

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
聚簇定期 interspaced 短回文重复/CRISPR 相关蛋白 9 (CRISPR/Cas9) 系统为基因工程提供了一个有前途的工具, 并开辟了转基因目标整合的可能性。我们描述了一种基于同源介导的端接 (HMEJ) 策略, 用于有效的 DNA 靶向集成体内和使用 CRISPR/Cas9 的靶向基因治疗。
Abstract
CRISPR/Cas9 系统作为一种有前景的基因组编辑平台, 具有很好的遗传操作能力, 特别是转基因靶向整合。然而, 由于同源重组 (HR) 的效率低下, 以及非同源端连接 (NHEJ) 的各种 indel 突变, 在无分裂细胞中,体内基因组编辑仍然是一个巨大的挑战。在这里, 我们描述了一个基于同源介导的端接 (HMEJ) 的 CRISPR/Cas9 系统, 用于有效的体内精确目标集成。在该系统中, 靶向基因组和包含同源臂 (约 800 bp) 的单导 RNA (sgRNA) 靶序列的施主载体被 CRISPR/Cas9。这种基于 HMEJ 的策略在小鼠受精卵以及肝细胞体内实现了有效的转基因集成。此外, 以 HMEJ 为基础的策略提供了一种有效的方法来矫正肝细胞中的 fumarylacetoacetate 水解酶 () 突变, 并抢救缺陷致肝功能衰竭小鼠. 结合起来, 专注于目标整合, 这种基于 HMEJ 的战略为各种应用提供了一个有前景的工具, 包括基因改良动物模型的生成和靶向基因治疗。
Introduction
精确的, 有针对性的基因组编辑通常需要生产转基因动物模型和临床治疗。为有效的靶向基因组编辑制定了各种策略, 如锌指核酸酶 (ZFN)、转录激活剂样效应核酸 (TALENs) 和 CRISPR/Cas9 系统。这些策略在基因组中创建目标 dna 双链断裂, 并利用内在的 dna 修复系统, 如同源重组 (HR)1,2, microhomology 介导的最终加入 (MMEJ)3,4,5和非同源端连接 (NHEJ)6,7,8 , 以诱导目标集成的转基因1,9。基于 HR 的策略是目前最常用的基因组编辑方法, 它在细胞系中非常有效, 但由于其在晚期 S/G2 阶段的受限发生而无法轻易进入非分裂细胞。因此, 基于 HR 的策略不适用于体内基因组编辑。最近, NHEJ 的策略被开发为有效的基因敲入在小鼠组织8。然而, 基于 NHEJ 的方法通常会在连接处引入 indels, 使得很难生成精确的基因组编辑, 尤其是在试图构建帧内融合基因8时。基于 MMEJ 的目标整合能够精确地进行基因组编辑。但是, 在以前的报告5中, 它只适度地提高了目标集成效率。因此, 在广泛的治疗应用程序3中迫切需要提高精确目标集成在体内的效率。
在最近发布的工作中, 我们演示了一种基于同源介导的端接 (HMEJ) 策略, 它在所有报告的策略中都显示了目标集成效率最高的体外和体内10。在这里, 我们描述了建立 HMEJ 系统的一个协议, 并构建了针对感兴趣的基因的单导 RNA (sgRNA) 向量, 以及窝藏 sgRNA 目标点的施主载体和800同源臂的 bp (图 1).在本协议中, 我们还描述了生成 DNA 敲除小鼠的详细步骤以及在组织中体内的目标集成的简要步骤。此外, 基于 HMEJ 的策略的概念验证研究表明, 它有能力纠正 "突变" 的变异并抢救/肝功能衰竭小鼠, 进一步揭示了其治疗潜力.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
在上海生物科学研究院 (CAS) 的生物医学研究伦理学委员会批准了包括动物实验在内的所有程序。
1. 捐助者质粒的设计
-
sgRNA 的选择
- 使用在线 CRISPR 设计工具预测目标区域上的 sgRNAs11,12,13,14,15。对于Cdx2轨迹, 设计六个不同的 sgRNAs (Cdx2-sgRNA1~Cdx2sgRNA6), 其周围有更高级别和较低潜在目标 (图 1)16的 stop 密码子。
- 进行线性化2µg 的 Cas9-CMV-EGFP 表达载体和 sgRNA BbsI 消化 (µL 1 BbsI 2 h 在37°c 在最后集中 1 U/µL 在容量20µL)。然后用1% 琼脂糖凝胶在1×泰缓冲器中纯化产品。
- 将一对 sgRNA 寡核苷酸混合在10µL 的 1× T4 DNA 连接酶缓冲液中, 最终浓度为50µM. 用温度梯度从95°c 孵化到25°c, 温度变化率为5摄氏度/5 分钟 (95 摄氏度为5分钟) 孵育寡聚体溶液;, 然后90°c 为5分钟, 85 °c 5 分钟,等.), 这将退火的寡核苷酸。
- 将退火产物的4µL、2µL 的线性化向量与1µL T4 dna 连接酶在µL 1× dna T4 缓冲区的10连接酶中, 然后结扎22摄氏度 1-2 h(图 1 B).
-
sgRNA 的测量核酸酶测定
注: sgRNA 试验用的靶向效率由测量员核酸酶化验 (也称为 T7 限制性 I (T7EI) 化验) 17 进行评估.选择 sgRNA 的高 DNA 解切效率和低距离之间的 sgRNA 切割点和停止密码子。- 染 Cas9-sgRNA-EGFP 表达载体到 N2a 细胞系中培养 DMEM 补充10% 胎牛血清, 1% 和1% 非必需氨基酸的转染套件 (见材料表)。在 5% CO2中孵育转染的细胞在37°c。
- 在48小时的孵化后, 通过荧光活化细胞分类收集5000转染细胞 (GFP+), 并使用非转染细胞作为控制。
- 消化收集的细胞在2-5 µL oflysis 缓冲 (0.1% 海卫 X-100, 0.1% 吐温 20, 100 µg/毫升蛋白酶 k) 在56摄氏度30分钟, 然后热灭活蛋白酶 k 在95°c 10 分钟。
- 使用制造商的协议, 通过嵌套 PCR (表 1) 放大示例。PCR 产品的大小设置为 300-500 bp。
- 将裂解产物的1µL 与 DNA 聚合酶和一对外引物混合, 识别 sgRNA 靶点周围的序列 (0.1 µM, 最终浓度) (表 1), 并在20µL 的体积内执行主 PCR。
- 激活 DNA 聚合酶在95°c 5 分钟, 并执行主要 PCR 为30个周期在95°c 为三十年代, 60 °c 为三十年代和72°c 为二十四年代 (1 min/1 kb), 以最后的引伸在72°c 为5分钟。
- 采用1µL 的初级 pcr 产物和一对嵌套内引物进行二次 pcr。
- 在20µL 1× T7EI 反应缓冲器 (50 毫米氯化钠) 中变性和重新退火300-600 的纯化 PCR 产物, 10 毫米的三盐酸, 10 毫米氯化镁2, 1 毫米的有效率的 pH 值 7.9) 使用温度梯度从95°c 到25°c 以率5°c/5 分钟。
- 在退火的 PCR 产物中加入1µL 的 T7EI 酶, 并在37摄氏度上消化2小时。然后在1×TAE 缓冲区中的2% 琼脂糖凝胶上运行消化产物120伏40分钟, 直到碎片分离 (参见材料表)。
- 使用 ImageJ 确定切割和未切割 DNA 的带强度。使用以前报告的9 (图 1C) 中的方法计算 indel 频率。
-
捐赠载体的构建
注意: 为Cdx2基因生成 HMEJ 供体向量, 构造一个供方 DNA (800 bp HAL-p2A-mCherry-800 bp), 两端有 23 nt Cdx2-sgRNAs 目标序列 (图 1D和图 1E)。靶序列的 PAM 与同源臂的末端相邻。吉布森大会建议 HMEJ 捐赠者克隆。- 放大 800 bp 左同源臂 (HAL) 与前 primer-1F (包含 15-20 nt 重叠序列从向量, 23 nt Cdx2-sgRNA 目标序列, 约 20 nt 序列从 HAL) 和反向 primer-1R (包含 15-20 bp 重叠序列从p2A-mCherry 和大约 20 nt 序列从 HAL) 在0.1 µM 最后浓度使用小鼠基因组 DNA 在 200 ng/µL (图 1D,表 1)。
- 放大 p2A-mCherry 插入片段与前 primer-2F (包含 15-20 nt 重叠序列从 HAL 和大约 20 nt 序列从插入片断) 和反向 primer-2R (包含 15-20 nt 重叠序列从哈和大约 20 nt 序列从插入片段) 在0.1 µM 最终浓度使用基因组 DNA 或质粒与记者序列在 100 ng/µL 或 30 ng/µL (图 1D,表 1)。
- 放大 800 bp 右同源臂 (哈) 与前 primer-3F (包含 15-20 nt 重叠序列从向量, 23 nt Cdx2-sgRNA 目标序列, 约 20 nt 序列从哈) 和反向 primer-3R (包含 15-20 nt 重叠序列从p2A-mCherry 和大约 20 nt 序列从哈) 在0.1 µM 最后浓度使用小鼠基因组 DNA 在 200 ng/µL (图 1D,表 1)。
- 根据制造商的说明 (表 1), 在1×TAE 缓冲器上运行所有的 pcr 产品, 并在1% 琼脂糖凝胶上使用凝胶萃取试剂盒纯化预期尺寸的 pcr 产品。
- 用 KpnI 和 XbaI 消化50-100 的构造载体。混合2µL 的线性化向量在 30-40 ng/µL 与三 PCR 扩增片段 (1 µL 为每, 100-200 ng/µL) 在2x 吉布森混合。添加 H2O 可将最终卷调整为10µL. 孵育50°c 的混合物60分钟。
- 根据制造商的指示, 用所有组装的产品转换合格的大肠杆菌细胞, 并提取质粒结构。通过 DNA 测序验证 HMEJ 的捐献者。
2. 利用 HMEJ 方法对小鼠胚胎进行基因组编辑
-
Cas9 mRNA 的生产
- 对于 Cas9 mRNA 的制备, 通过使用表 1中列出的适当底漆对, 将 T7 启动程序序列添加到 Cas9 编码区域。在最终浓度为0.1 µM 和 20 Cas9 表达载体的 Cas9 中加入底漆, 1×高保真 DNA 聚合酶组合。用 H2O 将最终卷调整为50µL。
- 激活 DNA 聚合酶在95°c 5 分钟, 并执行 PCR 为36个周期在95°c 为三十年代, 60 °c 为三十年代和68°c 为4分钟 (1 min/1 kb), 以最后的引伸在68°c 为10分钟。
- 纯化 T7-Cas9 PCR 产品的体外转录 (IVT), 然后转录 0.5-1 µg DNA 的 mRNA 转录试剂盒在37°c 8 小时的总容积20µL, 根据制造商的说明 (见材料表)。
- 添加1µL 的 DNase 的混合物, 以删除 DNA 模板在37°c 15 分钟. 在37°c 添加一个聚 a 尾45分钟, 并通过 RNA 纯化试剂盒恢复 Cas9 mRNA, 根据制造商的说明 (请参阅材料表)。
-
sgRNA 生产
- 生成 sgRNA 模板驱动的 T7 启动者与高保真 DNA 聚合酶如上所述。选择一个包含矢量的 sgRNA 脚手架作为模板。使用的引物列在表 1中。
- 纯化 T7-sgRNA PCR 产品, 并使用 0.5-1 µg DNA 作为体外转录的模板, sgRNA 使用短 RNA 转录试剂盒, 在20µL 的总容积中, 在37°c 为6小时, 按制造商的说明 (参见材料表/c11>)。
- 添加1µL 的 DNase 的混合物, 并继续孵化在37°c 15 分钟删除 DNA 模板。用 RNA 纯化试剂盒净化 sgRNAs (见材料表)。
- 将 sgRNA 稀释至500µL RNase 水中, 并将样品保存在−80°c, 长达3月。
注: CRISPR ribonucleoproteins (RNPs) 是替代替代品, 具有更好的切割效率18,19,20。
-
胚胎采集、显微注射和体外培养
- Superovulate 雌 B6D2F1 (C57BL/6 x DBA2J) 小鼠 (7-8 周大), 孕母马血清促性腺激素 (PMSG), 其次为人绒毛膜促性腺激素 (hCG) 48 小时后。在 hCG 注射以后, 议院女性与 B6D2F1 男性过夜。
- 牺牲女性由 CO2麻醉, 24 h 在 hCG 注射以后。在 M2 培养基中收集受精胚胎的输卵管 (每个雌性的 30-50 胚)。
- 将受精的胚胎 (一天注射大约300颗鸡蛋) 放入 KSOM 培养基 (5.55 克/升氯化钠, 0.19 克/升氯化钾, 0.05 克/升2PO4, 0.05 克/升 MgSO47H2O, 0.04 克/升葡萄糖, 1.12 克/升乳酸钠, 2.1 克/升 NaHCO3, 0.02 克/l-丙酮酸钠, 0.25 克/升 CaCl2· 2H2O, 0.004 克/升 EDTA, 0.146 克/升谷氨酰胺, 1 克/升牛血清白蛋白), 在37摄氏度的孵化器与 5% CO2。
- 混合 Cas9 mRNA (100 ng/µL)、sgRNA (50 ng/µL) 和 HMEJ 捐助向量 (100 ng/µL), 并添加 H2O 将最终卷调整为10µL. 把混合物放在冰上。
- 拉毛细管针 (外径1.0 毫米, 内径0.78 毫米与灯丝) 使用微拉出器 (参数: 热, 74; 拉, 60; 速度, 80; 时间/延迟, 200; 压力, 300。请参阅材料表)。商业针头可以替代微注射。
- 在含有5µg/毫升细胞松弛素 B 的 HEPES-CZB 介质液滴中, 将混合物的可能体积注入受精卵的胞浆中, 并使用具有恒定流设置的 microinjector(图 2 a) (参见 材料表)21。
注: 每组受精卵应在20-30 分钟内注射细胞松弛素 B 可提高注射后小鼠受精卵的存活能力。或者, 显微注射可以与压电系统一起操作, 如前所述22。 - 培养注射受精卵在 KSOM 培养基在37°c 在 5% CO2之下直到胚泡阶段在3.5 天以后为荧光观察 (图 2B和 2C)。
-
小鼠胚胎移植及生成的研究
- 输精管切除术雄性小鼠与注射剂在同一天发情的雌性小鼠交配。
- 培养注射受精卵入2细胞阶段在37°c 在 5% CO2之下, 并且转移 25-30 2 细胞胚胎到 pseudopregnant 的输卵管在0.5 天后 coitum (dpc)。接受的母亲送幼崽在 19.5 dpc。
-
鼠标基因分型
- 根据制造商的说明 (请参阅材料表), 使用 DNA 提取试剂盒从脚趾或尾部样品中提取小鼠基因组 dna。
- 用紫外/可见光谱测量的 200-400 ng 基因组 DNA, 确定 5 ' 和 3 ' 的敲接事件, 作为模板进行 PCR amplification。
- 激活 DNA 聚合酶在95°c 5 分钟, 并执行 PCR 为38个周期在95°c 为三十年代, 60 °c 为三十年代和72°c 为1分钟 (1 min/1 kb), 以最后的引伸在72°c 为10分钟。对于 5 ' 连接, 使用前底漆在 HAL 的上游, 与反向一个在敲入片段 (p2A-mCherry)。至于 3 ' 结, 使用前底漆在敲入片段 (p2A-mCherry), 与反向一个在下游的哈 (表 1)。
- 运行6µL 的 PCR 产品1% 琼脂糖凝胶在1×TAE 缓冲和检查预期的片段大小。然后通过 DNA 测序验证它们 (图 2D)。
3. 肝细胞中基于 HMEJ 的体内基因组编辑
- 将收件人 C57BL/6J 鼠标 (8 周) 放在一个限制装置中, 并将尾部通过狭缝。
- 在2毫升盐水溶液中混合 HMEJ 供体载体 (30 µg) 和 spCas9 表达载体 (30 µg)。对于控制实验, 在2毫升盐水溶液 (图 3A) 中挂起 HMEJ 供体向量 (30 µg)。
- 用70% 乙醇清洁鼠标尾部。将针插入尾静脉, 并在5-7 秒内注入质粒 DNA 溶液. 取出针头, 从抑制装置中松开鼠标。
- 在注射后5-9 天后通过 CO2麻醉来牺牲老鼠。灌注小鼠 transcardially 0.9% 生理盐水, 然后用蠕动泵4% 多聚甲醛, 并在4摄氏度一夜之间修复肝脏。
- 在一夜之间用30% 蔗糖脱水组织, 直到它沉到管子的底部。
- 切片的冷冻组织在厚度为10µm 的肝脏样本。
- 冲洗三次在0.1 米磷酸盐缓冲 (PB) 和孵化他们的主要抗体: 兔抗 mCherry (稀释在 5%) 隔夜在4摄氏度。
- 在 PB 中洗涤三次, 然后用二级抗体孵化: Cy3-AffiniPure 山羊抗兔 IgG, 在室温下在眼眶上进行2小时的免疫。
- Counterstain 部分与 DAPI 20 分钟, 并在玻璃幻灯片上安装甘油, 以进一步荧光观察 (图 3B)。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
在小鼠胚胎中基于 HMEJ 的基因组编辑:为了定义小鼠受精卵中基于 HMEJ 的方法的敲除效率, 我们将 Cas9 mRNA、sgRNA 靶向Cdx2基因和 HMEJ 捐献者放入鼠标受精卵中, 该基因被设计用来将 p2A-mCherry 报告器中的一个子项与 Cdx2 的最后一个密码子结合起来. 基因 (图 2A)。注射的受精卵发展成囊胚在文化中。为了评价 mCherry 荧光的有效性, 我们用荧光显微镜对其进行了分析, 发现12.9% 的囊胚接受 HMEJ 捐献者是阳性的 mCherry, 这在滋养外胚层中严格表达 (图2B、2C)。通过对 PCR 阳性小鼠进行排序, 我们还发现所有检测到的集成事件都是 5 ' 和 3 ' 连接点的精确帧内集成 (图 2D)。
基于 HMEJ 的基因组编辑在成人组织和 HMEJ 介导的基因治疗:为了研究 HMEJ 基因组的编辑是否可以应用于成人组织, 我们在Actb基因的停止密码子前插入 mCherry 盒, 传感ActbHMEJ 构造 C57/B6J 小鼠肝的尾静脉流体力学注入 (图 3A)。经过7天的注射, 我们发现近半数的转染肝细胞表达 mCherry 染色在肝脏部分 (图 3B)。
为了探讨使用 HMEJ 的基因治疗策略的可能性, 我们使用了 fumarylacetoacetate 水解酶 () 缺陷小鼠.tyrosinemia 鼠标是一种成熟的遗传性类型 I (HTI) 鼠标模型, 它在基因的外显子5中放置一个插入片段, 导致以下序列23中的 frameshift 突变. 为了维护 trifluoromethylbenzoyl /小鼠, 我们用酪氨酸分解通路上游的抑制剂, 2-(2-硝基-4-)-13-cyclohexanedione (NTBC) 24 处理了.在这里, 我们出发去看看 MMEJ 和 HMEJ 介导的基因矫正是否能拯救/鼠标中的. 突变. 我们水力注入 Cas9 构造和-MMEJ 或 HMEJ 构造, 旨在将5到14的子显式 cDNA 插入遗传基因的内含子 4, 到. - 小鼠肝脏 ( 图 3C). 注射后一周, NTBC 被撤回以诱发肝脏损伤 (图 3C)。在 NTBC 退出后, HMEJ 小鼠接收的纠正肝细胞比基于 MMEJ 的方法显示更有效的扩散 (图 3 D ).

图 1: HMEJ 介导的目标集成体外.
(A) 选择 sgRNAs 的实验方案: 六个不同的 sgRNAs (Cdx2-sgRNA1~Cdx2sgRNA6) 基于在线 Cdx2 设计选择了具有较高秩和非目标电位的CRISPR轨迹的终止密码子。工具.protospacer 相邻的母题 (PAM) 序列为红色。(B) 实验设计: 将 sgRNA、Cas9 和 GFP 表达的 Cas9-CMV-GFP 表达质粒引入 N2a 细胞。GFP+单元格在3天被排序为验船师化验。(C) 对Cdx2目标进行测量的检测: 6 种不同的 sgRNAs 是为测量师设计的。正常 N2a 细胞基因组 DNA 作为控制。*, 用于Cdx2的 sgRNA 2 mCherry 敲入实验。(D) 使用吉布森组件构造 HMEJ 捐助者的示意图概述。(E) HMEJ 介导的基因靶向策略在Cdx2轨迹中的示意图概述。哈尔, 左/右同源臂;三角形, sgRNA 目标站点;/或, 外向前/反向底漆;IF/IR, 内前/反向底漆。从上一个报表10修改的图。请单击此处查看此图的较大版本.

图 2: 通过 HMEJ 介导的目标集成对小鼠胚胎进行基因组编辑
(A) 微注射实验方案: 将 Cas9 mRNA (100 ng/µL)、sgRNA (50 ng/µL) 和供体质粒 (100 ng/µL) 的混合物注射到小鼠受精卵中。(B) 由 HMEJ 策略编辑的小鼠胚胎的代表荧光图像。酒吧, 20 µm. (C) 按 mCherry+囊胚百分比表示的敲出效率。数字在每个酒吧之上, 总囊胚计数。(D) 在Cdx2轨迹上进行基因编辑的小鼠序列分析。PCR 产品从 5 ' 和 3 ' 结点被放大了测序。上部, 同源臂;紫色, p2A;红色, mCherry;或哈尔, 右或左同源的手臂。虚线标记为清晰而省略的区域。从上一个报表10修改的图。请单击此处查看此图的较大版本.

图 3: HMEJ 介导的目标集成在体内.
(A) 水动力尾静脉注射示意图概述。通过水动力尾静脉注入的方式, 表达了供体序列和 sgRNA 的质粒, 以及表达 spCas9 的质粒。(B) 代表肝细胞免疫荧光图像。肝切片采集7天后注射。缩放条, 50 µm, 转染的细胞。(C) MMEJ 或 HMEJ 介导的基因置换策略的质粒, 旨在将5到14的外显子的发 cDNA 插入到发基因的内含子4中, 并通过水动力注射将其传递到了该方法. NTBC on: NTBC - 小鼠保存在水上;NTBC: 撤退 NTBC 水 (NTBC 撤退的第一天被定义了0天, 是注射之后的第七天)。(D) MMEJ 的免疫组化染色, 其肝脏部分由 ./-小鼠注射而成, 其为 HMEJ 质粒。缩放条, 100 µm 从以前的报告修改过的图5,10。请单击此处查看此图的较大版本.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
HMEJ 供体质粒的构建最关键的步骤是: (1) sgRNA 的选择, 其 DNA 解切效率高, sgRNA 切割部位与停止密码子之间的距离小, (2) HMEJ 供体的适当构造。CRISPR/Cas9-mediated 对两种转基因供体载体 (包含 sgRNA 靶点和800的 bp 同源臂) 和靶向基因组在体内进行高效和精确靶向整合是必要的.采用 HMEJ 法生成敲鼠的最关键步骤是: (1) 制备高质量的 Cas9 mrna 和 sgRNA (Cas9 mrna 和 sgRNA 中不存在变性), (2) 制备高质量 HMEJ 供体质粒。质粒对胚胎发育无毒性作用。
最近, 还报告了一种基于 NHEJ 的方法, 用于有效的体内基因组编辑8。然而, 如前几份报告8所述, 不同类型的 indel 突变通常是在结点引起的, 因此很难实现精确的集成。在这里, 我们上面描述的基于 HMEJ 的策略显示了精确的目标集成, 几乎没有任何 indel 突变。因此, 一个基于 HMEJ 的策略可以是一个理想的平台, 用正确的方法替换突变序列 (如点突变), 而不适用于基于 NHEJ 的算法。
嵌合体是胚胎基因编辑的一个主要问题。在早期胚胎阶段注射 Cas9 蛋白代替 mRNA, 可以在没有嵌合体的情况下, 在一个细胞阶段实现转基因撞击。对于临床应用, 将 CRISPR/Cas9 系统送到成人组织中仍然具有挑战性。
基于 HMEJ 的基因组编辑有许多未来的潜在用途。它可以用来产生基因改良的动物模型。考虑到它在胚胎中的高敲效率, 这种方法可以显著减少产生基因改良动物模型所需的动物数量, 特别是开辟了生成非人类灵长类遗传模型的可能性。基于 HMEJ 的基因组编辑可以沿袭在成人组织中追踪单个细胞类型, 这对于动物模型特别有用, 因为缺乏可用的动物模型, 如非人类灵长类。它可以用于靶向基因治疗: HMEJ 策略最有吸引力的应用是基因治疗的临床使用。在本研究中, 我们修正了遗传性 tyrosinemia I. 型小鼠的出突变, 并通过流体动力学注入指示载体. 然而, CRISPR/Cas9 系统进入成人组织仍然是临床使用的主要技术挑战, 因为水动力注射不太可能在患者中执行。目前, 在将这种 HMEJ 的方法转化为临床前, 迫切需要进一步完善配送策略。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
作者没有什么可透露的。
Acknowledgments
这项工作得到了中科院战略优先研究项目 (XDB02050007, XDA01010409), 国家高技术研发 & 开发计划 (863 计划; 2015AA020307), 国家自然科学基金 (自然科学基金资助 31522037, 31500825, 31571509,31522038), 中国青年千人才项目 (以 hy), 突破中国科学院工程, 上海市科技委员会项目 (16JC1420202), 中国科学技术部 (大部分; 2016YFA0100500)。
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
遗传学 问题 133 CRISPR/Cas9 目标整合 同源介导的末端联接,在体内 胚胎 转基因小鼠 水动力注射Erratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
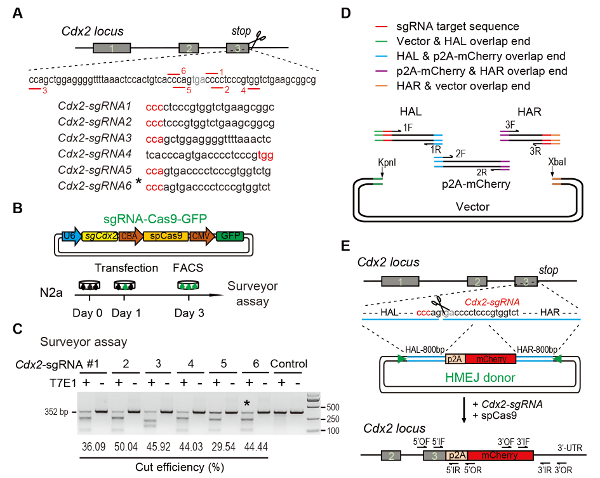
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
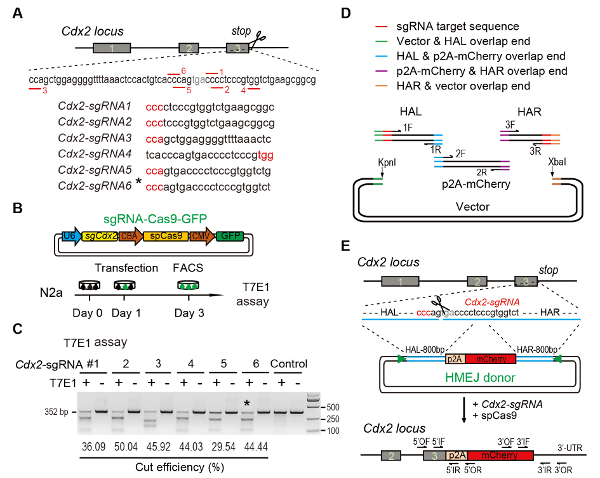
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
