

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
O cluster regularmente intercaladas curta palíndromo repetições/CRISPR associado da proteína 9 (CRISPR/Cas9) sistema fornece uma ferramenta promissora para a engenharia genética e abre a possibilidade de integração específica de transgenes. Descrevemos um fim mediada por homologia, juntar-se (HMEJ)-baseado estratégia eficiente DNA alvo integração na vivo e alvo de terapias do gene usando CRISPR/Cas9.
Abstract
Como uma promissora plataforma de edição do genoma, o sistema CRISPR/Cas9 tem um grande potencial para manipulação genética eficiente, especialmente para a integração específica de transgenes. No entanto, devido a baixa eficiência de recombinação homóloga (HR) e várias mutações indel de fim não-homóloga ingressar (NHEJ)-com base em estratégias em células não-divisão, na vivo genoma edição permanece um grande desafio. Aqui, descrevemos um fim mediada por homologia, juntar-se (HMEJ)-baseado sistema CRISPR/Cas9 para integração eficiente na vivo precisos alvo. Neste sistema, o alvo do genoma e o doador vector contendo armas de homologia (~ 800 bp) ladeado por alvo de RNA (sgRNA) único guia sequências são clivadas por CRISPR/Cas9. Esta estratégia baseada em HMEJ alcança a integração do transgene eficiente em zigotos de rato, bem como em hepatócitos na vivo. Além disso, uma estratégia baseada em HMEJ oferece uma abordagem eficiente para correção de fumarylacetoacetate hidrolase (Fah) mutação nos hepatócitos e resgata Fah-deficiência induzida ratos de insuficiência hepática. Tomados em conjunto, focalizando direcionados a integração, esta estratégia baseada em HMEJ fornece uma ferramenta promissora para uma variedade de aplicações, incluindo a geração de modelos de animais geneticamente modificados e terapias do gene alvo.
Introduction
Edição de genoma preciso, alvo é muitas vezes necessária para a produção de modelos de animais geneticamente modificados e terapias clínicas. Muito esforço foi feito para desenvolver várias estratégias para eficiente genoma alvo de edição, tais como nuclease dedo de zinco (ZFN), nucleases efetoras como ativador de transcrição (TALENs) e CRISPR/Cas9 sistemas. Estas estratégias criar quebras de dobro-costa de DNA alvo (DSB) no genoma e tirar proveito dos sistemas de reparo de DNA intrínsecos, como recombinação homóloga (HR)1,2, microhomology-mediada final juntar-se (MMEJ)3 , 4 , 5e não-homólogas final juntando (NHEJ)6,7,8 , para induzir a integração específica de transgenes1,9. A estratégia baseada no HR é atualmente o mais usado do genoma edição de abordagem, que é muito eficiente em linhas celulares, mas não é facilmente acessível de células não-dividindo devido a sua ocorrência restrita na fase tardia do S/G2. Assim, a estratégia baseada no HR não é aplicável para na vivo edição do genoma. Recentemente, a estratégia baseada em NHEJ foi desenvolvida para gene eficiente bata no rato tecidos8. No entanto, o método baseado em NHEJ geralmente introduz puntuais nos cruzamentos, tornando difícil gerar edição genoma preciso, especialmente quando tentando construir em-frame fusão de genes8. Alvo de integração baseada em MMEJ é capaz de genoma preciso de edição. No entanto, apenas moderadamente aumenta a eficiência de integração orientada em anteriores relatórios5. Por conseguinte, melhorando a eficiência da integração alvo precisos na vivo é urgentemente necessária para aplicações terapêuticas amplo3.
Em um trabalho recentemente publicado, demonstrámos um fim mediada por homologia, juntar-se (HMEJ)-com base em estratégia, que mostrou a mais alta eficiência de integração orientada em tudo relatado estratégias ambos in vitro e in vivode10. Aqui, descrevemos um protocolo para o estabelecimento do sistema de HMEJ, e também a construção de vetores de RNA (sgRNA) o single-guia como alvo o gene de interesse e o doador vetores abrigar sites de destino sgRNA e ~ 800 bp de braços de homologia (Figura 1) . Este protocolo, também descrevemos as etapas detalhadas para geração de DNA bater-em ratos e breves etapas de integração alvo em tecidos na vivo. Além disso, um estudo de prova de conceito da estratégia baseada em HMEJ demonstrou sua capacidade de corrigir a mutação Fah e resgatar Fah- / - fígado falha ratos, que revelou ainda mais o seu potencial terapêutico.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Todos os procedimentos, incluindo assuntos animais foram aprovados pelo Comitê de ética biomédica pesquisas nos institutos Shanghai para ciências biológicas (CAS).
1. projeto de plasmídeos de doador
-
Seleção de sgRNA
- Use ferramentas de design on-line CRISPR para prever sgRNAs sobre o destino região11,12,13,14,15. Para o locus Cdx2 , design seis sgRNAs diferentes (Cdx2-sgRNA1 ~Cdx2-sgRNA6) em torno da paragem códon com maior potencial de rank e inferior fora-alvos (Figura 1A)16.
- Linearizar 2 µ g de vetores de expressão Cas9-CMV-EGFP e sgRNA por digestão BbsI (1 µ l de BbsI por 2 h a 37 ° C, a uma concentração final de 1 U / µ l em um volume de 20 µ l). Em seguida, purifica o produto pelo kit de purificação de gel com um gel de agarose 1% em tampão TAE 1 ×.
- Mistura um par de oligonucleotídeos de sgRNA em 10 µ l de 1 × T4 DNA ligase buffer uma concentração final de 50 µM. Incubar a solução de oligo usando um gradiente de temperatura de 95 ° C e 25 ° C com uma taxa de alteração de temperatura de 5 ° C/5 min (95 ° C por 5 min em seguida, 90 ° C por 5 min, 85 ° C por 5 min, etc.), que será recoze o oligos.
- Mix 4 µ l de recozido produto, 2 µ l do vetor tornado linear com 1 μL de T4 DNA ligase em 10 µ l de 1 × T4 DNA ligase do buffer de e em seguida ligate a 22 ° C, durante 1-2 h (Figura 1B).
-
Ensaio de nuclease Surveyor de sgRNA
Nota: A eficiência de direcionamento do sgRNA usado para o experimento de bater-no é avaliada por ensaio de nuclease surveyor (também conhecido como T7 endonuclease (T7EI) ensaio)17. Selecione o sgRNA com alta eficiência de clivagem de DNA e uma baixa distância entre o local de corte de sgRNA e o códon de parada.- Transfect vetores de expressão Cas9-sgRNA-EGFP em linhas de células N2a cultivadas em DMEM suplementado com 10% de soro fetal bovino, 1% PSG e 1% não-essenciais aminoácidos pelo transfection kit (veja a Tabela de materiais). Incube a 37 ° C em 5% CO2células transfectadas.
- Após 48 h de incubação, coletar 5.000 células transfectadas (GFP+) por fluorescência-ativado da pilha (FACS) de classificação usando células não transfectadas como um controle.
- Digerir as células coletadas em 2-5 µ l oflysis buffer (0,1% Triton X-100, 0.1% Tween 20 e 100 µ g/mL Proteinase K) a 56 ° C por 30 min, e em seguida o calor inativar proteinase K a 95 ° C por 10 min.
- Amplifica a amostra por PCR aninhado (tabela 1) usando o protocolo do fabricante. O tamanho dos produtos PCR é definido a 300-500 bp.
- Misturar 1 µ l de produto de Lise com DNA polimerase e um par de primers exteriores, reconhecendo a sequência ao redor do local de destino de sgRNA (0,1 µM, concentração final) (tabela 1) e realizar o PCR primária em um volume de 20 µ l.
- Ativar o DNA polimerase a 95 ° C por 5 min e realizar o PCR primária para 30 ciclos a 95 ° C por 30 s, 60 ° C por 30 s e 72 ° C por 24 s (1min/1 kb), com uma extensão final a 72 ° C por 5 min.
- Realize o PCR secundária usando 1 µ l de produto PCR primário e um par de primers internas aninhadas.
- Desnaturar e re-recoze 300-600 ng do produto PCR purificado em 20 µ l de 1 × T7EI amortecedor da reação (50mm NaCl, 10 mM Tris-HCl, 10 mM MgCl2, pH de 1 mM DTT 7,9) usando um gradiente de temperatura de 95 ° C a 25 ° C com uma taxa de 5 ° C/5 min.
- Adicione 1 µ l da enzima T7EI para os produtos PCR recozidos e digerir a 37 ° C por 2 h. Em seguida, execute o produto da digestão em gel de agarose 2% em tampão TAE de 1 × 120 V por 40 min, até que os fragmentos são separados (veja a Tabela de materiais).
- Use o ImageJ para determinar as intensidades de banda de corte e DNA sem cortes. Calcule a frequência de indel usando os métodos como anteriormente relatado9 (Figura 1C).
-
Construção do vetor de doador
Nota: Para gerar vetores de doador HMEJ do gene Cdx2 , construir um doador de DNA (bp bp HAL-p2A-mCherry-800 800 HAR) ladeado com 23 nt Cdx2-sgRNAs segmentação sequência em ambas as extremidades (Figura 1D e 1 FiguraE). O PAM da sequência alvo era adjacente à extremidade do braço homóloga. Recomenda-se o assembly de Gibson para doador HMEJ clonagem.- Amplificar o braço de homologia esquerdo bp 800 (HAL) com o avançar da primeira demão-1F (que contém a sequência de sobreposição de nt de 15-20 de vetor, 23 nt Cdx2-sgRNA sequência de direcionamento e cerca de 20 nt sequência de HAL) e inverter a primeira demão-1R (que contém a sequência de sobreposição de bp de 15-20 de p2A-mCherry e cerca de 20 sequência de nt de HAL) na concentração final de 0,1 µM usando DNA genômico de rato em 200 ng / µ l (Figura 1D, tabela 1).
- Amplificar o fragmento de inserção p2A-mCherry com o avançar da primeira demão-2F (contendo a sequência de sobreposição de nt de 15-20 de HAL e cerca de 20 nt sequência de fragmento de inserção) e inverter (que contém a sequência de sobreposição de nt de 15-20 de HAR e cerca de 20 nt sequência da primeira demão-2R o fragmento de inserção) na concentração de final 0.1 µM usando genômica DNA ou plasmídeo com sequências de repórter em 100 ng / µ l ou 30 ng / µ l (Figura 1D, tabela 1).
- Amplificar o braço de certa homologia bp 800 (HAR) com o avançar da primeira demão-3F (que contém a sequência de sobreposição de nt de 15-20 de vetor, 23 nt Cdx2-sgRNA sequência de direcionamento e cerca de 20 nt sequência de HAR) e inverter a primeira demão-3R (que contém a sequência de sobreposição de nt de 15-20 de p2A-mCherry e cerca de 20 sequência de nt de HAR) na concentração final de 0,1 µM usando DNA genômico de rato em 200 ng / µ l (Figura 1D, tabela 1).
- Executar todos os produtos PCR em gel de agarose a 1% em tampão TAE 1 × e purificar os produtos PCR de tamanho esperado pelo kit de extração do gel, de acordo com as instruções do fabricante (tabela 1).
- Digeri a 50-100 ng de um vetor de construção com KpnI e XbaI. Misture 2 µ l do vetor tornado linear em 30-40 ng / µ l, com três PCR amplificados fragmentos (1 µ l de cada um, 100-200 ng / µ l) na 2 x mistura de Gibson. Adicione H2O para ajustar o volume final de 10 µL.Incubate a mistura a 50 ° C por 60 min.
- Competentes de Escherichia coli de transformar células com todo o produto montado e extrair o plasmídeo constrói pelo kit de extração de DNA de acordo com as instruções do fabricante. Verifique se o doador HMEJ por sequenciamento de DNA.
2. genoma edição em embriões de rato usando o método baseado em HMEJ
-
Produção de mRNA Cas9
- Para a preparação de mRNA Cas9, adicione a sequência de promotor T7 para o Cas9 região de codificação por amplificação por PCR usando o par de primer apropriado listado na tabela 1. Adicionar o primer Cas9 F/R em uma concentração final de 0,1 µM e 20 ng de Cas9 expressando o vetor para 1 × alta fidelidade mistura de DNA polimerase. Ajustar o volume final de 50 µ l com H2O.
- Ativar o DNA polimerase a 95 ° C por 5 min e realizar o PCR para 36 ciclos a 95 ° C por 30 s, 60 ° C por 30 s e 68 ° C por 4 min (1min/1 kb), com uma extensão final a 68 ° C por 10 min.
- Purificar o produto do PCR T7-Cas9 em vitro transcrição (IVT) e em seguida transcrever 0.5-1 µ g de DNA pelo kit de transcrição do mRNA em 37 ° C, durante 8 h em um volume total de 20 µ l, de acordo com as instruções do fabricante (ver Tabela de materiais).
- Adicione 1 µ l de DNase à mistura para remover o molde do ADN a 37 ° C durante 15 min. adicionar uma cauda poli-A por 45 min a 37 ° C e recuperar o mRNA Cas9 pelo kit de purificação de RNA, de acordo com as instruções do fabricante (ver Tabela de materiais).
-
Produção de sgRNA
- Gere o modelo de sgRNA, impulsionado por um promotor T7 com alta fidelidade DNA polimerase, como acima. Escolha um andaime de sgRNA contendo o vetor como o modelo. Os primers utilizados são listados na tabela 1.
- Purificar o produto do PCR T7-sgRNA e use 0.5-1 µ g de DNA como o modelo para a transcrição em vitro de sgRNA usando um kit de transcrição do RNA curto a 37 ° C por 6 horas em um volume total de 20 µ l, de acordo com as instruções do fabricante (ver tabela de materiais de < / c11 >).
- Adicione 1 µ l de DNase à mistura e continue a incubação a 37 ° C por 15 min remover o modelo de DNA. Purificar o sgRNAs pelo kit de purificação de RNA, como acima (ver Tabela de materiais).
- Diluir o sgRNA a 500 ng / µ l em água livre de RNase e armazenar as amostras em −80 ° C por até 3 meses.
Nota: Ribonucleoproteínas CRISPR (RNPs) são uma substituição alternativa com uma melhor eficiência de19,corte18,20.
-
Cultura de coleção, microinjeção e in vitro de embriões
- Superovulate B6D2F1 (C57BL/6 × DBA2J) ratos fêmeas (7-8 semanas de idade) por gonadotropina de soro de égua grávida (PMSG), seguidos de gonadotrofina coriônica humana (hCG) 48 h mais tarde. Após a injeção de hCG, fêmeas de casa com machos de B6D2F1 durante a noite.
- Sacrifica as fêmeas por CO2 anestesia, 24h após a injeção de hCG. Recolha os embriões fertilizados de seus ovidutos (com 30-50 embriões para cada fêmea) médio/M2.
- Lugar os embriões fertilizados (cerca de 300 ovos por injeção de um dia) em meio KSOM (5,55 g/L de NaCl, KCl, 0,05 g/L KH2PO4, 0,05 g/L MgSO4•7H2O, 0,04 g/L de glicose, 1,12 g/L de sódio de 0,19 g/L de lactato, 2,1 g/L NaHCO3 , piruvato de sódio 0,02 g/L, 0,25 g/L CaCl2•2H2O, 0,004 g/L EDTA, 0,146 g/L, L-glutamina e albumina de soro bovino de 1 g/L) a 37 ° C numa incubadora com 5% de CO2.
- MRNA Cas9 Mix (100 ng / µ l), sgRNA (50 ng / µ l), doador HMEJ vector (100 ng / µ l) e adicionar o H2O para ajustar o volume final de 10 µ l. Coloque a mistura no gelo.
- Puxe capilares agulhas (diâmetro exterior de 1,0 mm, diâmetro interno 0,78 mm com filamento) usando um extrator de micropipeta (parâmetros: calor, 74; puxar, 60; velocidade, 80; retardo, 200, pressão, 300. Consulte tabela de materiais). Agulhas comerciais seria uma substituição alternativa para o microinjection.
- Injetar um provável volume da mistura no citoplasma de zigotos com pró-núcleos bem definidos em uma gota de meio de HEPES-CZB contendo 5 µ g/mL citocalasina B usando um microinjector com constante fluxo de configurações(Figura 2)(ver Tabela de materiais)21.
Nota: Cada grupo de zigotos deve ser injectado dentro de 20-30 min. Cytochalasin B poderia aumentar a viabilidade do zigoto rato após a injeção. Alternativamente, microinjeção pode ser operada com o sistema de piezo, conforme descrito anteriormente,22. - Cultura de zigotos injetados no meio KSOM a 37 ° C, menos de 5% de CO2 até o blastocisto estágio após 3,5 dias para observação da fluorescência (figuras 2B e 2C).
-
Transferência de embriões e a geração de ratos
- Meu estral ratos fêmeas do ICR com vasectomizado camundongos machos do ICR no mesmo dia como injeção.
- Cultura de zigotos injetados na fase 2-célula a 37 ° C sob 5% CO2e a transferência de 25-30 2-célula de embriões em ovidutos das pseudopregnant fêmeas ICR em 0,5 dia post coitum (dpc). Mães de destinatários entregam filhotes na dpc 19,5.
-
Genotipagem de mouse
- Extrair DNA genômico de rato do dedo do pé ou cauda amostras usando um kit de extração de DNA, de acordo com as instruções do fabricante (ver Tabela de materiais).
- Identifica a junção de 5' e 3' do TOC-em eventos usando 200-400 ng de DNA genômico, medido por espectrometria de UV/vis, como um modelo para realizar o PCR amplification.
- Ativar o DNA polimerase a 95 ° C por 5 min e realizar o PCR para 38 ciclos a 95 ° C por 30 s, 60 ° C por 30 s e 72 ° C, durante 1 minuto (1 min/1 kb), com uma extensão final a 72 ° C por 10 min. Para 5' junção, use o primer para a frente na jusante do HAL, com o reverso no TOC-no fragmento (p2A-mCherry). Quanto à 3' junção, use o primer para a frente no TOC-no fragmento (p2A-mCherry), com o reverso na jusante do HAR (tabela 1).
- Execute 6 µ l do produto de PCR em gel de agarose 1% 1 × TAE buffer e verifique o tamanho do fragmento esperado. Em seguida, verificá-los por (Figura 2D) de sequenciamento de DNA.
3. HMEJ-baseado no Vivo genoma edição em hepatócitos
- Coloque destinatário C57BL/6J rato (8 semanas) em um dispositivo de restrição e coloque a cauda através da fenda.
- Misture vetores de doador HMEJ (30 µ g) e vetores de expressão spCas9 (30 µ g) em 2 mL de solução salina. Para o experimento de controle, suspenda vetores de doador HMEJ (30 µ g) em 2 mL de solução salina(Figura 3).
- Limpe o rabo de rato com etanol a 70%. Inserir a agulha na cauda da veia e injetar a solução de DNA de plasmídeo dentro de 5-7 s. Retire a agulha e solte o mouse do dispositivo de restrição.
- Sacrificar os ratos pelo CO2 anestesia depois de 5-9 dias após a injeção. Perfundir o transcardially de ratos com soro fisiológico a 0,9%, seguido por paraformaldeído 4%, utilizando uma bomba peristáltica e corrigir o fígado durante a noite a 4 ° C.
- Desidrate o tecido usando 30% de sacarose durante a noite, até que ele afunda até o fundo do tubo.
- Secção do tecido congelado em uma espessura de 10 µm para amostras de fígado.
- Enxagúe as seções três vezes em tampão fosfato 0,1 M (PB) e incube-os com anticorpo primário: Rabbit anti-mCherry (diluído em 5% NGS) durante a noite a 4 ° C.
- Lave as seções três vezes no PB e incube-os então com anticorpo secundário: Cy3-AffiniPure cabra anti-coelho IgG para 2 h à temperatura ambiente em um agitador orbital.
- Counterstain as seções com DAPI por 20 min e montagem com glicerina em lâminas de vidro para observação de fluorescência posterior (Figura 3B).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Edição de genoma HMEJ-baseado em embriões do rato: Para definir a eficiência de bater-do método baseado em HMEJ de zigotos de rato, nós entregamos Cas9 mRNA, sgRNA, tendo como alvo o gene Cdx2 e o doador HMEJ em zigotos de rato, que foi projetado para fundir um gene repórter de p2A-mCherry para o último códon do Cdx2 gene(Figura 2). Os zigotos injetados desenvolveram em blastocistos na cultura. Para avaliar a eficiência de bater-no, analisamos a fluorescência de mCherry com um microscópio fluorescente, e achamos que 12,9% de receber doadores HMEJ os blastocistos foram positivas para mCherry, que era estritamente expresso na trofectoderma (figuras 2B 2 C). Por sequenciamento os ratos PCR positivos, nós também achamos que todos examinaram eventos de integração foram precisos em-frame integrações nos cruzamentos tanto 5' e 3' (Figura 2D).
Edição de genoma HMEJ-baseado em tecidos adultos e terapia gênica mediada por HMEJ: Para investigar se a edição baseada em HMEJ genoma poderia ser aplicado em tecidos adultos, inserimos a gaveta de mCherry antes do códon de parada do gene Actb por transducing Actbconstruções de - HMEJ de fígado de rato C57/B6J por cauda-veia injeção hidrodinâmica(Figura 3). Após 7 dias de injeções, nós encontramos que quase metade dos hepatócitos transfectadas expressas mCherry manchado nas secções hepáticas (Figura 3B).
Para explorar a possibilidade de utilizar uma estratégia baseada em HMEJ de terapia genética, usamos fumarylacetoacetate hidrolase (Fah)-ratos deficientes. O Fah- / - mouse é uma bem-estabelecida tirosinemia hereditária tipo I modelo do rato (HTI), que abriga um fragmento de inserção no exon 5 do gene Fah , causando mutações frameshift na seguinte sequência de23. Para manter o Fah- / - ratos, tratamos os Fah- / - ratos com um inibidor do montante da via catabólica de tirosina, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Aqui, partimos para ver se MMEJ e HMEJ-mediada por correção de gene poderia resgatar Fah mutação no Fah- / - mouse. Injetamos direccionado Cas9 construção juntamente com o Fah- MMEJ ou construções de Fah- HMEJ, projetadas para inserir Fah do cDNA do exon 5 a 14 em intron 4 do gene Fah , Fah - / - mouse (fígados Figura 3 C). uma semana após a injeção, NTBC foi retirada para induzir lesão hepática (Figura 3C). Após a retirada do NTBC, Fah-corrigidos hepatócitos dos ratos recebendo Fah- HMEJ e construções Cas9 Fah- / - mostraram proliferação mais eficaz do que o método baseado em MMEJ (Figura 3D ).

Figura 1 : HMEJ-mediada alvo integração em vitro.
(A) esquema Experimental para a seleção de sgRNAs: seis sgRNAs diferentes (Cdx2-sgRNA1 ~Cdx2-sgRNA6) em torno do códon de parada do locus Cdx2 com um potencial mais alto rank e o alvo foram escolhidos baseados em design CRISPR online ferramenta. A sequência de motivo adjacentes (PAM) de protospacer é em vermelho. (B) desenho Experimental: plasmídeo de expressão The Cas9-CMV-GFP expressando GFP, Cas9 e sgRNA foram introduzido em células N2a. Células GFP+ foram classificadas no dia 3 para o ensaio do agrimensor. (C) topógrafo do ensaio para o direcionamento de Cdx2 : 6 sgRNAs diferentes foram projetados para o ensaio do agrimensor. N2a normal célula DNA genômico serve como controle. *, o sgRNA usado para Cdx2-2A-mCherry batida-no experimento. (D) visão geral esquemática da construção dos doadores HMEJ usando o assembly de Gibson. (E) visão geral esquemática do gene HMEJ-mediada visando estratégia em Cdx2 locus. Braço de homologia HAL/HAR, esquerda/direita; triângulos, sgRNA sites de destino; DE / OR, cartilha de avanço/retrocesso exterior; IF / IR, cartilha de avanço/retrocesso interna. Figura modificada do anterior relatório10. Clique aqui para ver uma versão maior desta figura.

Figura 2 : Edição de genoma em embriões de rato através de mediada por HMEJ direcionado a integração
(A) esquema Experimental da microinjeção: uma mistura de mRNA Cas9 (100 ng / µ l), sgRNA (50 ng / µ l) e o doador plasmídeos (100 ng / µ l) foram injetados nos zigotos de rato. (B) imagens de fluorescência representativa de embriões de rato, editados pela estratégia HMEJ. Bar, 20 µm. (C) Knock-em eficiência indicada pela percentagem de mCherry+ blastocistos. Número acima de cada bar, totais blastocistos contados. (D) análise de ratos gene-editado em Cdx2 locus de sequências. Produtos PCR amplificados de 5' e 3' sites de junção foram sequenciados. Superior, braço de homologia; roxo, p2A; vermelho, mCherry; HAR ou HAL, braço direito ou esquerdo homólogo. Linhas tracejadas marcam a região omitida para clareza. Figura modificada do anterior relatório10. Clique aqui para ver uma versão maior desta figura.

Figura 3 : HMEJ-mediada alvo integração na vivo.
(A) visão geral esquemática da injeção de veia de cauda hidrodinâmica. Uma mistura de plasmídeo expressando sgRNA e sequência de doador e plasmídeo expressando spCas9 foram entregues para o fígado através da injeção de veia de cauda hidrodinâmica. (B) imagens de imunofluorescência representativa dos hepatócitos. As seções de fígado foram coletados 7 dias pós injeção. Barra de escala, 50 µm. GFP, transfectadas células. (C) plasmídeos de qualquer MMEJ ou HMEJ-mediada por gene estratégia de substituição concebidos para inserir intron 4 do gene Fah Fah do cDNA do exon 5 a 14 foram entregues em fígado de rato Fah- / - por injeção hidrodinâmica. NTBC em: Fah- / - os ratos foram mantidos em água NTBC; NTBC fora: retirada de água do NTBC (o primeiro dia da retirada do NTBC foi definido como dia 0, que é o 7º dia após a injeção). (D) Fah immunohistochemistry coloração de fígado seções do Fah- / - ratos injetados com plasmídeos MMEJ ou HMEJ. Barra de escala, 100 µm. figura modificada de anteriores relatórios5,10. Clique aqui para ver uma versão maior desta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Os passos mais críticos na construção de plasmídeos de doador HMEJ são: (1) seleção da sgRNA com alta eficiência de clivagem de DNA e baixa distância entre local de corte de sgRNA e stop códon e (2) a boa construção de doador HMEJ. Clivagem CRISPR/Cas9-mediada em ambos vetor de doador do transgene (contendo sites de destino sgRNA e braços de homologia ~ 800 bp) e genoma alvo é necessária para a integração de alvo eficiente e precisa in vivo. Os passos mais importantes da geração de batida-em ratos usando o método baseado em HMEJ são: (1) a preparação da alta qualidade de mRNA Cas9 e sgRNA (nenhuma degeneração existe no mRNA Cas9 e sgRNA) e (2) a elaboração do plasmídeo de doador HMEJ de alta qualidade. O plasmídeo mostra sem efeitos tóxicos sobre o desenvolvimento embrionário.
Recentemente, um método baseado em NHEJ também tinha sido relatado para eficiente na vivo genoma edição8. No entanto, vários tipos de mutações indel geralmente foram induzidos nos cruzamentos, conforme descrito em anteriores relatórios8, tornando difícil conseguir a integração precisa. Aqui, a estratégia baseada em HMEJ, que descrevemos acima mostrou integração alvo precisa com quase nenhum mutações indel. Assim, uma estratégia baseada em HMEJ poderia ser uma plataforma ideal para substituir uma sequência mutante (como um ponto de mutação) com uma correta, que não é aplicável para o método baseado em NHEJ.
Mosaicismo é um grande problema para edição de genes em embriões. Injeção de proteína Cas9 em vez de mRNA em uma fase embrionária no início pode alcançar transgene bater-em uma fase de célula sem mosaicismo. Para aplicações clínicas, a entrega dos sistemas CRISPR/Cas9 em tecidos adultos é ainda um desafio.
Existem muitos usos potenciais futuros de edição baseada em HMEJ do genoma. Ele pode ser usado para gerar modelos de animais geneticamente modificados. Considerando sua alta batida-em eficiência em embriões, este método pode reduzir significativamente o número de animais necessário para a geração de modelos de animais geneticamente modificados e particularmente, abre a possibilidade de gerar modelos genéticos de primatas não humanos. Edição baseada em HMEJ do genoma pode tipos de célula individual de rastreamento de linhagem em tecidos adultos, que é particularmente útil para modelos animais, uma vez que há uma falta de modelos animais disponíveis, tais como os primatas não-humanos. Ele pode ser usado para terapias alvo gene: A aplicação mais atraente de uma estratégia baseada em HMEJ é a terapia gênica para clínica usa. Neste estudo, corrigimos a mutação Fah de tirosinemia hereditária tipo eu ratos por injeção hidrodinâmica dos vetores indicados. No entanto, a entrega do sistema CRISPR/Cas9 em tecidos adultos ainda é o grande desafio técnico para uso clínico, como injeção hidrodinâmica é improvável ser realizada em pacientes. Atualmente, mais melhoria da estratégia de entrega é urgentemente necessária antes de traduzir este método baseado em HMEJ para a clínica.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores não têm nada para divulgar.
Acknowledgments
Este trabalho foi apoiado por CAS estratégica pesquisa programa prioritário (XDB02050007, XDA01010409), o nacional Hightech R & D programa (programa 863; 2015AA020307), Fundação Nacional de ciências naturais da China (NSFC concede 31522037, 31500825, 31571509, 31522038), programa mil jovens talentos de China (para HY), Break através do projeto da Academia Chinesa de Ciências, Xangai cidade Comissão de ciência e tecnologia de projeto (16JC1420202 para HY), o Ministério da ciência e tecnologia da China (maioria; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Genética edição 133 CRISPR/Cas9 alvo integração mediada por homologia final juntar-se In Vivo embrião modificado geneticamente ratos injeção hidrodinâmicaErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
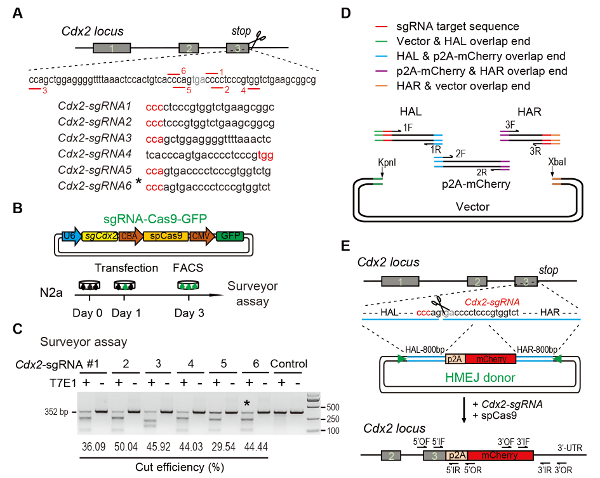
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
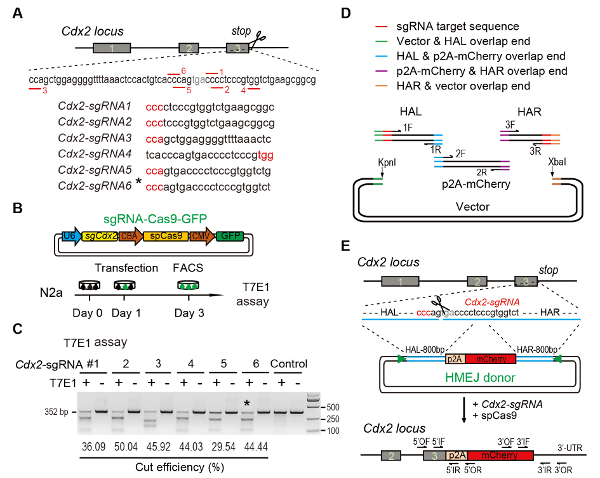
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
