

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
El cluster regularmente otro proteínas cortas repeticiones palindrómico/CRISPR asociado 9 (CRISPR/Cas9) sistema proporciona una herramienta prometedora para la ingeniería genética y abre la posibilidad de integración dirigida de los transgenes. Describimos una mediada por homología se terminan uniendo (HMEJ)-estrategia por turnos para ADN eficiente objetivo integración en vivo y dirigida terapias genéticas usando CRISPR/Cas9.
Abstract
Como una plataforma de edición de genoma prometedora, el sistema CRISPR/Cas9 tiene gran potencial para la manipulación genética eficiente, especialmente para la integración dirigida de los transgenes. Sin embargo, debido a la baja eficiencia de recombinación homóloga (HR) y las varias mutaciones indel de no homóloga se terminan uniendo (NHEJ)-basado en estrategias en las células no se dividen, en vivo genoma edición sigue siendo un gran desafío. Aquí, describimos una mediada por homología se terminan uniendo (HMEJ)-sistema CRISPR/Cas9 eficiente en vivo precisa integración específica. En este sistema, el genoma de destino y el donante vector que contiene los brazos de homología (~ 800 bp) flanqueado por destino de RNA (sgRNA) solo guía secuencias son troceadas por CRISPR/Cas9. Esta estrategia basada en HMEJ logra integración eficiente del transgen en cigotos de ratón, así como en hepatocitos en vivo. Por otra parte, una estrategia basada en HMEJ ofrece un enfoque eficaz para la corrección del fumarylacetoacetate hidrolasa (Fah) mutación en los hepatocitos y rescata Fah-deficiencia inducida por ratones de insuficiencia hepática. Tomados en conjunto, centrándose en el objetivo integración, esta estrategia basada en la HMEJ proporciona una herramienta prometedora para una variedad de aplicaciones, incluyendo la generación de modelos animales modificados genéticamente y terapias genéticas específicas.
Introduction
La edición del genoma, precisa, específica es a menudo necesaria para la producción de modelos animales modificados genéticamente y tratamientos clínicos. Se ha realizado mucho esfuerzo para desarrollar diversas estrategias para el genoma objetivo eficiente de edición, como nucleasas de dedos de zinc (ZFN), nucleasas efectoras como activador de transcripción (TALENs) y sistemas de CRISPR/Cas9. Estas estrategias crean roturas de doble cadena DNA específicas (OSD) en el genoma y toman ventaja de intrínsecos sistemas de reparación del ADN, como la recombinación homóloga (HR)1,2, microhomology-mediada se terminan uniendo (MMEJ)3 , 4 , 5y no homólogas terminan uniendo (NHEJ)6,7,8 para inducir la integración dirigida de los transgenes1,9. La estrategia de recursos humanos es actualmente el más de uso general genoma edición de enfoque, que es muy eficiente en las líneas celulares, pero no fácilmente accesibles a las células no se dividen por su ocurrencia restringida en la fase tardía de la S/G2. Así, la estrategia de recursos humanos no es aplicable para en vivo la edición del genoma. Recientemente, la estrategia basada en NHEJ se desarrolló para eficaz de genes knock-en tejidos de ratón8. Sin embargo, el método basado en NHEJ usualmente presenta indels en las uniones, lo que es difícil generar la edición del genoma exacto, especialmente cuando se trata de construir en el marco de la fusión de genes8. Integración dirigida basada en MMEJ es capaz de la edición del genoma exacto. Sin embargo, sólo modestamente aumenta la eficiencia de integración específicas en informes anteriores5. Por lo tanto, mejorar la eficiencia de integración específicas precisa en vivo se necesita urgentemente para amplias aplicaciones terapéuticas3.
En un trabajo publicado recientemente, hemos demostrado una mediada por homología se terminan uniendo (HMEJ)-basado en la estrategia, que mostró la mayor eficiencia de integración específica en todo divulgado estrategias, tanto in vitro e in vivo10. Aquí, describimos un protocolo para el establecimiento del sistema HMEJ, y también la construcción de los vectores de RNA (sgRNA) solo guía al gen de interés y el donante vectores propios sitios de destino sgRNA y ~ 800 bp de brazos de homología (figura 1) . En este protocolo, también se describen los pasos detallados para la generación de ADN knock-en ratones y breves pasos de integración específica en tejidos en vivo. Por otra parte, un estudio de prueba de concepto de la estrategia basada en HMEJ demostrado su capacidad para corregir la mutación de la Fah y rescatar a Fah- / - hígado insuficiencia ratones, que además revelaron su potencial terapéutico.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Todos los procedimientos incluyendo temas animales han sido aprobados por el Comité de ética de investigación biomédica en los institutos de Shangai para la ciencia biológica (CAS).
1. diseño de donante plásmidos
-
Selección de sgRNA
- Utilizar herramientas de diseño en línea CRISPR para predecir sgRNAs en la región de destino de12,11,13,14,15. Para el locus Cdx2 , diseño seis sgRNAs diferentes (Cdx2-sgRNA1 ~Cdx2-sgRNA6) alrededor de la parada codon con mayor rango y menor potencial de-objetivos (figura 1A)16.
- Alinear 2 μg de vectores de expresión Cas9-CMV-EGFP y sgRNA por digestión BbsI (1 μl de BbsI por 2 h a 37 ° C a una concentración final de 1 U/μl de un volumen de 20 μl). Luego purificar el producto kit de purificación de gel con un gel de agarosa al 1% en Tampón TAE 1 x.
- Mezcla un par de oligonucleótidos sgRNA en 10 μl de 1 × T4 ADN ligasa buffer una concentración final de 50 μm. Incubar la solución oligo utilizando un gradiente de temperatura de 95 ° C a 25 ° C con una tasa de cambio de temperatura de 5 ° C/5min (95 ° C durante 5 min. entonces de 90 ° C por 5 min, 85 º C por 5 min, etcetera.), que hibridan los oligos.
- Mezcle 4 μL de recocido producto, 2 μl del vector linearizado con 1 μl de T4 ADN ligasa en 10 μl de tampón de 1 × T4 DNA ligasa y luego ligar a 22 ° C durante 1-2 h (figura 1B).
-
Ensayo de nucleasa de topógrafo de sgRNA
Nota: Se evaluó la eficacia dirigida a de sgRNA utilizado para el experimento de golpe por análisis de nucleasa de topógrafo (también conocido como endonucleasa T7 I (T7EI) ensayo)17. Seleccione lo sgRNA con alta eficiencia de clivaje de ADN y una baja distancia entre el sitio de corte sgRNA y el codón de parada.- Transfectar vectores de expresión Cas9-sgRNA-EGFP en líneas de células N2a cultivadas en DMEM suplementado con suero bovino fetal 10%, 1% PSG y el 1% no esenciales aminoácidos por la transfección kit (véase Tabla de materiales). Incube las células transfected a 37 ° C en 5% CO2.
- Después de 48 h de incubación, recoger 5.000 células transfected (GFP+) por celular activado por fluorescencia (FACS) de clasificación usando las células transfectadas no como control.
- Digerir las células recogidas en 2-5 μl de tampón de oflysis (0,1% Tritón X-100, 0,1% Tween 20 y 100 μg/mL proteinasa K) a 56 ° C durante 30 minutos y luego calor inactivar la proteinasa K a 95 ° C durante 10 minutos.
- Amplificar la muestra por PCR anidada (tabla 1) utilizando el protocolo del fabricante. El tamaño de los productos PCR se establece en 300-500 bp.
- Mezclar 1 μl del producto de la lisis con polimerasa de la DNA y un par de cebadores externos reconociendo la secuencia alrededor del sitio de destino sgRNA (0,1 μm, concentración final) (tabla 1) y realizar la primaria PCR en un volumen de 20 μl.
- Activar el ADN polimerasa a 95 ° C por 5 min y realizar la PCR primaria por 30 ciclos a 95 ° C por 30 s, 60 ° C durante 30 s y 72 ° C por 24 s (1 minuto/1 kb), con una extensión final a 72 ° C durante 5 minutos.
- Llevar a cabo la polimerización en cadena secundario usando 1 μl del producto PCR primario y un par de cebadores internos anidados.
- Desnaturalizar y volver a cocer 300-600 ng del producto PCR purificado en 20 μl de tampón de reacción T7EI (50 mM NaCl, 10 mM Tris-HCl, 10 mM de MgCl2, 1 mM TDT pH 7.9) de × 1 utilizando un gradiente de temperatura de 95 ° C a 25 ° C con una tasa de 5 ° C/5min.
- Añadir 1 μl de enzima de T7EI a los productos de PCR recocidos y digerir a 37 ° C por 2 h. Luego ejecute el producto de la digestión en gel de agarosa al 2% en Tampón TAE de 1 × 120 V durante 40 minutos hasta que los fragmentos están separados (véase Tabla de materiales).
- Utilizar ImageJ para determinar las intensidades de la banda de corte y de la DNA sin cortar. Calcular la frecuencia de indel usando los métodos previamente registrados9 (figura 1C).
-
Construcción del vector de donantes
Nota: Para generar vectores de donantes HMEJ de gen Cdx2 , construir un donante de ADN (800 bp HAL-p2A-mCherry-800 bp HAR) flanqueado por 23 nt Cdx2-sgRNAs a la secuencia en ambos extremos (figura 1D yEde la figura 1). El PAM de la secuencia de destino estaba adyacente al extremo del brazo homólogo. Asamblea de Gibson se recomienda para donantes HMEJ clonación.- Amplificar el brazo de homología izquierda bp 800 (HAL) con avance cartilla-1F (que contiene 15-20 nt superposición secuencia de vectores, 23 nt Cdx2-sgRNA secuencia dirigida a y cerca de 20 secuencia de nt de HAL) y cartilla-1R (contiene la secuencia de 15-20 bp superposición de p2A-mCherry y cerca de 20 secuencia de nt de HAL) en concentración final de 0.1 μm usando la DNA genomic de ratón a 200 ng/μl (D,figura 1, tabla 1).
- Amplificar el fragmento de inserción p2A-mCherry con avance cartilla-2F (contiene 15-20 nt superposición secuencia de HAL y cerca de 20 secuencia de nt del fragmento de inserción) y revertir la cartilla-2R (contiene 15-20 nt superposición secuencia de HAR y secuencia de nt cerca de 20 de el fragmento de inserción) en concentración final de 0,1 μm utilizando genomic ADN o plásmido con secuencias de reportero a 100 ng/μl o 30 ng/μl (D,figura 1, tabla 1).
- Amplificar el brazo de derecho homología bp 800 (HAR) con avance cartilla-3F (que contiene 15-20 nt superposición secuencia de vectores, 23 nt Cdx2-sgRNA secuencia dirigida a y cerca de 20 secuencia de nt de HAR) y cartilla-3R (contiene secuencia de superposición de nt de 15-20 de p2A-mCherry y cerca de 20 secuencia de nt de HAR) en concentración final de 0.1 μm usando la DNA genomic de ratón a 200 ng/μl (D,figura 1, tabla 1).
- Ejecutar todos los productos PCR en gel de agarosa al 1% en Tampón TAE 1 x y purificar los productos PCR del tamaño esperado kit de extracción de gel, según las instrucciones del fabricante (tabla 1).
- Digerir el 50-100 ng de un vector de construcción con KpnI y XbaI. Mezcle 2 μl del vector linearizado en 30-40 ng/μl con tres PCR amplifica fragmentos (1 μl de cada uno, 100-200 ng/μL) en 2 x mezcla de Gibson. Agregar H2O para ajustar el volumen final de 10 µL.Incubate la mezcla a 50 ° C durante 60 minutos.
- Competentes de e. coli de transformar las células con el producto ensamblado y extracto el plásmido construye por kit de extracción de ADN según las instrucciones del fabricante. Secuenciación de ADN para comprobar al donante HMEJ.
2. genoma edición en embriones de ratón utilizando el método basado en la HMEJ
-
Producción de mRNA de Cas9
- Para la preparación de Cas9 mRNA, agregue la secuencia de promotor de T7 a la Cas9 codificación región por amplificación por PCR utilizando el par de cartilla apropiados listado en la tabla 1. Añadir la cartilla Cas9 F/R en una concentración final de 0.1 μm y 20 ng de Cas9 expresando el vector 1 × alta fidelidad mezcla de ADN polimerasa. Ajustar el volumen final de 50 μL con H2O.
- Activar el ADN polimerasa a 95 ° C por 5 min y realizar la PCR para 36 ciclos a 95 ° C por 30 s, 60 ° C durante 30 s y 68 ° C por 4 minutos (1 minuto/1 kb), con una extensión final a 68 ° C durante 10 minutos.
- Purificar el producto de PCR de Cas9 T7 en vitro transcripción (IVT) y luego transcribir 0.5-1 μg de ADN por kit de transcripción de mRNA a 37 ° C durante 8 horas en un volumen total de 20 μl, según las instrucciones del fabricante (véase Tabla de materiales).
- Añadir 1 μl de DNasa a la mezcla para quitar la plantilla de ADN a 37 º C durante 15 minutos agregar una cola poly-A 45 min a 37 ° C y recuperar el mRNA Cas9 por kit de purificación de RNA, según las instrucciones del fabricante (véase Tabla de materiales).
-
Producción de sgRNA
- Generar la plantilla sgRNA conducida por un promotor T7 con polimerasa de la DNA de la alta fidelidad como arriba. Elegir un andamio sgRNA que contienen vectores como plantilla. Los cebadores utilizados se enumeran en la tabla 1.
- Purificar el producto PCR de T7-sgRNA y utilizar 0.5-1 μg de DNA como plantilla para la transcripción en vitro de sgRNA usando un kit de transcripción de RNA corto a 37 ° C durante 6 h en un volumen total de 20 μl, según las instrucciones del fabricante (ver tabla de materiales de < / c11 >).
- Añadir 1 μl de DNasa a la mezcla y continuar la incubación a 37 ° C durante 15 min eliminar la plantilla de la DNA. Purificar el sgRNAs por kit de purificación de RNA, como anteriormente (véase Tabla de materiales).
- Diluir el sgRNA a 500 ng/μl de agua libre de ARNasa y almacenar las muestras en el ° C −80 hasta 3 meses.
Nota: Ribonucleoproteínas CRISPR (RNPs) son una substitución de la alternativa con mejor corte eficiencia18,19,20.
-
Cultivo de colección, microinyección y en vitro de embriones
- Superovulate femeninos B6D2F1 (C57BL/6 × DBA2J) ratones (7-8 semanas) de gonadotropina de suero de yegua preñada (PMSG), seguidos de gonadotropina coriónica humana (hCG) 48 h más tarde. Después de la inyección de hCG, mujeres de casa con los varones B6D2F1 durante la noche.
- Sacrificio de las hembras por anestesia de CO2 , 24 h después de la inyección de hCG. Recogen los embriones fecundados de sus oviductos (con 30-50 embriones para cada mujer) en medio de M2.
- Lugar los embriones fertilizados (cerca de 300 huevos para la inyección de un día) en medio KSOM (5,55 g/L de NaCl, KCl, 0.05 g/L de KH2PO4, 0,05 G/l MgSO4•7H2O, 0.04 g/L glucosa, 1,12 g/L sodio de 0,19 g/L lactato, 2,1 g/L NaHCO3 , piruvato de sodio 0.02 g/L, 0,25 g/L CaCl2•2H2O, 0,004 g/L de EDTA, 0,146 gr/L glutamina y 1 g/L albúmina de suero bovino) a 37 ° C en una incubadora con 5% CO2.
- Mezcla Cas9 mRNA (100 ng/μL), sgRNA (50 ng/μL) y donantes HMEJ vector (100 ng/μL) y agregar H2O para ajustar el volumen final de 10 μl. poner la mezcla en hielo.
- Tire las agujas capilares (diámetro externo de 1,0 mm, diámetro interno 0,78 mm con filamento) utilizando un extractor de micropipeta (parámetros: calor, 74, tirón, 60; velocidad, 80; retardo, 200 presión, 300. Véase tabla de materiales). Agujas comerciales sería una sustitución alternativa para la microinyección.
- Inyectar un volumen probable de la mezcla en el citoplasma de cigotos con pronúcleos definidas en una gota de medio de CZB HEPES que contiene 5 μg/mL citocalasina B utilizando un microinyector con constante flujo de configuración (figura 2A) (ver Tabla de materiales)21.
Nota: Se debe inyectar cada grupo de cigotos dentro de 20-30 min Cytochalasin B podría aumentar la viabilidad del zigoto de ratón después de la inyección. Por otra parte, microinyección puede funcionar con el sistema piezoeléctrico, como se describió anteriormente22. - La cultura los cigotos inyectados en medio KSOM a 37 ° C debajo del 5% de CO2 hasta blastocisto etapa después de 3,5 días para observación de la fluorescencia (figuras 2B y 2C).
-
Transferencia de embriones y la generación de ratones
- Compañero de estros ratones hembras ICR con vasectomía ratones machos ICR en el mismo día de la inyección.
- La cultura los cigotos inyectados en la fase de 2 células a 37 ° C con 5% CO2y embriones de 2 células de transferencia 25-30 en oviductos de las hembras ICR seudopreñadas en 0,5 días post coitum (dpc). Las madres beneficiarias entregan cachorros en dpc 19,5.
-
Genotipificación de ratón
- Extracto de ADN genómico de ratón de dedo del pie o cola muestras utilizando un kit de extracción de ADN, según las instrucciones del fabricante (véase Tabla de materiales).
- Identificar a la Unión 5' y 3' de knock-eventos de uso de 200-400 ng de ADN genómico medido por espectrofotometría de UV/vis como plantilla para realizar la polimerización en cadena amplification.
- Activar el ADN polimerasa a 95 ° C por 5 min y realizar la PCR para 38 ciclos a 95 ° C por 30 s, 60 ° C durante 30 s y 72 ° C por 1 minuto (1 min/1 kb), con una extensión final a 72 ° C durante 10 minutos. Para el cruce de 5', use el primer avance en el upstream de la HAL, con el reverso en el fragmento de knock (p2A-mCherry). En cuanto a 3' cruce, utilice el primer avance en el fragmento de knock (p2A-mCherry), con el reverso en el abajo del HAR (tabla 1).
- Ejecutar 6 μl del producto PCR en gel de agarosa al 1% en 1 x TAE buffer y compruebe el tamaño del fragmento esperado. Luego verificar por secuencia (figura 2D) de la DNA.
3. HMEJ-basado en Vivo genoma edición en hepatocitos
- Coloque el receptor ratón C57BL/6J (8 semanas) en un dispositivo de restricción y poner la cola a través de la ranura.
- Mezcla de vectores donantes HMEJ (30 μg) y vectores de la expresión de la spCas9 (30 μg) en 2 mL de solución salina. Para el experimento control, suspender vectores donantes HMEJ (30 μg) en 2 mL de solución salina (figura 3A).
- Limpiar la cola de ratón con etanol al 70%. Insertar la aguja en la cola de la vena e inyectar la solución de ADN de plásmido dentro de 5-7 s. Retire la aguja y suelte el ratón desde el dispositivo que refrena.
- Sacrificio de ratones por CO2 anestesia después de 5-9 días después de la inyección. Inundar lo ratones transcardially con solución salina al 0.9%, seguido de paraformaldehído al 4% utilizando una bomba peristáltica y fijar el hígado durante la noche a 4 ° C.
- Deshidratar el tejido con sacarosa al 30% durante la noche, hasta que se hunde hasta el fondo del tubo.
- Sección el tejido congelado en un espesor de 10 μm para las muestras de hígado.
- Enjuague las secciones tres veces con tampón fosfato 0,1 M (PB) e incubar con el anticuerpo primario: conejo anti-mCherry (diluido en 5% NGS) durante la noche a 4 ° C.
- Lávese las secciones tres veces en PB y luego los incubar con el anticuerpo secundario: Cy3-AffiniPure de cabra anti-conejo IgG por 2 h a temperatura ambiente en un agitador orbital.
- Contratinción las secciones con DAPI por 20 min y montar con glicerina en portaobjetos de vidrio para mayor observación de fluorescencia (figura 3B).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Edición del genoma basado en HMEJ en embriones de ratón: Para definir la eficiencia knock-in del método basado en la HMEJ de cigotos de ratón, entregamos Cas9 mRNA, sgRNA contra el gen Cdx2 y el donante HMEJ en cigotos de ratón, que fue diseñado para fundir un gen reportero de p2A-mCherry al codón de último de la Cdx2 gen (figura 2A). Los cigotos inyectados convirtieron en blastocistos en la cultura. Para evaluar la eficacia de la knock-in, se analizó la fluorescencia mCherry con un microscopio de fluorescencia, y encontramos que el 12,9% de los blastocistos recibir donantes HMEJ eran positivo para mCherry, que se expresaba terminantemente en el trophectoderm (figuras 2B 2 C). Mediante la secuenciación de los ratones positivos de PCR, también se encontró que todos los examinaron eventos de integración fueron integraciones en el marco preciso en las ensambladuras 5' y 3' (figura 2D).
Edición del genoma basado en HMEJ en tejidos adultos y terapia génica mediada por HMEJ: Para investigar si la edición del genoma basado en HMEJ podría aplicarse en tejidos adultos, nos inserta el casete mCherry justo antes del codón de parada del gen Actb transducing Actbconstrucciones de - HMEJ de hígado de ratón C57/B6J por cola-vena inyección hidrodinámica (figura 3A). Después de 7 días de inyecciones, se encontró que casi la mitad de los hepatocitos transfected expresado mCherry como manchados en las secciones de hígado (figura 3B).
Para explorar la posibilidad de usar una estrategia basada en HMEJ para la terapia génica, se empleó la hidrolasa del fumarylacetoacetate (Fah)-ratones deficientes. El ratón Fah- / - es un bien establecido Tirosinemia hereditaria tipo I modelo de ratón (HTI), que alberga un fragmento de inserción en el exón 5 del gen Fah , causando mutaciones del mutágeno ' frameshift ' en la siguiente secuencia23. Para mantener el Fah- / - ratones, tratamos el Fah- / - ratones con un inhibidor de la aguas arriba de la vía catabólica de la tirosina, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24. Aquí salimos a ver si la corrección génica mediada MMEJ y HMEJ podría rescatar Fah mutación en el ratón Fah- / . Inyectamos hidrodinámico Cas9 construir junto con Fah- MMEJ o Fah- HMEJ construcciones, diseñadas para insertar Fah cDNA del exón 5 a 14 en el intrón 4 del gen de la Fah , Fah- / - ratón (hígados Figura 3 C). una semana después de la inyección, Nebt se retiró para inducir daños en el hígado (figura 3C). Después de la retirada de NTBC, Fah-hepatocitos corregidas de los Fah- / - ratones recibiendo Fah- HMEJ y construcciones Cas9 demostraron la proliferación más eficaz que el método basado en MMEJ (figura 3D ).

Figura 1 : HMEJ-mediada dirigida integración en vitro.
(A) esquema Experimental para la selección de sgRNAs: seis sgRNAs diferentes (Cdx2-sgRNA1 ~Cdx2-sgRNA6) en el codón de parada del locus Cdx2 con un potencial más alto rango y objetivo elegido se basaban en diseño CRISPR online herramienta. La secuencia de protospacer motivo adyacente (PAM) está en rojo. Diseño Experimental (B): plásmidos de expresión la Cas9-CMV-GFP expresión sgRNA, Cas9 y GFP fueron introducidos en las células N2a. Las células GFP+ fueron clasificadas en el día 3 para ensayo de topógrafo. Topógrafo (C) ensayo de Cdx2 targeting: 6 sgRNAs diferentes fueron diseñados para el análisis del topógrafo. ADN genómico de células N2a normal sirve como control. *, el sgRNA utilizado para Cdx2-2A-mCherry knock-en experimento. (D) Descripción esquemática de la construcción de los donantes HMEJ usa Gibson. (E) Descripción esquemática del gen HMEJ-mediada contra estrategia en locus Cdx2 . Brazo de homología HAL/HAR, izquierda/derecha; triángulos, sitios de destino sgRNA; DE / OR, cartilla de avance/retroceso externa; IF / IR, interno primer avance/retroceso. Figura modificada del anterior informe10. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2 : Edición del genoma en embriones de ratón vía mediada por HMEJ objetivo integración
(A) esquema Experimental de microinyección: una mezcla de Cas9 mRNA (100 ng/μL), plásmidos sgRNA (50 ng/μL) y de los donantes (100 ng/μL) fueron inyectadas en cigotos de ratón. (B) imágenes de fluorescencia representante de embriones de ratón creados por estrategia HMEJ. Bar, 20 μm. (C) Knock-in eficiencia indica porcentaje de mCherry+ blastocistos. Número encima de cada barra, blastocistos total contados. (D) análisis de ratones gene-editado en Cdx2 locus de la secuencia. Productos PCR amplificados de sitios de unión 5' y 3' fueron ordenados. Superior, brazo de homología; púrpura, p2A; rojo, mCherry; HAR o HAL, brazo homólogo derecho o izquierdo. Las líneas punteadas marcan la región omitida para mayor claridad. Figura modificada del anterior informe10. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3 : HMEJ-mediada dirigida integración en vivo.
(A) Descripción esquemática de la inyección en la vena hidrodinámica cola. Una mezcla de plásmidos expresar sgRNA y secuencia de donantes y plásmidos expresar spCas9 fueron entregados al hígado a través de inyección en la vena hidrodinámica cola. (B) imágenes de inmunofluorescencia representante de hepatocitos. Las secciones de hígado fueron recogidas 7 días después de la inyección. Barra de escala 50 μm. GFP, transfected las células. (C) plásmidos de cualquier estrategia de reemplazo de gene MMEJ o HMEJ-mediada por diseñado para insertar Fah cDNA del exón 5 a 14 en el intrón 4 del gen de la Fah fueron entregados en hígado de ratón Fah- / - por inyección hidrodinámica. NTBC en: Fah- / - ratones se mantuvieron en agua NTBC; NTBC off: retiro del agua NTBC (el primer día de retiro NTBC fue definido como día 0, que es el 7 º día después de la inyección). (D) Fah inmunohistoquímica tinción de secciones hepáticas de Fah- / - ratones inyectados con plásmidos MMEJ o HMEJ. Barra de escala, 100 μm. figura modificada de anteriores informes5,10. Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Los pasos más críticos en la construcción de plásmidos de donantes HMEJ son: (1) selección de lo sgRNA con alta eficiencia de clivaje de ADN y baja distancia entre sgRNA corte y codón de parada y construcción (2) adecuada de donantes HMEJ. Escote CRISPR/Cas9-mediada en tanto vector de donante transgénico (contiene ~ 800 bp homología brazos y sitios de destino sgRNA) y genoma específica es necesario para la integración específica eficiente y precisa en vivo. Los pasos más críticos de generación de golpe en ratones, utilizando el método basado en la HMEJ son: (1) la preparación de alta calidad de Cas9 mRNA y sgRNA (ninguna degeneración existe en Cas9 mRNA y sgRNA) y (2) la preparación del plásmido de donantes HMEJ de alta calidad. El plásmido muestra sin efectos tóxicos sobre el desarrollo embrionario.
Recientemente, un método basado en NHEJ también se ha divulgado para el eficiente en vivo genoma edición8. Sin embargo, varios tipos de mutaciones indel generalmente fueron inducidos en las ensambladuras, como se ha descrito en anteriores informes8, lo que hace difícil lograr la integración precisa. Aquí, la estrategia de HMEJ que describimos anteriormente mostró precisa integración dirigida con apenas ninguna mutaciones indel. Así, una estrategia basada en HMEJ podría ser una plataforma ideal para la sustitución de una secuencia mutada (como una mutación de punto) con la correcta, que no es aplicable para el método basado en NHEJ.
Mosaicismo es un problema importante para la edición de genes en embriones. Inyección de Cas9 proteína en vez de mRNA en una anterior etapa embrionaria puede lograr transgénicos knock en una etapa de célula sin mosaicismo. Para usos clínicos, entrega de los sistemas CRISPR/Cas9 en tejidos adultos es todavía un reto.
Hay muchos usos potenciales futuros de la edición del genoma basado en HMEJ. Puede utilizarse para generar modelos animales modificados genéticamente. Teniendo en cuenta su alta knock-in eficiencia en embriones, este método podría reducir significativamente el número de animales necesario para la generación de modelos animales modificados genéticamente y sobre todo abre la posibilidad de generar modelos genéticos de primates no humanos. La edición del genoma basado en HMEJ puede tipos de la célula individual de seguimiento del linaje en tejidos adultos, que es especialmente útil en modelos animales, ya que hay una falta de modelos animales disponibles, como primates no humanos. Puede ser utilizado para terapias genéticas específicas: la aplicación más atractiva de una estrategia basada en HMEJ es la terapia génica para clínica utiliza. En este estudio, hemos corregido la mutación de la Fah de Tirosinemia hereditaria tipo I ratones por inyección hidrodinámica de los vectores indicados. Sin embargo, entrega del sistema CRISPR/Cas9 en tejidos adultos sigue siendo el mayor reto técnico para el uso clínico, como inyección hidrodinámica es poco probable que realizar en los pacientes. En la actualidad, más mejora de la estrategia de entrega es urgente antes de traducir este método HMEJ basado en la clínica.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
Este trabajo fue apoyado por CAS estratégicas prioritarias programa de investigación (XDB02050007, XDA01010409), el nacional Hightech R & D programa (programa 863; 2015AA020307), la Fundación Nacional de Ciencias naturales de China (NSFC concede 31522037, 31500825, 31571509, 31522038), programa de talentos China juvenil de mil (en HY), rotura a través del proyecto de la Academia China de Ciencias, Shanghai ciudad Comité de ciencia y tecnología proyecto (16JC1420202 a HY), el Ministerio de ciencia y tecnología de China (más; 2016YFA0100500).
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
Genética número 133 CRISPR/Cas9 integración específica mediada por la homología se terminan uniendo en Vivo embrión genéticamente ratones inyección hidrodinámicaErratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
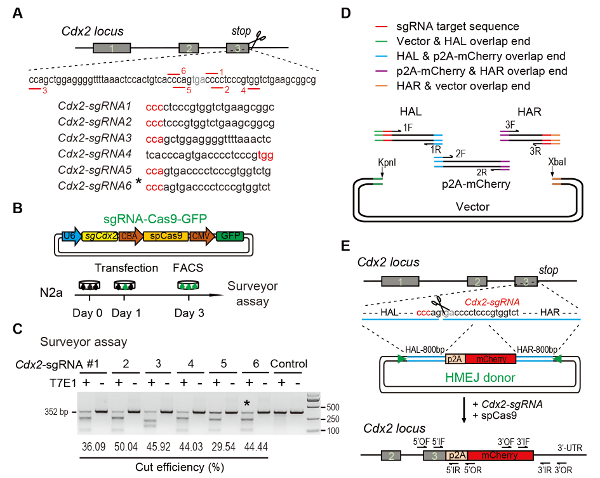
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
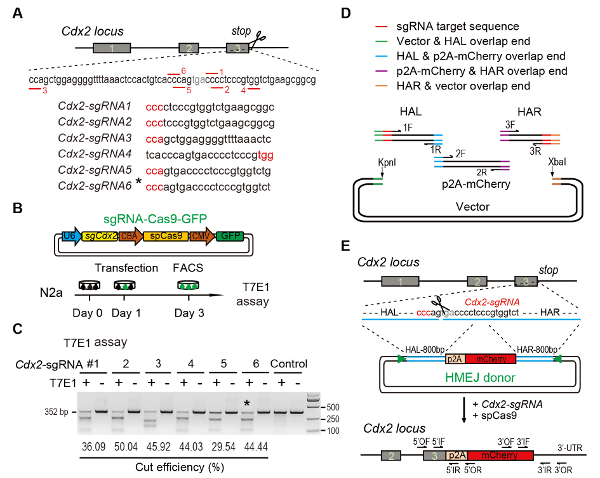
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
