach 3,5 Tagen für Fluoreszenz-Beobachtung (Abbildungen 2 b und 2C).
-
Embryo-Transfer und Generierung von Mäusen
- Mate estrous ICR weibliche Mäuse mit vasektomierten ICR männliche Mäuse am selben Tag als Injektion.
- Kultur der injizierten Zygoten in das 2-Zell-Stadium bei 37 ° C unter 5 % CO2und Transfer 25-30 2-Zellen Embryonen in den Eileiter pseudopregnant ICR-Weibchen bei 0,5 Tag Post Coitum (Dpc). Empfängers Mütter liefern Welpen zu 19,5 Dpc.
-
Maus-Genotypisierung
- Extrahieren Sie Maus genomischer DNA aus Zehe oder Schweif Proben mit einem DNA-Extraktion-Kit, gemäß den Anweisungen des Herstellers (siehe Tabelle der Materialien).
- Identifizieren der 5' und 3' Kreuzung von Knock-in Veranstaltungen mit 200-400 ng genomic DNA als Vorlage durch UV/Vis-Spektrometrie gemessen, die PCR Lautverstärkung durchzuführen.
- DNA-Polymerase bei 95 ° C für 5 min zu aktivieren, und führen Sie die PCR für 38 Zyklen bei 95 ° C für 30 s, 60 ° C für 30 s und 72 ° C für 1 min (1 min/1 kb), mit der letzten Erweiterung bei 72 ° C für 10 Minuten. Verwenden Sie für die 5' Kreuzung die forward Primer auf den vorgelagerten HAL, mit der Rückseite auf die Knock-in-Fragment (p2A-mCherry). 3' Kreuzung nutzen Sie die vorwärts Grundierung auf die Knock-in-Fragment (p2A-mCherry), mit der Rückseite eines auf die nachgelagerten HAR (Tabelle 1).
- 6 µL des PCR Produktes auf 1 % Agarose-Gel auf 1 x TAE-Puffer und prüfen, ob die erwarteten Fragmentgröße ausgeführt. Dann überprüfen sie durch DNA-Sequenzierung (Abbildung 2D).
3. HMEJ-basierte In-Vivo Genom Bearbeitung in Hepatozyten
- Empfängers C57BL/6J Maus (8 Wochen) in eine Haltevorrichtung legen Sie und das Ende durch den Schlitz.
- Mischen Sie HMEJ Spender Vektoren (30 µg) und spCas9 Expressionsvektoren (30 µg) in 2 mL Kochsalzlösung. Auszusetzen Sie für das Kontrollexperiment HMEJ Spender Vektoren (30 µg) in 2 mL Kochsalzlösung(Abbildung 3).
- Reinigen Sie die Maus-Rute mit 70 % Ethanol. Setzen Sie die Nadel in das Heck Vene und injizieren die Plasmid-DNA-Lösung innerhalb von 5-7 s. die Nadel entfernen und lassen Sie die Maustaste aus der Haltevorrichtung.
- Opfern Sie Mäuse durch CO2 Narkose nach 5-9 Tage nach der Injektion. Die Mäuse Transcardially mit 0,9 % Kochsalzlösung durchspülen, gefolgt von 4 % Paraformaldehyd mittels einer peristaltischen Pumpe und beheben die Leber über Nacht bei 4 ° C.
- Das Gewebe mit 30 % Saccharose über Nacht, bis es auf den Boden des Rohres sinkt zu entwässern.
- Abschnitt des gefrorenen Gewebes bei einer Dicke von 10 µm für Leber Proben.
- Spülen Sie die Abschnitte dreimal in 0,1 M Phosphat gepuffert (PB) und inkubieren sie mit primären Antikörper: Kaninchen-Anti-mCherry (in 5 % verdünnt NGS) über Nacht bei 4 ° C.
- Waschen Sie Abschnitte dreimal in PB und inkubieren sie dann mit Sekundärantikörper: Cy3-AffiniPure Ziege IgG Anti-Kaninchen für 2 h bei Raumtemperatur auf einem Orbitalschüttler.
- Gegenfärbung die Abschnitte mit DAPI für 20 min und Mount mit Glycerin auf Objektträgern für weitere Fluoreszenz Beobachtungen (Abbildung 3B).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
HMEJ-basierte Genom-Bearbeitung in Mausembryonen: Um die Effizienz Knock-in der HMEJ-basierte Methode Maus Zygoten definieren, lieferten wir Cas9 mRNA, SgRNA gezielt das Gen Cdx2 und der HMEJ Spender in Maus Zygoten, die entworfen wurde, um ein Reportergen p2A-mCherry zu den letzten Codon Cdx2 Sicherung gen (Abbildung 2A). Die injizierten Zygoten entwickelte sich zu Blastozysten in der Kultur. Um die Knock-in-Effizienz zu bewerten, haben wir die mCherry Fluoreszenz mit einem Fluoreszenz-Mikroskop analysiert, und wir fanden, dass 12,9 % der Blastozysten empfangen HMEJ Spender positiv für mCherry, die streng in den Trophektoderm zum Ausdruck kam (Zahlen 2 b 2 C). Durch Sequenzierung PCR positiv Mäuse, fanden wir auch, dass alle Integrationsveranstaltungen untersuchten präzise in-Frame-Integrationen an 5' und 3' Kreuzungen (Abbildung 2D).
HMEJ-basierte Genom-Bearbeitung in adulten Geweben und HMEJ-vermittelte Gentherapie: Um zu untersuchen, ob HMEJ-basierte Genom-Bearbeitung in adulten Geweben angewendet werden könnte, legten wir die mCherry Kassette direkt vor das Stopcodon Actb Gens durch transducing Actb- HMEJ Konstrukte, C57/B6J Maus Lebern von Heck-Vene hydrodynamische Injektion(Abbildung 3). Nach 7 Tagen von Injektionen fanden wir, dass fast die Hälfte der transfizierten Hepatozyten mCherry zum Ausdruck gebracht, wie auf die Leber Abschnitte (Abbildung 3B) befleckt.
Um die Möglichkeit der Verwendung einer HMEJ-basierte Strategie für die Gentherapie zu erkunden, wir Beschäftigten Fumarylacetoacetate Hydrolase (Fah)-mangelhafte Mäuse. Die Fah- / - Maus ist eine gut etablierte erbliche Tyrosinemia Typ I (HTI) Maus-Modell, das eine Einfügung Fragment im Exon 5 des Gens Fah , verursacht Frameshift-Mutationen in der folgenden Sequenz23birgt. Um Fah- / - Mäusen zu halten, haben wir die Fah- / - Mäuse mit einem Inhibitor von den vorgelagerten Tyrosin katabolen Weg, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24behandelt. Hier wollten wir sehen, ob MMEJ - und HMEJ-vermittelten gen Korrektur Fah Mutation in der Fah- / - Maus retten konnte. Wir injiziert hydrodynamisch Cas9 Konstrukt zusammen mit Fah- MMEJ oder Fah- HMEJ Konstrukte, Fah cDNA Exon 5 bis 14 in Intron 4 Fah Gens einfügen soll, Fah- / - Maus Lebern () Abbildung 3 ( C). eine Woche nach der Injektion NTBC wurde zurückgezogen, Leberschäden (Abb. 3C) induzieren. Nach dem Abzug der NTBC, Fah-korrigierte Hepatozyten der Fah- / - Mäuse erhalten Fah- HMEJ und Cas9 Konstrukte zeigten eine wirksamere Verbreitung als MMEJ basierende Methode (Abbildung 3D ).

Abbildung 1 : HMEJ-vermittelten gezielte Integration in-vitro-.
(A) experimentelle Schema für die Auswahl der SgRNAs: sechs verschiedene SgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) um das Stopcodon Cdx2 Locus mit einem höheren Rang und Ziel waren ausgewählt basierend auf Online-CRISPR Design Werkzeug. Die Protospacer angrenzenden Motiv (PAM) Sequenz ist rot markiert. (B) experimentelles Design: The Cas9-CMV-GFP Ausdruck Plasmide mit dem Ausdruck SgRNA, Cas9 und GFP in N2a Zellen eingeführt wurden. GFP+ Zellen wurden am 3. Tag für Vermesser-Assay sortiert werden. (C) Vermesser assay für Cdx2 targeting: 6 verschiedene SgRNAs für Landvermesser Assay entwickelt wurden. Normale N2a Zelle genomischer DNA dient als Kontrolle. *, der SgRNA zur Cdx2-2A-mCherry-Knock-in-Experiment. (D) schematische Übersicht des Aufbaus der HMEJ Spender mit Gibson Assembly. (E) schematische Übersicht über HMEJ-mediated Gene targeting-Strategy bei Cdx2 Locus. HAL/HAR, links/rechts Homologie Arm; Dreiecke, SgRNA Ziel-Sites; DER / OR, äußeren vorwärts/rückwärts-Primers; IF / IR, innere vorwärts/rückwärts-Primer. Abbildung von vorherigen Bericht10geändert. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2 : Genom-Bearbeitung in Mausembryonen über HMEJ-vermittelten gezielte Integration
(A) experimentelle Schema der Mikroinjektion: eine Mischung aus Cas9 mRNA (100 ng/µL), SgRNA (50 ng/µL) und Spender Plasmide (100 ng/µL) wurden in Maus Zygoten injiziert. (B) repräsentative Fluoreszenzbilder von Mäuseembryonen herausgegeben von HMEJ Strategie. Bar, 20 µm. (C) Knock-in Effizienz durch Prozentsatz der mCherry+ Blastozysten angezeigt. Anzahl über jede Bar, total Blastozysten gezählt. (D) Analyse des Gens bearbeitet Mäuse am Cdx2 Locus Sequence. PCR-Produkte verstärkt von 5' und 3' Kreuzung Sites wurden sequenziert. Homologie Oberarm; lila, p2A; rot, mCherry; HAR oder HAL, rechten oder linken homologe Arm. Gestrichelte Linien markieren die Region, die aus Gründen der Übersichtlichkeit weggelassen. Abbildung von vorherigen Bericht10geändert. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 3 : HMEJ-vermittelten gezielte Integration in Vivo.
(A) schematische Übersicht der hydrodynamischen Schweif Vene Injektion. Eine Mischung aus Plasmide Spender Sequenz und SgRNA zum Ausdruck bringen und mit dem Ausdruck spCas9 Plasmide wurden nach der Leber durch hydrodynamische Schweif Vene Injektion geliefert. (B) repräsentative Immunfluoreszenz Bilder von Hepatozyten. Die Leber Abschnitte wurden gesammelt 7 Tage Post Injektion. Maßstabsleiste, 50 µm. GFP, transfizierten Zellen. (C) Plasmide entweder MMEJ oder HMEJ-vermittelte gen Ersatz Strategie Fah cDNA Exon 5 bis 14 in Intron 4 Fah -gen eingefügt wurden durch hydrodynamische Injektion in Fah- / - Maus Lebern geliefert. NTBC auf: Fah- / - Mäuse wurden beibehalten, auf NTBC Wasser; NTBC aus: Rückzug der NTBC Wasser (der erste Tag der NTBC Rückzug wurde definiert als Tag 0, ist der 7. Tag nach der Injektion). (D) Fah immunhistochemische Färbung der Leber Abschnitte aus Fah- / - Mäusen injiziert mit MMEJ oder HMEJ Plasmide. Maßstabsleiste, 100 µm. Abbildung von vorherigen Berichten5,10geändert. Bitte klicken Sie hier für eine größere Version dieser Figur.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Die wichtigsten Schritte beim Bau von HMEJ Spender Plasmide sind: (1) Wahl des SgRNA mit hoher DNA Spaltung Effizienz und geringe Abstand zwischen SgRNA schneiden Website und Stopp-Codon und (2) die ordnungsgemäße Errichtung der HMEJ Spender. CRISPR/Cas9-vermittelte Spaltung auf beide Transgen-Spender-Vektor (mit SgRNA-Ziel-Sites und ~ 800 bp Homologie Arme) und gezielte Genom ist notwendig für eine effiziente und präzise gezielte Integration in-vivo. Die wichtigsten Schritte der Generation von Knock-in Mäusen mit der HMEJ-basierte Methode sind: (1) die Vorbereitung der hohen Qualität der Cas9 mRNA und SgRNA (keine Degeneration existiert in Cas9 mRNA und SgRNA) und (2) die Vorbereitung des hochwertigen HMEJ Spender Plasmids. Das Plasmid zeigt keine toxischen Wirkungen auf die embryonale Entwicklung.
Vor kurzem wurde eine NHEJ-basierte Methode auch für effiziente in Vivo Genom Bearbeitung8berichtet. Dennoch wurden verschiedene Arten von Indel Mutationen in der Regel an den Verbindungsstellen induziert, wie beschrieben in den vorherigen Berichten8, macht es schwierig, genaue Integration zu erreichen. Hier zeigte die HMEJ-basierte Strategie, die, der wir oben beschrieben, genau gezielte Integration mit kaum Indel Mutationen. So könnte eine Grundstrategie des HMEJ eine ideale Plattform für den Austausch einer mutierten Sequenz (z. B. einer Punktmutation) mit dem richtigen Namen, die nicht für NHEJ basierende Methode anwendbar ist.
Evtl. ist ein großes Problem für die gen Bearbeitung in Embryonen. Injektion von Cas9 Protein anstelle von mRNA in einem früher embryonalen Stadium kann Transgen Knock-in Zelle irgendwann ohne somatisches erreichen. Für klinische Anwendungen ist die Lieferung der CRISPR/Cas9-Systeme in adulten Geweben noch anspruchsvoll.
Es gibt viele zukünftige Einsatzmöglichkeiten des HMEJ-basierte Genom-Bearbeitung. Es kann verwendet werden, um gentechnisch veränderte Tiermodelle zu generieren. In Anbetracht seiner hohen Knock-in Effizienz in Embryonen diese Methode könnte erheblich reduzieren Sie die tierische Anzahl benötigt zur Erzeugung gentechnisch veränderter Tiermodelle und besonders eröffnet die Möglichkeit der Generierung von nichtmenschlichen Primaten genetische Modelle. HMEJ-basierte Genom-Bearbeitung können Linie Spur einzelner Zelltypen in adulten Geweben, die eignet sich besonders für Tiermodelle, da gibt es ein Mangel an verfügbaren Tiermodellen, wie nicht-menschlichen Primaten. Es eignet sich für gezielte Gentherapien: die attraktivste Anwendung einer HMEJ-basierte Strategie ist Gentherapie Klinik verwendet. In dieser Studie haben wir die Fah -Mutation des erblichen Tyrosinemia Typ korrigiert ich Mäuse durch hydrodynamische Injektion der angegebenen Vektoren. Lieferung des Systems CRISPR/Cas9 in adulten Geweben ist jedoch immer noch die größte technische Herausforderung für die klinische Anwendung als hydrodynamische Injektion ist unwahrscheinlich, dass bei Patienten durchgeführt werden. Aktuell, weitere Verbesserung der Lieferung Strategie dringend vor dieser HMEJ basierende Methode in der Klinik zu übersetzen.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben nichts preisgeben.
Acknowledgments
Diese Arbeit wurde unterstützt durch CAS strategische Priorität Research Program (XDB02050007, XDA01010409), der nationalen Hightech-R & D-Programm (863 Programm; 2015AA020307), der National Natural Science Foundation of China (NSFC Zuschüsse, 31522037, 31500825, 31571509, 31522038), China Youth tausend Talente Programm (HY), Pause durch Projekt der chinesischen Akademie der Wissenschaften, Projekt Shanghai City Committee von Wissenschaft und Technik (16JC1420202, HY), das Ministerium of Science and Technology of China (die meisten; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Genetik Ausgabe 133 CRISPR/Cas9 gezielte Integration Homologie-vermittelte Ende kam In Vivo Embryo veränderte genetisch Mäuse hydrodynamische InjektionErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
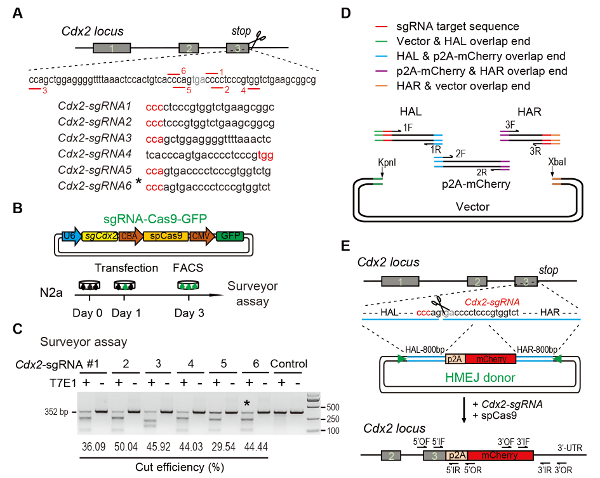
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
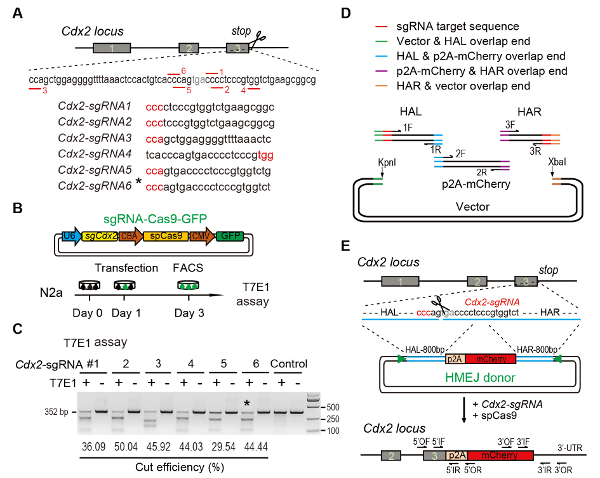
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
