

Summary
Biología sintética permite la ingeniería de proteínas con propiedades sin precedentes mediante la inserción co-translational de aminoácidos no-canónico. Aquí, se presenta como una variante espectralmente rojo-cambiado de puesto de un fluoróforo tipo GFP con propiedades espectroscópicas fluorescencia novela, llamado proteína fluorescente de "oro" (GdFP), se produce en e. coli mediante incorporación de presión selectiva (SPI).
Abstract
Proteínas fluorescentes son herramientas fundamentales para las ciencias biológicas, en particular para microscopía de fluorescencia de las células vivas. Mientras que las variantes tipo salvaje y de ingeniería de la proteína fluorescente verde de Aequorea victoria (avGFP), así como homólogos de otras especies ya cubren grandes partes del espectro óptico, un espacio espectral permanece en la región del infrarrojo cercano, por que fluoróforos basada en avGFP no están disponibles. Variantes de la proteína fluorescente rojo-cambiado de puesto (FP) ampliaría considerablemente el toolkit para unmixing espectral de múltiples especies moleculares, pero los FPs rojo-cambiado de puesto naturalmente derivados de corales o anémonas de mar tienen menor rendimiento cuántico de fluorescencia y foto inferior-estabilidad en comparación con las variantes avGFP. Más manipulación y posible expansión del sistema conjugado del cromóforo hacia la región espectral far-red está limitado también por el repertorio de 20 aminoácidos canónicos prescritos por el código genético. Para superar estas limitaciones, la biología sintética puede lograr aún más espectral rojo cambio a través de la inserción de aminoácidos no-canónicos en la tríada del cromóforo. Describimos el uso de SPI a ingeniero avGFP variantes con novedosas propiedades espectrales. Expresión de la proteína se realiza en un auxotrófica triptófano de e. coli cepa y complementando los medios de crecimiento con precursores de indole adecuado. Dentro de las células, estos precursores son convertidos en los correspondientes análogos de triptófano e incorporados en las proteínas de la maquinaria ribosomal en respuesta a codones UGG. La sustitución de Trp-66 en la variante "cian" mejorada del avGFP (ECFP) por un electrón-donar 4-aminotryptophan resultados en GdFP con un cambio de Stokes 108 nm y una emisión fuertemente rojo-cambiado de puesto máximo (574 nm), mientras que termodinámicamente más estable que su predecesor PLCE. Incorporación de residuos específicos del aminoácido no-canónico se analiza por espectrometría de masas. Las propiedades espectroscópicas de GdFP se caracterizan por espectroscopia de fluorescencia de tiempo resuelto como una de las aplicaciones valiosas de FPs genéticamente codificados en Ciencias de la vida.
Introduction
Desde el descubrimiento de la proteína verde fluorescente en las medusas Aequorea victoria (avGFP) en 19621 y la primera expresión heteróloga en 19942 en otras células eucariotas, son proteínas fluorescentes de la familia GFP herramientas muy valiosas y objetivos en Ciencias de la vida. Ingeniería molecular y genética extensa incluida la regulación de la utilización de codones específicos, aceleración de plegable mejor maduración, mayor brillo, prevención de la Oligomerización y adaptación de las propiedades espectrales y fotoquímicas incluyendo la capacidad reversible photoswitch3,4,5,6. GFP debe su fluorescencia de su 4-(p- hydroxybenzylidene) cromóforo imidazolidin-5-one (HBDI). Este último está autocatalytically formado por la tríada llamado cromóforo de aminoácidos (Ser-65/Tyr-66/Gly-67 en avGFP) después de la formación de un enlace covalente adicional dentro de la espina dorsal del peptide bajo la influencia del oxígeno molecular7. El sistema conjugado resonantly estabilizado interactúa dinámicamente con su entorno molecular, permitiendo la absorción en el rango visible y fluorescencia verde característica de estas proteínas.
Dentro de la tríada del cromóforo, es obligatoria la presencia de un aminoácido aromático. Sin embargo, el repertorio estándar aminoácido consta de sólo cuatro residuos aromáticos (His, Phe, Trp y Tyr). Esto limita los enfoques convencionales de mutagénesis para lograr variantes avGFP rojo-cambiado de puesto substancialmente más con respecto a las FPs natural más rojo-cambiado de puesto como DsRed8 de coralimorphs striata Discosoma o mKate/mNeptune9 de la anémona de mar Entacmaea quadricolor. Por lo tanto, la parte far-red y de infrarrojo cercano del espectro óptico por encima de 600 nm está escasamente cubierta por variantes de la GFP. Esto es, por supuesto, una limitación severa para los acercamientos microscópicos fluorescencia que requiere demultiplexar espectral de varias especies de fluoróforo al mismo tiempo. Por ejemplo, marcadores de longitud de onda larga son necesarios para hacer uso del régimen de baja absorción de tejido de la piel entre 700-1.000 nm en configuración para tejido profundo imagen10.
Proteínas fluorescentes procedentes de avGFP se dividen en varias clases basadas en las propiedades espectroscópicas y naturaleza química de sus cromóforos11. Con su tríada 65/Ser-Tyr-Gly-66/67, el cromóforo de tipo salvaje existe como una mezcla equilibrada entre la forma neutra, fenólica (λmax = 395 nm, ε = 21.000 M-1cm-1) y la forma aniónica phenolate (λmax = 475 nm, ε = 7.100 M -1cm-1), y el espectro de emisión presenta un solo pico en 508 nm. El grupo del oxhidrilo del Ser-65 es de importancia crítica, como dona un H-bond a Glu-222 en las proximidades del cromóforo (distancia: 3.7 Å), que promueve la ionización de este carboxilato. Clase se caracterizan por un cromóforo aniónico phenolate, como EGFP (64-Phe-Leu/65-Ser-Thr; λmax = 488 nm, ε = 35.600 M-1cm-1, λem = 509 nm). Debido a la substitución de Ser-65-Thr(Ala,Gly), se suprime el pico de excitación nm 395 de la forma neutral del fenol y el 470-475 nm pico de phenolate aniónico es cinco a seis veces mejorado y cambiado de puesto a 490 nm. Clase II compone de proteínas con un cromóforo fenólico neutral, al igual que en zafiro-GFP. Aquí, la sustitución de Thr-203-Ile suprime casi totalmente la excitación nm 475, dejando sólo el pico en 399 nm. Ya que el cromóforo aniónico no puede correctamente solvatados, su forma neutral es el favorito. Clase III comprende las variantes fluorescentes "amarillas" (EYFP; Ser-65-Gly/Val-68-Leu/ser-72-ala/THR-203-Tyr; Λ εmax = 514 nm, ε = 84, 600 M-1cm-1, λem = 527 nm) con interacción de apilamiento π de una cadena lateral aromática y phenolate, como provocada por las sustituciones del Thr-203-His(Trp,Phe,Tyr), que conducen a 20 nm máximos de emisión rojo-cambiado de puesto (Thr-Tyr-203). Sustitución más (Gln-69-Lys) resulta en otro cambio roja de 1-2 nm a 529 nm, la variante más rojo-cambiado de puesto del avGFP conocido11. El cambio del fenol de un indol (Tyr-66-Trp) crea clase IV, como en el PLCE cian fluorescente (Ser-65-Thr/Tyr-66-Trp; λmax1 = 434 nm, ε = 24.800 M-1cm-1, λmax2 = 452 nm, ε = 23.600 M-1cm-1 ; Λem1 = 477 nm, λem2 = 504 nm). El alojamiento de lo voluminoso indol probablemente está habilitado por otros, mutaciones compensatorias. Los máximos de excitación y emisión de ECFP caen entre las de proteínas con cromóforos neutros o aniónicos. Las proteínas de la clase V del puerto un imidazol en lugar de fenol (Tyr-66-su), por ej., azul fluorescente proteínas como EBFP. Clase VI se produce por un intercambio de fenol-fenil favoreciendo la forma neutral del cromóforo, que por lo tanto lleva a las más azul-cambiado de puesto de excitación y emisión pico posiciones (360 nm y 442 nm, respectivamente).
Mutagénesis sitio-dirigida clásica es especialmente conveniente para la producción de variantes de cromóforo novela avGFP, por la permutación del tripéptido y residuos interactúan en el marco de los 20 aminoácidos canónicos de 65-67. Estas posibilidades pueden ampliarse más cuando se introdujeron variantes no-canónico de aminoácidos aromáticos durante la síntesis de proteínas12. En principio, hay dos maneras de lograr esto. La primera estrategia se basa en la tolerancia del substrato de la maquinaria de traducción de proteínas, especialmente de aminoacil-tRNA sintetasas (aaRSs) hacia análogos de aminoácidos relacionados. Para lograr esto con eficacia alta, auxotrófica cepas de expresión de e. coli se emplean que son incapaces de sintetizar el aminoácido natural correspondiente. Esto permite la sustitución de éste mediante la adición de aminoácidos no-canónico adecuados (ncAAs) o precursores de ella al medio de cultivo. Esta estrategia, también conocido como incorporación de presión selectiva (SPI)13,14permite reemplazos específicos de residuos, que resultan en la incorporación global de la ncAA. La segunda estrategia usa codón ARNt supresor que se encargan de la ncAA por ingeniería aaRS enzimas. Esto resulta en la readthrough de codones de parada en el marco y permite la incorporación específica de ncAA. Por lo tanto, este método de supresión del codón de parada (SCS) lleva a la expansión del código genético15. Mediante mutagénesis, se coloca un codón de parada en el gene de la blanco en el sitio deseado. En principio, SPI puede utilizarse también para crear péptidos recombinantes y proteínas teniendo una única instalación de ncAA, dado que raro aminoácidos canónicos como la Met o Trp son escogidos para la sustitución. Con Trp, enfoques SPI se ha demostrado que trabajar con una gran variedad de análogos como 4 - F - y 5 - F - y 6-F-Trp, 7-aza-Trp, 4-OH - y 5-OH-Trp, así como 4 - 5-NH2- Trp o incluso β (thienopyrrolyl) alanina derivados16 ,17,18,19,20. Así, SPI podría ser altamente ventajoso para la sustitución de aminoácidos aromáticos de cromóforos GFP por variantes no-canónico para explorar la posibilidad de adaptar más espectros y Stokes shift de estos FPs. En cuanto a cualquier modificación de la secuencia de la proteína, la compatibilidad con la maduración de plegamiento y el cromóforo FP debe probarse experimentalmente.
En este trabajo, utilizamos la clase IV ECFP21, que lleva en lugar de la avGFP de tipo salvaje Tyr, un residuo de Trp en su tríada de cromóforo. Con el SPI, esta Trp-66 (y Trp-57, el sólo otro residuo Trp en PLCE) es sustituido por 4-amino-TRP. La presencia del grupo amino electrón-donar de 4-amino-Trp en el cromóforo favorece la estabilización de la resonancia de una transferencia de protón del estado excitado mucho rojo-cambiado de puesto (ESPT) dotada de un cambio de Stokes 108 nm. Esta proteína fluorescente "oro" (GdFP) constituye la variante con el cambio de red más grande de la fluorescencia máxima (574 nm) entre todas las proteínas derivadas del avGFP. Describimos el método de producción de proteína GdFP SPI y proporcionar los protocolos para el análisis obligatorio de las proteínas modificadas resultantes por espectroscopía de masas. Además, mostramos cómo GdFP puede ser utilizada y analizada en métodos de espectroscopia de fluorescencia de tiempo resuelto.
Protocol
1. transformación de e. coli de Trp auxotrófica
- Transformar químicamente o espectro de células (50 μL) de una tensión auxotrófica Trp de e. coli , por ejemplo. ATTC 49980 (WP2, mutante derivada de la cepa de e. coli B/R22), con 1 μl de una solución acuosa de 1 ng/μl del plásmido de His6 ECFP pQE - 80 L con shock térmico o electroporación, respectivamente. Por favor consulte la base de datos educación ciencia de JoVE23,24 para más detalles.
Nota: El vector de expresión pQE - 80L His6-ECFP codifica un N-terminal 6 x su etiquetado ECFP21 impulsado por un promotor T5 bacteriano con el operador de lac. Además lleva un marcador de selección AmpR y un origen de replicación de colE1 (la secuencia de pQE - 80 L vector columna vertebral puede encontrarse en: https://www.qiagen.com/mx/resources/resourcedetail?id=c3b71572-4d82-4671-a79b-96357fe926d1&lang=en & autoSuggest = true). El peso molecular teórico de la proteína de tipo salvaje de His6 ECFG (después de la maduración del cromóforo25) es Da 28303.92. La secuencia de la proteína objetivo traducido es como sigue (su-etiqueta subrayada, secuencias derivadas de vectores en negrita): MRGSHHHHHHGSMVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTLTWGVQCFSRYPDHMK
QHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYISHNVYITADKQKNGIKANFKIRHNIEDGS
VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYK. - Las células transformadas en placas de LB-agar (tabla 1) suplidas con glucosa 10 g/L, 100 ampicilina μg/mL de la placa e incubar las placas a 37 ° C durante la noche.
2. expresión de la proteína recombinante
-
La cultura de e. coli ATCC 49980 pQE - 80 L His6-PLCE
- Preparar 5 mL de medio LB (tabla 1; complementado con glucosa 10 g/L, 100 ampicilina μg/mL) en un 14 mL estéril poliestireno cultura tubo de crecimiento aeróbico e inocular con un solo colonias de una placa de agar utilizando un asa de punta o inoculación de pipeta estéril.
Nota: Se recomienda el uso de colonias de células recién transformadas. Las placas con colonias de bacterias (en el paso 1.2.) pueden almacenarse a 4 ° C por varios días. - Incubar durante una noche las células a 37 ° C en un agitador orbital a 200-250 rpm.
- Preparar 5 mL de medio LB (tabla 1; complementado con glucosa 10 g/L, 100 ampicilina μg/mL) en un 14 mL estéril poliestireno cultura tubo de crecimiento aeróbico e inocular con un solo colonias de una placa de agar utilizando un asa de punta o inoculación de pipeta estéril.
-
Expresión de tipo salvaje ECFP
- Inocular el medio LB fresco 10 mL (tabla 1; complementado con glucosa 10 g/L, 100 ampicilina μg/mL) con 100 μl de la cultura durante la noche en un erlenmeyer de 100 mL. Incube el frasco a 37 ° C en un agitador orbital a 200 rpm.
Nota: Opcionalmente, este paso puede realizarse en 10 mL NMM19 suplementado (tabla 1) con 100 μg/L ampicilina y 0.5 mM de L-triptófano (alternativamente, puede utilizarse las indol). - Medir la densidad óptica a 600 nm (OD600) cada 20 minutos preferentemente medir densidad de la célula determinando la extinción de 600 nm (OD600) en un espectrofotómetro usando una cubeta con una longitud de ruta de acceso de 1 cm. siempre realizar una referencia medición en el medio de cultivo correspondiente. Diluir las muestras y mezclar las muestras bien para obtener un valor de 0.1-0.8, entonces calcular OD600 usando el factor de dilución. Para obtener más información, consulte publicación anterior 26.
- Al llegar a un valor de600 OD de 0.5-0.8 (aproximadamente 2-3 h después de la inoculación), tomar muestra "antes de la inducción" para SDS-PAGE (electroforesis en gel de poliacrilamida sodio dodecil sulfato, paso 4).
- Inducir la expresión de la proteína objetivo ajustando la cultura líquida a 0,5 mM de IPTG (Isopropil β-D-1-tiogalactopiranósido, de solución de 1 M) e incubar a 30 ° C en un agitador orbital a 200 rpm durante 4-8 h.
Nota: Comúnmente se expresan proteínas fluorescentes cian a temperaturas inferiores a 37 ° C27. - Tomar la muestra "después de la expresión" para SDS-PAGE (paso 4.).
- La cosecha de las células bacterianas por centrifugación 10 min a 5.000 x g a 4 ° C.
- Eliminar el sobrenadante por decantación y la congelación de los pellets de células a-20 ° C o -80 ° C hasta la purificación de proteínas objetivo.
- Inocular el medio LB fresco 10 mL (tabla 1; complementado con glucosa 10 g/L, 100 ampicilina μg/mL) con 100 μl de la cultura durante la noche en un erlenmeyer de 100 mL. Incube el frasco a 37 ° C en un agitador orbital a 200 rpm.
-
SPI para producir GdFP
- Inocular 10 mL de medio de NMM19 (tabla 1) complementado con 100 de μg/mL ampicilina, triptófano μm 15 y 10 μl del cultivo durante la noche en un erlenmeyer de 100 mL e incubar el frasco de cultura durante la noche a 30 ° C en un agitador orbital a 200 rpm.
Nota: Existe una variedad de medios químicamente definidos para el cultivo de e.coli y SPI. Además de NMM usado aquí, MOPS medio28, glucosa y minerales sales medio29, Davis media mínima30, M9 media mínima de31o GMML32 puede ser utilizado. - Al día siguiente, medir OD600 cada 30 minutos hasta que el valor sólo cambia por menos de 0.05 sobre 30 minutos. El valor de la meseta debe ser aproximadamente 1.
Nota: Las desviaciones de ± 0,3 unidades son aceptables. Dependiendo de la cepa bacteriana y el medio utilizado, la concentración de triptófano inicial (paso. 2.3.1) puede ser necesario ajustar. - Tomar muestra "antes de la inducción" para SDS-PAGE (paso 4.).
- La cosecha de las células bacterianas por centrifugación 10 min a 5.000 x g a 4 ° C. Eliminar el sobrenadante por decantación.
- Resuspender las células en 10 mL de medio de NMM19 con 100 ampicilina μg/mL en un matraz Erlenmeyer de 100 mL y agregar 4-amino-indole a una concentración final de 1 mM con solución madre de 50 mM. Continuar la incubación durante 30 min a 30 ° C en un agitador orbital a 200 rpm.
Nota: Este paso se recomienda debido a la baja estabilidad química de la ampicilina y asegura la absorción celular de 4-amino-indole. - Inducir la expresión de la proteína blanco añadiendo IPTG a una concentración final de 0,5 mM, con valores de 1 M e incubar la muestra durante la noche a 30 ° C en un agitador orbital a 200 rpm.
Nota: Comúnmente se expresan proteínas fluorescentes cian a temperaturas inferiores a 37 ° C27. - Al día siguiente, medir OD600.
- Tomar la muestra "después de la expresión" para SDS-PAGE (paso 4.).
- La cosecha de las células bacterianas por centrifugación 10 min a 5.000 x g y 4 ° C y descarte el sobrenadante por decantación.
- En caso de que dicha embarcación no fue utilizada para la centrifugación, transferir el precipitado de células en un tubo de poliestireno de cónico de 50 mL con una espátula. Congelar el precipitado de células a-20 ° C o -80 ° C hasta la purificación de proteínas objetivo.
- Inocular 10 mL de medio de NMM19 (tabla 1) complementado con 100 de μg/mL ampicilina, triptófano μm 15 y 10 μl del cultivo durante la noche en un erlenmeyer de 100 mL e incubar el frasco de cultura durante la noche a 30 ° C en un agitador orbital a 200 rpm.
3. blanco purificación de proteínas mediante cromatografía de afinidad de iones metálicos inmovilizados (IMAC)
-
Lisis de la célula bacteriana
- Descongelar el precipitado de células en hielo durante 10-20 min.
- Resuspender el precipitado de células en un tubo de poliestireno de cónico de 50 mL con 5 mL de tampón de unión fría (tabla 1) en el hielo.
- Añadir 20 μl de lisozima de 50 mg/mL, 20 μl de 1 mg/mL de DNasa I, 20 μl de 1 mg/mL Rnasa A. cerrar el tubo, mezclar suavemente por inversión 5 veces y mantenerlo en hielo durante 30 minutos.
Nota: La interrupción parcial de la célula ocurre como catalizada por la lisozima. - Lyse las células de sonificación utilizando una punta de homogeneizador de ultrasonidos mediante tres ciclos de 3 minutos en un tubo de poliestireno de 15 mL refrigerado por hielo granizado con 2 s de pulso, 4 s de amplitud de la pausa y el 45%.
Nota: Como alternativa, homogeneización de alta presión puede usarse, por ej., 20 ciclos a 14.000 psi. Si es necesario, diluir con tampón de Unión para alcanzar el volumen mínimo del instrumento. Por otra parte, reactivos de extracción de proteína pueden utilizarse para la interrupción de la célula. Véase tabla de materiales por ejemplo. - Centrifugue la muestra durante 30 min a 15.000-18.000 x g, 4 º C.
- Transferir el sobrenadante a un tubo nuevo y anotar el volumen de líquido.
- Filtrar la solución con un filtro de jeringa de 0.45 μm utilizando una jeringa con cierre Luer plástico 5 mL y un fluoruro del polivinilideno (PVDF) filtro de la jeringuilla.
- Tomar muestra "lisado" para SDS-PAGE (paso 4.).
- Resuspender el precipitado de desechos de células en ddH2O (a igual volumen como ex lisado).
- Tomar "pellets" de muestra para SDS-PAGE (paso 4.).
-
Purificación de IMAC
- Utilice un 1 mL preenvasados o llena la columna IMAC FPLC (cromatografía líquida rápida de proteínas) según las instrucciones del fabricante. Uso de tampón de unión (tabla 1) para el equilibrado de la columna así como para la etapa de lavado que sigue después de que el lisado celular se ha aplicado a la columna.
- Piscina y recoger fracciones de eluato con GdFP que puede ser identificado por el color dorado luz visible.
Nota: Opcionalmente, la proteína diana puede ser eluida utilizando un gradiente lineal imidazol (0-250 mM) utilizando un sistema automatizado de FPLC. - Determinar la concentración de proteína usando el valor de la literatura para el coeficiente de extinción a 466 nm (ɛ466 nm = 23.700 M-1 cm-1)33 con tampón de elución como referencia. Para obtener más información sobre el procedimiento, consulte publicación anterior26.
- Tome el "efluente" de muestra para SDS-PAGE y use 1-10 μg de proteína por carril en el caso de tinción de Coomassie.
Nota: Cantidades de muestra de SDS pueden variar dependiendo del método de coloración y tinte sensibilidad. - Dializarse una alícuota de las fracciones de eluato contra diálisis buffer o tampón de MS con una membrana de un peso molecular límite (MWCO) 5.000-10, 000. Preparar según las instrucciones del fabricante de la membrana de diálisis. Dializan de una muestra de 1 mL tres veces contra 100 mL de tampón durante al menos 2 h. Para obtener más información sobre este procedimiento, consulte la publicación anterior34.
- Para el almacenamiento, congelar la muestra de proteína en el buffer de diálisis a-80 ° C.
Nota: Alícuotas deben ser estables durante al menos 6 meses.
4. preparación de la muestra SDS-PAGE del extracto de células completas de e. coli
- Transferir una suspensión equivalente a 1 mL de OD600 = 1 suspensión (ej. 500 μl de OD600= 2) en un tubo de microcentrífuga de 1,5 mL.
- Recoger las células por centrifugación durante 10 minutos a 5000 x g, temperatura ambiente. Deseche el sobrenadante mediante pipeteo.
- Añadir 80 μl de ddH2O y 20 μl de SDS 5 x cargando buffer de tinte (tabla 1) para el precipitado de células y mezclar mediante pipeteo.
- Desnaturalizar las células por calentamiento a 95 ° C por 5 min en un baño o calor bloque. Posteriormente, enfriar las muestras a temperatura ambiente.
- Utilice 10 μL para la tinción de Coomassie SDS-PAGE según la anterior publicación35.
Nota: Cantidades de muestra de SDS pueden variar dependiendo del método de coloración y tinte sensibilidad.
5. Análisis de la masa intacta de la proteína por cromatografía líquida de alto rendimiento (HPLC) acoplado a Electrospray ionización tiempo de vuelo espectrometría de masas (LC-ESI-TOF-MS)
Nota: Reservas, ajustes y HPLC gradiente pueden variar dependiendo de la columna de la separación y el instrumento utilizado. Ver tabla de materiales para equipo ejemplar.
- Determinar la concentración de proteína de una muestra se dializaron contra MS buffer como se describió anteriormente (paso 3.2.3.) con MS buffer (ver la tabla de materiales) como referencia.
- Diluir la muestra de proteína usando MS buffer para un volumen final de 80 μl de 0,1 mg/mL, mezclar mediante pipeteo cuidadoso, transferir la solución a un vial de muestreadores de MS con inserto de vidrio y cerrar con una tapa. Quite las burbujas de aire agitando el frasco.
- Llenar un segundo frasco muestreador automático sin inserto de vidrio (buffer en blanco) con 1 mL de tampón de MS.
- Que el instrumento se caliente. Realizar la calibración del instrumento. Asegúrese de que hay suficientes cantidades de disolventes cromatografía líquido-grado (> 100 mL).
- Un gradiente HPLC lineal 20 min del 5% al 80% buffer un (0,1% ácido fórmico en ddH2O), combinado con el almacenador intermediario B del programa (el ácido fórmico 0,1% en acetonitrilo).
- Iniciar HPLC a un flujo de 0,3 mL/min y esperar hasta que la presión de la columna es estable.
- Un volumen de inyección de muestreador automático de 5 μL para el método LC-ESI-TOF-MS, crear una lista de tareas para ejecutar en blanco y después un funcionamiento de la muestra y asignar el correspondiente inyector automático vial posiciones. Ejecutar la lista de trabajo.
- Una vez terminada la lista de tareas, abra el archivo de datos generado. Seleccionar un rango en el ion total actual (TIC) parcela deconvolución y deconvolucionar el espectro MS utilizando el algoritmo de deconvolución de máxima entropía.
Nota: Dependiendo de las condiciones experimentales, otras especies pueden ocurrir de FP no madurado o aductos de iones del tampón.
6. fluorescencia mediciones de toda la vida y los espectros asociados a caries (DAS) de GdFP
Nota: Para la instrumentación de la espectroscopia de fluorescencia de tiempo resuelto, consulte Tabla de materiales de equipo ejemplar. La absorbancia como excitación de fluorescencia y espectros de emisión de proteínas fluorescentes puede también grabar usando espectrofotómetros UV/Vis y fluorescencia de laboratorio.
-
Medición de vida resueltos de longitud de onda de la fluorescencia de GdFP
- Preparar 2 mL de 1 solución μm de GdFP por dilución en tampón PBS (tabla 1) a pH 7. Llene la solución en una cubeta de cuarzo de 1 cm.
- Instalar ps-pulsada 470 nm láser de excitación de la muestra y el filtro de emisión de pase de largo 488 nm y ajustar la rejilla del correlación de tiempo y longitud de onda fotón contando detector de36 (TWCSPC) para la adquisición del régimen de longitud de onda de 600 L/mm 500-700 nm.
- Adquirir la emisión de fluorescencia a una velocidad de recuento de 200 x 103 fotones/s hasta 103 cuentas se acumulan en el máximo de adquisición de las curvas de decaimiento de la fluorescencia con software de conteo de fotones individuales.
-
Medición de la respuesta instrumental función37 (IRF)
- Vuelva a colocar la cubeta de muestra con una cubeta de cuarzo de 1 cm con sílice coloidal 1 g/L (~ 220 m2/g) en buffer de PBS a pH 7.
Nota: La suspensión de sílice está preparada con una suspensión acuosa de 400 g/L. - Quitar 488 nm emisión long-pass filtro y filtros de inserto gris para ajustar la tasa de conteo en el detector de TWCSPC por debajo de 100 x 103 cuentas/s.
- Ajuste la rejilla para la adquisición de 470 fotones nm en canal 8 del detector TWCSPC de 16 canales.
- Adquirir el IRF hasta aproximadamente 10 x 103 cuentas se acumulan en la emisión máxima.
- 38 programa de guarnición convertir curvas de decaimiento de la fluorescencia y IRF a archivos de datos ASCII con la global.
- Conducta Global encajan según un modelo de una suma de tres componentes exponenciales con cursos de la vida como parámetros enlazados.
- Parcela caries asociadas espectros (DAS) distribuciones de la amplitud de los componentes individuales del decaimiento en dependencia de la longitud de onda con software de análisis de datos.
- Vuelva a colocar la cubeta de muestra con una cubeta de cuarzo de 1 cm con sílice coloidal 1 g/L (~ 220 m2/g) en buffer de PBS a pH 7.
Representative Results
Usando la técnica de incorporación de presión selectiva, Trp-66 en la tríada del cromóforo de PLCE (y Trp-57, el sólo otro residuo Trp en ECFP) puede sustituirse por 4-amino-Trp, generando la GdFP rojo-cambiado de puesto con propiedades espectrales distintas. Espectrometría de masas debe utilizarse para demostrar la integración estequiométrica deseada del aminoácido no-canónicos en la proteína, con resultados que se muestran en la figura 1. Luego, nos proporcionan datos de microscopía, espectroscopia de absorción UV-Vis, espectroscopia de fluorescencia de estado estacionario y resueltos de tiempo y longitud de onda para caracterizar las propiedades del fluoróforo GdFP con énfasis en la dependencia del pH de la espectros.
Para confirmar el cambio de los dos residuos de Trp en ECFP de 4-amino-Trp, se realizan análisis de espectrometría de masa. La figura 1 muestra un espectro representativo de ESI-MS deconvoluted de GdFP. Mientras que ECFP de tipo salvaje tiene una masa calculada de proteína de 28,283.9 Da después de la maduración del cromóforo, la masa correspondiente de GdFP es Da 28,313.9. El espectro ESI-MS deconvoluted de GdFP exhibe un pico masivo principal en 28,314.1 ± 0,1 Da, que se desvía del valor teórico por menos de 10 ppm. Estar dentro del rango de exactitud típica para este tipo de análisis25, esto confirma la incorporación de la ncAA por SPI (valor experimental para ECFP tipo: Da 28,283.7).
La figura 2 muestra la proyección de imagen de microscopía de fluorescencia confocal imágenes (CFIM) de las células bacterianas que expresan ECFP, EGFP, EYFP y GdFP a la resuspensión de bacterias en tampón PBS. Todas las imágenes fueron adquiridas en un microscopio equipado con una excitación de objetivo y láser UV en la misma energía para cada muestra.
Figura 3A muestra una superposición de imágenes CFIM de bacteria e. coli expresan varios FPs incluyendo GdFP, siempre vigilado con la energía de excitación muy similar (longitudes de onda como en la figura 2). Figura 3B muestra las estructuras del cromóforo de las variantes FP que se muestra. En relación con el brillo de GdFP comparado con PLCE (fluorescencia cuántica rendimiento φ = 0.4), EGFP (φ = 0,6) y EYFP (φ = 0,6) es importante tener en cuenta que para GdFP, una gama más amplia de la adquisición de la luz de la fluorescencia (30 nm) fue utilizado en contraste con 20 nm utilizado para otros spe cies, con el fin de ajustar la intensidad de las imágenes a valores similares. Con un coeficiente de extinción ligeramente menor y un rendimiento cuántico reducida como consecuencia de propiedades fotofísicas única, el brillo de GdFP es menor en comparación con los otros FPs se muestra.
El espectro de absorción de PLCE (figura 3) tiene dos máximos característicos a 434 nm y 452 nm. Por el contrario, GdFP se caracteriza por una banda de absorción ancha rojo-cambiado de puesto con el máximo a 466 nm. La absorción de EGFP es más rojo-cambiado de puesto a 488 nm. Sin embargo, debido al más grande cambio de Stokes de GdFP (108 nm) en comparación con el PLCE (41 nm) y EGFP (20 nm), el espectro de emisión de GdFP es el más rojo-cambiado de puesto de los tres GFP derivados del investigado aquí (figura 3D). Mientras que la emisión de fluorescencia de ECFP muestra dos máximos característicos a 475 nm y 505 nm, EGFP tiene una amplia emisión principal banda alcanzando un máximo de 508 nm (λmax) con un hombro leve a 540 nm. La fluorescencia de GdFP aparece en aproximadamente 565 nm (λmáx.) (Figura 3D). Su espectro de emisión contiene una pequeña contribución de ECFP de tipo salvaje que también es visible como un pequeño hombro a 475 nm. Esta pequeña fracción de PLCE se sintetiza antes de la inducción durante el procedimiento SPI, como descrito33.
Figura 3E muestra los cambios de pH-dependiente en el espectro de absorción de GdFP. Para un cambio de pH de 8 a 5, la emisión máxima cambia ligeramente al rojo y se observa un leve ensanchamiento de la banda de absorción. Sin embargo, la reducción de la amplitud de la absorción es sólo alrededor del 10% entre pH 8 y pH 5, indicando que las propiedades del suelo estado del cromóforo GdFP muy débilmente modificado por pH.
El tiempo resuelve la emisión de fluorescencia por recuento de fotones individuales se muestra en la figura 4. Las curvas de decaimiento en los canales espectrales en 550 nm y 600 nm (Figura 4A) muestra un ligeramente más rápido decaimiento de la fluorescencia a 600 nm frente a la decadencia a 550 nm. Los resultados de un ajuste global de la fluorescencia del decaimiento curvas con dos componentes exponenciales como resultado dos componentes de decaimiento espectral distinguible de la fluorescencia con constantes de tiempo de 1.0 ns y 3.3 ns (figura 4 y D).
La emisión de fluorescencia de GdFP depende del pH, como es típico de muchas variantes de la proteína fluorescente de la familia GFP. Figura 4B se compara la emisión de fluorescencia de GdFP entre pH 5 y pH 8, que muestra claramente una disminución en la intensidad de fluorescencia en el pH más bajo, mientras que las características espectrales permanecen constante.
Los espectros asociados a caries (DAS)39 de GdFP (figura 4 y D) se caracterizan por dos bandas de emisión distintos. La contribución de la lenta 3.3 componente de ns es más pronunciada en el rango de longitud de onda corta alrededor 550 nm (60%) con menor contribución del componente más rápido (40%). A 600 nm, ambos componentes tienen sobre la misma amplitud. A un cambio de pH 7 (figura 4) a pH 6 (figura 4), las características espectrales del DAS apenas cambia y las constantes de tiempo de la rutina de instalación global también son los mismos (la precisión de las constantes de tiempo DAS es de ± 0,15 ns). Sin embargo, la diferencia de las amplitudes absolutas de los dos componentes DAS es claramente evidente que totalmente representa la amplitud de la emisión de fluorescencia reducida sobre el mismo cambio de pH en la Figura 4B.
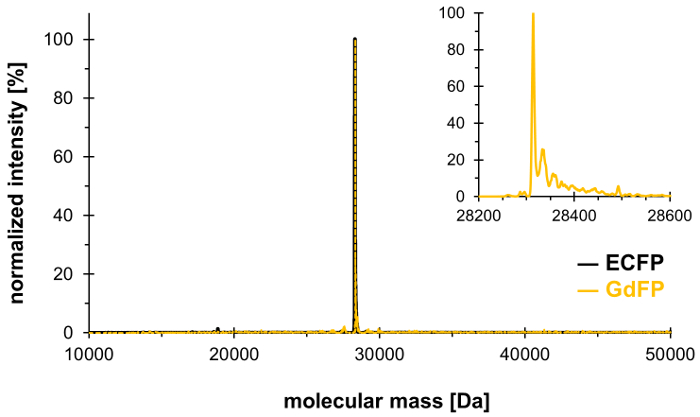
Figura 1: espectro de ESI-MS deconvoluted representante de GdFP. El espectro ESI-MS de GdFP (color oro, magnifica parcela muestra como detalle) muestra un pico principal en Da 28314.1 (calcula el valor 28313.9 Da). En negro se muestra el espectro para ECFP de tipo salvaje. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: imágenes de microscopía de fluorescencia Confocal de poblaciones bacterianas que expresan diferentes FPs. Los valores siguientes de longitud de onda fueron utilizados para la adquisición de la imagen: ECFP (λex = 457 nm, detección: 461-480 nm), EGFP (λex = 488 nm, detección: 495-515 nm), GDFP (λex = 476 nm, detección: 560-590 nm), EYFP (λex = 514 nm, detección: 520-530 nm). Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: propiedades espectrales de GdFP. (A) CFIM imagen de una mezcla de células bacterianas que expresan ECFP, EGFP y GdFP después la resuspensión de bacterias en tampón PBS. (B) estructuras cromóforo GdFP (con 4-amino-Trp en lugar de residuos 66), los padres ECFP (con Trp en la posición 66) y EFGP (con Tyr en la posición 66). (C) comparación de los espectros de absorción normalizados de GdFP, PLCE y EGFP, mientras que en (D), el espectro de emisión de fluorescencia normalizado de PLCE (excitación a 430 nm) es en comparación con los espectros de emisión de fluorescencia de EGFP y GdFP (ambos excitó a 450 nm). (E) pH-dependencia de los espectros de absorción (normaliza la absorción a 280 nm). Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: fluorescencia de tiempo resuelto de GdFP. (A) fluorescencia decaimiento de GdFP seguimiento por tiempo y longitud de onda-resuelto fotón contando en los canales espectrales en 550 nm y 600 nm (± 12,5 nm) después de excitación con pulsos de láser de 470 nm. La función de la respuesta instrumental (IRF) proporciona información sobre la resolución de tiempo de la configuración usada. (B) variación del espectro de emisión de GdFP dependiente de pH (excitación a 460 nm). (C, D) Asociada a caries espectros (DAS) de GdFP en pH 7 (C) y pH 6 (D) determinado después de deconvolución de fluorescencia resuelta en tiempo y longitud de onda se descompone y montaje global de caries en todos los canales por un sistema global de dos funciones exponenciales con constantes de tiempo enlazado. Haga clic aquí para ver una versión más grande de esta figura.

Figura 5: estructuras de la transferencia de carga intramolecular de PLCE (negro) y cromóforos GdFP (oro). El aumento de tamaño del sistema cromóforo por el donante del electrón buena de un grupo amino como parte de la ncAA permite la formación de estructuras más mesomérico para lograr la resonancia de estabilización del estado excitado. Se muestran los puntos de conexión para el andamio de la FP como semicírculos. Haga clic aquí para ver una versión más grande de esta figura.
| Solución madre de | concentración, solvente | Nota | |
| 20% D-glucosa | 200 g/L D-glucosa en ddH2O | esterilizar por filtración a través de un filtro de la jeringuilla del tamaño del poro de 0.45 μm | |
| indol | 50 mM de isopropanol | ||
| 4-amino-indole | 50 mM en 20% de etanol (20 mL de etanol en un volumen final de 100 mL llenado de ddH2O) | ||
| IPTG | 1 M en ddH2O | ||
| L-triptófano | 15 mM disuelto en ddH2O con 1 M HCl (agregar ácido clorhídrico gota a gota bajo agitación hasta que el polvo es dissoved) | ||
| lisozima | 50 mg/mL en ddH2O | ||
| DNasa I | 1 mg/mL en ddH2O | ||
| Rnasa A | 1 mg/mL en ddH2O | ||
| Amp100 | ampicilina 100 mg/mL en ddH2O | ||
| sodio-dodecylsulfate (SDS) | 200 g/L en el ddH2O | ||
| sulfato de amonio ((NH4)2SO4) | 1 M en ddH2O | esterilizar en autoclave | |
| fosfato de biácido del potasio (KH2PO4) | 1 M en ddH2O | esterilizar en autoclave | |
| di-potasio hidrogenofosfato (K2HPO4) | 1 M en ddH2O | esterilizar en autoclave | |
| sulfato de magnesio (MgSO4) | 1 M en ddH2O | esterilizar en autoclave | |
| D-glucosa | 1 M en ddH2O | esterilizar por filtración a través de un filtro de la jeringuilla del tamaño del poro de 0.45 μm | |
| cloruro de sodio (NaCl) | 5 M en ddH2O | esterilizar en autoclave | |
| cloruro de calcio (CaCl2) | 1 g/L | esterilizar por filtración a través de un filtro de la jeringuilla del tamaño del poro de 0.45 μm | |
| cloruro de hierro (II) (FECLAS2) | 1 g/L | esterilizar por filtración a través de un filtro de la jeringuilla del tamaño del poro de 0.45 μm | |
| tiamina | 10 g/L | esterilizar por filtración a través de un filtro de la jeringuilla del tamaño del poro de 0.45 μm | |
| biotina | 10 g/L | esterilizar por filtración a través de un filtro de la jeringuilla del tamaño del poro de 0.45 μm | |
| mezcla de elementos traza | sulfato de cobre (CuSO4), cinc cloruro (vivencias2), cloruro del manganeso (MnCl2), molibdato de amonio ((NH4)2MoO4); cada 1 mg/L en el ddH2O | esterilizar por filtración a través de un filtro de la jeringuilla del tamaño del poro de 0.45 μm | |
| mezcla de 19 aminoácidos | 1.) disolver 0.5 g de L-fenilalanina y L-tirosina de 0,5 g en 100 ml ddH2O con la adición gota a gota de 1 M de HCl, bajo agitación hasta que se disuelva el polvo. | ||
| 2.) pesa 0,5 g de cada uno de la restantes L-aminoácidos (excepto el L-triptófano). Mezclar con 22 mL fo 1 M KH2PO4 y 48 mL de 1 M K2HPO4. Añadir ddH2O hasta 800 mL. Revolver hasta que la solución llega a estar clara. | |||
| Agregar 3.) el disuelto L-fenilalanina y L-tirosina desde el paso 1.) y ajustar el volumen a 1 L con ddH2O. | |||
| 4.) esterilizar la mezcla del aminoácido por filtración de vacío con una unidad de filtro de botella. | |||
| Buffers y medios de comunicación | Composición y preparación | ||
| Concentrado de SDS carga del buffer de tinte, x 5 | 0.25 M Tris pH 6.8, glicerol al 50% v/v, 0,25% w/v bromofenol azul, didhiothreitol de 0,5 M (TDT; alternativomente 5% β-mercaptoetanol), 10% w/v sodio-dodecylsulfate (SDS) | ||
| tampón de Unión | 50 mM sodio dihydrogenphosphate (NaH2PO4), 500 mM NaCl, imidazol de 10 mM, pH 8 | ||
| tampón de elución | 50 mM sodio dihydrogenphosphate (NaH2PO4), 500 mM NaCl, imidazol de 250 mM, pH 8 | ||
| buffer de diálisis | 50 mM sodio dihydrogenphosphate (NaH2PO4), 150 mM NaCl, glicerol 100 mL/L, pH 8 | ||
| Buffer de MS | 10 mM Tris-HCl, pH 8 | ||
| nuevo medio mínimo que contiene 19 L-aminoácidos excepto L-triptófano (NMM19) | Mezclar todas las soluciones stock para obtener las siguientes concentraciones finales: 7,5 mM (NH4)2SO4, 1,7 mM NaCl, 22 m m KH2PO4, 50 m m K2HPO4, 1 mM MgSO4, 20 mM D-glucosa, 50 mg/L de mezcla de 19 aminoácidos, 1 μg/L CaCl2, 1 μg/L FECLAS2, tiamina μg/L 10, biotina 10 mg/L, mezcla de elementos traza de 0.01 mg/L | ||
| Medio LB | Composición: triptona 10 g/L, extracto de levadura 5 g/L, 10 g/L de NaCl, pH 7.0 en ddH2O | ||
| Preparación: | |||
| 1.) pese a 50 g de triptona, extracto de levadura 25 g, 5 g de NaCl en una botella de vidrio de 1 L. | |||
| 2.) Añadir ddH2O a ~ 800 mL y disolver componentes bajo agitación. | |||
| 3.) medir el pH y ajustar a pH 7 por adición gota a gota de 1 M de HCl o NaOH de M 1, si es necesario. Añadir ddH2O hasta 1 L. | |||
| 4.) esterilizar en autoclave, compruebe pérdida de volumen luego y añadir ddH estéril2O para compensar si es necesario. Almacenar a 4 ° C hasta su uso. | |||
| Placas de agar LB | Composición: triptona 10 g/L, extracto de levadura 5 g/L, 10 g/L de NaCl, agar-agar 15 g/L, pH 7.0 en ddH2O | ||
| Preparación: | |||
| 1.) pese a 50 g de triptona, extracto de levadura 25 g, 5 g NaCl, agar-agar 7,5 g en una botella de vidrio de 1 L. | |||
| 2.) Añadir ddH2O hasta 500 componentes mL y disolver con agitación. | |||
| 3.) medir el pH y ajustar a pH 7 por adición gota a gota de 1 M de HCl o NaOH de M 1, si es necesario. Añadir ddH2O hasta 1 L. | |||
| 4.) esterilizar en autoclave, compruebe pérdida de volumen luego y añadir ddH estéril2O para compensar, si es necesario. (Nota: LB agar puede almacenarse a 4 ° C hasta su uso para la preparación de las placas de agar LB. Cuidadosamente derretimiento solidificado agar utilizando un microondas) | |||
| 5.) cuando la solución esté aún caliente (30-40 ° C), añadir ampicilina a una concentración final de 100 μg/mL | |||
| 6.) Vierta aproximadamente 15 mL del líquido en el paso 5.) en un estéril 10 cm plato de Petri bajo condiciones estériles. Cuando el agar se solidifica, las placas pueden almacenarse durante 1 semana a 4 ° C hasta su uso. | |||
| tampón fosfato salino (PBS) | Composición: 137 mM NaCl, 2,7 mM KCl, 10 mM de Na2HPO4, 1,8 mM KH2PO4, 1 mM CaCl2, 0,5 mM MgCl2, pH 7. Esterilizar por autoclave o por filtración. | ||
Tabla 1: La solución Stock y buffer.
Discussion
Para lograr eficiencia de incorporación de ncAA muy alto, el método basado en la auxotrophy de SPI se basa en el uso de las células del huésped metabólicamente ingeniería, que no son capaces de sintetizar a la correspondiente contraparte natural de la ncAA. Para e. coli, estas cepas son fácilmente disponibles. Incluso la incorporación simultánea de múltiples ncAAs en la misma proteína es factible utilizando cepas multiauxotrophic. El modo específico de residuo de reemplazo y el repertorio químico está restringido a los análogos químicos similares puede considerarse como desventajas. Sin embargo, se puede producir un gran número de variantes de proteínas como el aparato de traducción bacteriana natural tolera numerosos análogos de aminoácidos. Por ejemplo, podrían incorporarse ncAAs más de 50 en las proteínas utilizando en vitro traducción, representa aproximadamente el 73% de todos los codones del código genético para reasignación40. Además, el SPI puede también permitir que etiquetado multisitio eficiente de la proteína blanco del41. En principio, la metodología SPI no se limita a e. coli, pero puede trabajar en cualquier otro host y para cada uno de los aminoácidos canónicos 20, siempre que las cepas auxotrófica y medios de cultivo definidos están disponibles. Por ejemplo, dos análogos de la metionina, azidohomoalanine (Aha) y homopropargylglycine (Hpg), son comercialmente disponibles y utilizados para el etiquetado de proteínas y proteomas en diversos organismos. Además, Aha puede ser producida intracelularmente y posteriormente incorporada en la proteína42. Este ncAA es especialmente conveniente para bioorthogonal conjugaciones como química del tecleo desarrollado por Tirrel y compañeros de trabajo: por ejemplo, en planta de tejido de Arabidopsis thaliana, mori del bómbice larvas43, Drosophila las células44, larvas de pez cebra45 así como células de mamíferos incluyendo neuronas46, las proteínas pueden estar etiquetadas con Aha47,48. Del mismo modo, con éxito se incorporaron péptidos antimicrobianos en cepas de Trp auxotrófica Lactococcus lactis 49Trp análogos. SPI es también útil para el campo de la xenobiología50,51, que explora las alternativas a la composición química básica de la vida. Por ejemplo, basado en trabajos anteriores en e. coli52 y53de B. subtilis, una cepa de e. coli fue desarrollada recientemente por una estrategia evolutiva con presión selectiva al utilizar thienopyrrole en vez de indol, dando por resultado la substitución de todo el proteoma del triptófano por thienopyrrole-alanina en el código genético54. Generalmente, el canónico aminoácido Trp, que es codificada por un único triplete (UGG), presenta un objetivo prometedor para la ingeniería de proteínas debido a las ricas facetas de la química del indol, que ofrece numerosas variaciones químicas. Recientemente y como una alternativa a la incorporación de SPI, una novela plataforma SCS capaces de incorporar análogos de Trp site-specifically en anfitriones bacterianos y eucariotas ha sido reportado55. Esto amplía aún más la caja de herramientas de en vivo ingeniería de proteínas basada en ncAA, incluyendo la alteración de propiedades espectrales.
Además del uso de los ejércitos de expresión auxotrófica, el protocolo SPI requiere condiciones de fermentación terminante, tanto en términos de tiempo de expresión de destino y la composición del medio para alcanzar alta eficiencia de incorporación de la ncAA y producción de la proteína blanco 56. cultivo se lleva a cabo utilizando medios mínima químicamente definidos, que básicamente contienen además sales principales las fuentes de nitrógeno (sal de amonio) y carbono (D-glucosa), vitaminas y oligoelementos. Aunque no estrictamente necesario en ausencia de otros auxotrophies, los restantes aminoácidos (20 -n, si n aminoácidos deben ser substituidos) comúnmente se añaden para promover el crecimiento bacteriano57. Durante una fase de crecimiento inicial antes de la inducción de expresión de la proteína objetivo, n aminoácidos canónicos a cambiarse se agregan en la limitación de las concentraciones. Crecimiento celular se desarrolla hasta los específicos aminoácidos esenciales están agotados, indicado como experimentalmente por un fijo OD600. Posteriormente, el medio de cultivo se sustituye por medio fresco carece del aminoácido empobrecido y que contiene la ncAA en concentraciones abundantes. Para la incorporación de ribosomal de análogos de triptófano como se muestra en el presente Protocolo, se alimenta un indol analógica, que se convierte en intracelular por convierte en el correspondiente derivado del triptófano triptófano sintasa58. A continuación, se induce expresión de la proteína objetivo. En esta etapa, las células están cerca del final de crecimiento logarítmico, como un equilibrio entre el número total de la célula y fitness. Como la presencia e incorporación de la canónica amino llevaría a producción de proteínas de tipo salvaje, es fundamental para asegurar que el aminoácido esencial está completamente agotado antes de la inducción. Asimismo, es obligatoria para examinar la eficacia de la incorporación de la ncAA en la proteína diana, comúnmente por espectrometría de masas. En caso de presencia de los aminoácidos canónicos, condiciones de cultivo deban ajustarse, p. ej., alterando la concentración de la amino acid(s) esenciales para la fase de crecimiento inicial o de la duración de este último. En caso de actividad de aaRS baja hacia la ncAA, la sobreexpresión de la enzima endógena o la expresión de un aaRS diferentes, que es más activo a la ncAA, puede ser llevado a cabo59.
El canónico aminoácido Trp está dotado de tres características: (i) su abundancia natural en las proteínas es baja; (ii) sus propiedades biofísicas y químicas son únicas (e.g., suele ser el origen dominante de la fluorescencia intrínseca de proteínas y péptidos) y (iii) contribuye a una variedad de interacciones bioquímicas y funciones incluyendo Apilamiento π, H-vinculación interacciones catión-π. Todas estas características se cambian radicalmente en sustitución de 4-amino-Trp Trp → duda GdFP. más allá, el diseño de una clase de "oro" de avGFPs es un notable ejemplo de ingeniería a medida autofluorescent proteínas. Con diferentes propiedades espectrales, FPs pueden ajustarse a ciertas ventanas espectrales mediante mutagénesis y ncAA la incorporación. En el caso de GdFP, esto se logra por un simple intercambio químico H → NH2 en el marco del anillo indol contenido en la tríada de cromóforo PLCE. Figura 5 muestra los efectos de la incorporación de la ncAA en el cromóforo. La introducción del grupo electrón-donar procedentes de 4-amino-indole (convertido intracelularmente a 4-amino-Trp) permite una variedad de estructuras mesomérico que puede explicar un estado excitado estabilizado. Espectroscópico, su cambio de Stokes ampliada y emisión de fluorescencia rojo-cambiado de puesto el resultado de estas propiedades distintas del sistema conjugado extendido. Como informó anteriormente, la transferencia de carga intramolecular mejorada en el cromóforo GdFP es inherentemente sensible al pH (Figura 4B) y acompañado por un cambio más grande en el momento de dipolo entre la S1 y S0 suelo excitado estado Comparado con ECFP33. Como grupos electrón-donar alternativos, podrían utilizarse análogos de triptófano llevan un anillo de indol sustituido por grupos hidroxis, según ha informado en un estudio comparativo con el modelo proteína barstar41.
Los espectros de absorción y fluorescencia de GdFP se ampliaron respecto a PLCE y EGFP (figura 3 y D). Ensanchamiento homogéneo de las bandas de absorción y fluorescencia es causada generalmente por modos vibracionales en el cromóforo y, además, por acoplamiento del cromóforo más vibracionales modos presentes en la proteína60. El acoplamiento al entorno de proteína local es apoyado por cargas localizadas en el cromóforo. Como la homogeneidad estructural de la proteína conduce a variaciones locales del espectro Noheda, tal acoplamiento entre los espectros vibrónico del cromóforo y el resto de la proteína están respaldados por deslocalización de la carga y mesomérico Estados como se indica en Figura 5. Este acoplamiento también es compatible con el gran cambio de Stokes y necesariamente reduce el rendimiento cuántico de fluorescencia. En comparación con otros FPs rojo-cambiado de puesto, GdFP incluso exhibe estabilidad mejorada de la proteína y una baja tendencia de agregación33,61,62. No sólo difiere en el color de otras variantes del punto de congelación, sino que también exhibe una termoestabilidad substancialmente creciente y mejorada cooperativa plegable33. Su intensidad de fluorescencia es conservado al calentar a 60 ° C, mientras que la fluorescencia de PLCE es reducida a alrededor del 30% al menos un 90%. En proteínas, aminoácidos aromáticos a menudo contribuyen a las redes de interacción cadenas laterales, que normalmente tienen un efecto estabilizador en la estructura terciaria de la proteína. avGFP alberga tal lado cadena red, que consiste en el cromóforo de sí mismo, tan bien como Phe-165, su-148 y Tyr-145. Estas cadenas de lado no sólo son muy rígidas en la estructura de GdFP33, pero lo importante es que forman contactos hidrofóbicos con el cromóforo. La característica más prominente de novela en GdFP que es el cromóforo aminated más proximal para Phe-165. Esta interacción es una característica no observada en otros avGFPs conocidos. Como los residuos de dos 3.2-4.5 Å aparte, interacciones amino aromático pueden ser también presente. Junto con la estabilización de resonancia inducida por aminación del cromóforo, estos probablemente estabilice esta red hidrofóbica de los aminoácidos de manera cooperativa. Una transferencia de carga intramolecular más eficaz podría ser apoyada por estas interacciones en el estado excitado en comparación con el estado del cromóforo, y al menos en parte responsable de los 108 nm Stokes shift33,62 .
En diseño racional de fluoróforo propiedades, se prevé un aumento en el tamaño del sistema π deslocalizado para dar lugar a una longitud de onda de excitación rojo-cambiado de puesto. Esta regla se obedece por la serie de aminoácidos en la posición 66 hacia neutro cromóforos: Phe (λmax = 355 nm) < su (λma x= 386 nm) < Tyr (λmax = 395 nm) < Trp (λmax = 436 nm)63. En la naturaleza, esta extensión del sistema conjugado del cromóforo de enlaces π se ha logrado mediante diferentes estrategias. Para DsRed de Discosoma striata, se extiende por la integración de un aminoácido adicional, así cambiando λmax a 573 nm64. El cromóforo de asFP595 (λmax = 595 nm) de Anemonia sulcata fue ampliado por un grupo imino, ampliando su sistema π65. Ya que el cromóforo de GdFP y otros avFPs es del mismo tamaño, un principio diferente ha de implicar una longitud de onda de emisión en el rango de la DsRed ampliado y asFP595 cromóforos. El profundo cambio de Stokes de 108 nm se atribuye a la distinta estructura de la GdFP cromóforo, que revela un nuevo principio fotofísicas en el diseño de proteínas autofluorescent. Cálculos preliminares (como se informó en el 62) han demostrado que el momento dipolo de la el estado excitado del cromóforo de GdFP es substancialmente más grande que en el estado de la tierra, en contraste con los valores respectivos de PLCE. Considerando que el momento dipolo de GdFP aumenta de ~ 3 D (Debye) en el estado de0 S a ~ 15 S1, el cambio para el cromóforo de PLCE fue bastante moderado (de D ~ 4 ~ 6 D). Por lo tanto, la fluorescencia dorada única de GdFP es causada por la transferencia de carga intramolecular substancial en el cromóforo, que aumenta la variedad de posibles estructuras mesomérico (ver figura 5) que permiten la estabilización de la resonancia. Esto reduce el nivel de energía de la que se produce la emisión. Como consecuencia del cambio profundo en el momento de dipolo a la excitación, la separación de carga intramolecular es la razón principal de los cambios en el potencial electrostático del entorno del cromóforo. La matriz circundante de la proteína, a su vez, se ajusta a los cambios en la distribución de la carga después de la excitación del cromóforo. La relajación estructural posterior disminuye el nivel de energía del cromóforo excitado, que cambia de puesto el espectro de fluorescencia al rojo debido a su carácter de transferencia de carga. Por lo mismo, como consecuencia de la gran cambio de Stokes y tasas mejoradas de radiationless procesos, el rendimiento cuántico de fluorescencia de GdFP es reducido en comparación con ECFP33.
El rendimiento cuántico alto y pequeño cambio de Stokes de PLCE y EGFP se atribuyen generalmente a un entorno de proteína rígida del cromóforo, que reduce los grados de libertad y, en consecuencia, conversión interna para favorecer la relajación radiativa del estado excitado 66. en consecuencia, el diseño molecular de cromóforos más rígido integrados con acoplamiento reducido a la matriz de proteína restante podría servir como una guía para producir rojo-cambiado de puesto más lejos los derivados GFP con rendimiento cuántico de fluorescencia alta. Por lo tanto, para ingeniería más enfoques para producir proteínas autofluorescent rojo-cambiado de puesto, ampliación del sistema de electrones π y una estructura rígida del cromóforo con débil acoplamiento con el medio ambiente de la proteína es altamente deseable. Dichas modificaciones también podrían introducirse directamente en cromóforos basada en GFP o por colocación de ncAAs deseada en las proximidades del cromóforo.
Disclosures
Los autores declaran que no tienen intereses financieros que compiten.
Acknowledgments
Este trabajo fue financiado por la Fundación alemana de investigación (grupo de excelencia "unificar conceptos en catálisis) T.F. y nota y por el Ministerio Federal de educación y ciencia (BMBF programa"HSP 2020", SynTUBio de WIMIplus de TU proyecto) a F. J.S.
Materials
| Name | Company | Catalog Number | Comments |
| Chemicals | |||
| 4-aminoindole | Sigma-Aldrich | 525022 | |
| acetonitrile | VWR | HiPerSolv CHROMANORM ULTRA for LC-MS, 83642 | LC-MS grade required |
| agar-agar | Carl Roth | 5210 | |
| ammonium molybdate ((NH4)2MoO4) | Sigma-Aldrich | 277908 | |
| ammonium sulfate ((NH4)2SO4) | Sigma-Aldrich | A4418 | |
| ampicillin sodium salt | Carl Roth | K029 | |
| biotin | Sigma-Aldrich | B4501 | |
| bromophenol blue | Sigma-Aldrich | B0126 | |
| calcium chloride (CaCl2) | Sigma-Aldrich | C5670 | |
| colloidal silica | Sigma-Aldrich | Ludox HS-40, 420816 | |
| Coomassie Brillant Blue R 250 | Carl Roth | 3862 | |
| copper sulfate (CuSO4) | Carl Roth | CP86.1 | |
| D-glucose | Carl Roth | 6780 | |
| di-sodium hydrogen phosphate (Na2HPO4) | Carl Roth | X987 | |
| di-potassium hydrogen phosphate (K2HPO4) | Carl Roth | P749.1 | |
| 1,4-dithiothreitol (DTT) | Carl Roth | 6908 | |
| DNase I | Sigma-Aldrich | D5025 | |
| ethanol | Carl Roth | 9065.1 | |
| formic acid | VWR | HiPerSolv CHROMANORM for LC-MS, 84865 | LC-MS grade required |
| glycerol | Carl Roth | 3783 | |
| imidazole | Carl Roth | X998 | |
| indole | Sigma-Aldrich | I3408 | |
| iron(II) chloride (FeCl2) | Sigma-Aldrich | 380024 | |
| isopropanol | Carl Roth | AE73.1 | |
| isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma-Aldrich | I6758 | |
| lysozyme | Sigma-Aldrich | L6876 | |
| magnesium chloride (MgCl2) | Carl Roth | KK36.1 | |
| magnesium sulfate (MgSO4) | Carl Roth | 8283.2 | |
| manganese chloride (MnCl2) | Sigma-Aldrich | 63535 | |
| β-mercaptoethanol | Carl Roth | 4227.3 | |
| potassium chloride (KCl) | Carl Roth | 6781.3 | |
| potassium dihydrogen phosphate (KH2PO4) | Sigma-Aldrich | P5655 | |
| RNase A | Carl Roth | 7156 | |
| sodium chloride (NaCl) | Carl Roth | P029 | |
| sodium dihydrogen phosphate (NaH2PO4) | Carl Roth | T879 | |
| sodium dodecyl sulphate (NaC12H25SO4) | Carl Roth | 0183 | |
| thiamine | Sigma-Aldrich | T4625 | |
| Tris(hydroxymethyl)-aminomethane (Tris) | Carl Roth | 5429 | |
| Tris hydrochloride (Tris-HCl) | Sigma-Aldrich | 857645 | |
| tryptone | Carl Roth | 8952 | |
| yeast extract | Carl Roth | 2363 | |
| zinc chloride (ZnCl2) | Sigma-Aldrich | 229997 | |
| Name | Company | Catalog Number | Comments |
| amino acids | |||
| L-alanine | Sigma-Aldrich | A7627 | |
| L-arginine | Sigma-Aldrich | A5006 | |
| L-asparagine | Sigma-Aldrich | A8381 | |
| L-aspartic acid | Sigma-Aldrich | A0884 | |
| L-cysteine | Sigma-Aldrich | C7352 | |
| L-glutamic acid | Sigma-Aldrich | G2128 | |
| L-glutamine | Sigma-Aldrich | G3126 | |
| L-glycine | Sigma-Aldrich | G7126 | |
| L-histidine | Sigma-Aldrich | H8000 | |
| L-isoleucine | Sigma-Aldrich | I2752 | |
| L-leucine | Sigma-Aldrich | L8000 | |
| L-lysine | Sigma-Aldrich | L5501 | |
| L-methionine | Sigma-Aldrich | M9625 | |
| L-proline | Sigma-Aldrich | P0380 | |
| L-phenylalanine | Sigma-Aldrich | P2126 | |
| L-serine | Sigma-Aldrich | S4500 | |
| L-threonine | Sigma-Aldrich | T8625 | |
| L-tryptophan | Sigma-Aldrich | T0254 | |
| L-tyrosine | Sigma-Aldrich | T3754 | |
| L-valine | Sigma-Aldrich | V0500 | |
| Name | Company | Catalog Number | Comments |
| Lab materials | |||
| 0.45 µm syringe filter with PVDF membrane | Carl Roth | CCY1.1 | |
| 1.5 mL microcentrifuge tubes | Eppendorf | 30120086 | |
| conical polystyrene (Falcon) tubes, 50 mL | Fisher Scientific | 14-432-22 | |
| Luer-Lock syringe 5 mL | Carl Roth | EP96.1 | |
| dialysis membrane, Molecular Weight Cut-Off (MWCO) 5,000 | Spectrum Medical Industries | Spectra/Por MWCO 5000 dialysis membrane, 133198 | |
| Immobilized Metal ion Affinity Chromatography (IMAC) column 1 mL, Ni-NTA | Macherey Nagel | Protino series, 745410.5 | |
| petri dishes (polystyrene, sterile) | Carl Roth | TA19 | |
| pQE-80L plasmid vector | Qiagen | no longer available | replaced by N-terminus pQE Vector set Cat No./ID: 32915 |
| protein extraction reagent BugBuster | EMB Millipore | 70921-4 | |
| round-bottom polystyrene tubes, 14 mL | Fisher Scientific | Corning Falcon, 14-959-1B | |
| Trp-auxotrophic E. coli strain | ATCC | ATCC 49980 | Bridges BA et al., Chem Biol Interact., 1972, 5(2):77-84; see main text for alternatives |
| Name | Company | Catalog Number | Comments |
| Mass Spectrometry equipment | |||
| mass spectrometer for LC-ESI-TOF-MS | Agilent | Agilent 6530 Accurate-Mass QTOF | coupled with Infinity LC system |
| mass spectrometry data analysis software | Agilent | MassHunter Qualitative Analysis software v. B.06.00 | |
| High-Performance Liquid Chromatography (HPLC) column for LC-ESI-TOF-MS | Sigma-Aldrich | Supelco Discovery BIO Wide Pore C5 HPLC column, 3 µm particle size, 10 cm x 2.1 mm | |
| HPLC autosampler vials 1.5 mL | Sigma-Aldrich | Supelco 854165 | with conical 0.1 mL glass inserts, screw caps and septa |
| Name | Company | Catalog Number | Comments |
| General equipment | |||
| benchtop centrifuge for 1.5 mL Eppendorf tubes | Eppendorf | 5427 R | |
| cooling centrifuge for 50 mL Falcon tubes | Eppendorf | 5810 R | |
| high pressure microfluidizer for bacterial cell disruption | Microfluidics | LM series with “Z” type chamber | |
| peristaltic pump for LC | GE Healthcare | P-1 | |
| Fast Protein Liquid Chromatography (FPLC) system | GE Healthcare | ÄKTA pure 25 L | |
| orbital shaker for bacterial cultivation | Infors HT | Minitron | |
| UV/Vis spectrophotometer | Biochrom | ULTROSPEC 2100 | |
| ultrasonic homogenizer for bacterial cell disruption | Omnilab | Bandelin SONOPULS HD 3200, 5650182 | with MS72 sonifier tip |
| Name | Company | Catalog Number | Comments |
| Fluorescence spectroscopy equipment | |||
| ps-pulsed laser 470 nm | Picoquant GmbH | PDL-470 | |
| time- and wavelength-correlated single photon counting (TWSPC) acquisition software | Picoquant GmbH | SymPhoTime 64 | |
| time- and wavelength-correlated single photon counting (TWSPC) detector | Picoquant GmbH | PML-16C | 16 spectral channels, to be selected by grating settings |
| single photon counting software | Picoquant GmbH | SPCM 9.75 | |
| global fitting software | Picoquant GmbH | SPC2Glo(R) | |
| fluorescence decay data analysis software | Picoquant GmbH | FluoFit program | |
| data analysis software | OriginLab Inc. | Origin 9.2 | |
| neutral density filter set | Schott | NG1 to NG11 | (400 - 650 nm, transmission 50 %, 20%, 10 %, 5 %) |
| 488 nm long-pass emission filter | AHF Analysentechnik | AHF-488 | |
| quartz cuvette | Thorlabs GmbH | CV10Q1400 | 1 cm pathlength |
References
- Shimomura, O., Johnson, F. H., Saiga, Y. Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan, Aequorea. J Cell Compar Physl. 59 (3), 223-239 (1962).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263 (5148), 802-805 (1994).
- Andresen, M., et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. P Natl Acad Sci USA. 102 (37), 13070-13074 (2005).
- Andresen, M., et al. Structural basis for reversible photoswitching in Dronpa. P Natl Acad Sci USA. 104 (32), 13005-13009 (2007).
- Brakemann, T., et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat Biotechnol. 29 (10), 942-947 (2011).
- Kremers, G. -J., Gilbert, S. G., Cranfill, P. J., Davidson, M. W., Piston, D. W. Fluorescent proteins at a glance. J Cell Sci. 124 (Pt 2), 157-160 (2011).
- Shimomura, O. Structure of the chromophore of aequorea 0. shimomura green fluorescent protein. FEBS Lett. 104 (2), 220-222 (1979).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 22 (12), 1567-1572 (2004).
- Shcherbo, D., et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 4 (9), 741-746 (2007).
- Shcherbakova, D. M., Subach, O. M., Verkhusha, V. V. Red fluorescent proteins: advanced imaging applications and future design. Angew Chem Int Edit. 51 (43), 10724-10738 (2012).
- Stepanenko, O. V., Verkhusha, V. V., Kuznetsova, I. M., Uversky, V. N., Turoverov, K. K. Fluorescent proteins as biomarkers and biosensors: throwing color lights on molecular and cellular processes. Curr Protein Pept Sc. 9 (4), 338-369 (2008).
- Wang, L., Xie, J., Deniz, A. A., Schultz, P. G. Unnatural amino acid mutagenesis of green fluorescent protein. J Org Chem. 68 (1), 174-176 (2003).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Sharma, N., Furter, R., Kast, P., Tirrell, D. A. Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett. 467 (1), 37-40 (2000).
- Liu, C. C., Schultz, P. G. Adding new chemistries to the genetic code. Annu Rev Biochem. 79, 413-444 (2010).
- Twine, S. M., Murphy, L., Phillips, R. S., Callis, P., Cash, M. T., Szabo, A. G. The Photophysical Properties of 6-Azaindole. J Phys Chem B. 107 (2), 637-645 (2003).
- Lepthien, S., Hoesl, M. G., Merkel, L., Budisa, N. Azatryptophans endow proteins with intrinsic blue fluorescence. P Natl Acad Sci USA. 105 (42), 16095-16100 (2008).
- Budisa, N., et al. Probing the role of tryptophans in Aequorea victoria green fluorescent proteins with an expanded genetic code. Biol Chem. 385 (2), 191-202 (2004).
- Ross, J. B., et al. Spectral enhancement of proteins: biological incorporation and fluorescence characterization of 5-hydroxytryptophan in bacteriophage lambda cI repressor. P Natl Acad Sci USA. 89 (24), 12023-12027 (1992).
- Soumillion, P., Jespers, L., Vervoort, J., Fastrez, J. Biosynthetic incorporation of 7-azatryptophan into the phage lambda lysozyme: Estimation of tryptophan accessibility, effect on enzymatic activity and protein stability. Protein Eng Des Sel. 8 (5), 451-456 (1995).
- Heim, R., Tsien, R. Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 6 (2), 178-182 (1996).
- Bridges, B. A., Mottershead, R. P., Rothwell, M. A., Green, M. H. L. Repair-deficient bacterial strains suitable for mutagenicity screening: tests with the fungicide captain. Chem Biol Interact. 5 (2), 77-84 (1972).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: The Heat Shock Method. J Vis Exp. , (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: Electroporation. J Vis Exp. , (2017).
- Grigorenko, B. L., Krylov, A. I., Nemukhin, A. V. Molecular modeling clarifies the mechanism of chromophore maturation in the green fluorescent protein. J Am Chem Soc. , (2017).
- JoVE Science Education Database. General Laboratory Techniques. Introduction to the Spectrophotometer. J Vis Exp. , (2017).
- Goedhart, J., et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun. 3, 751 (2012).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F. Culture medium for enterobacteria. J Bacteriol. 119 (3), 736-747 (1974).
- Hörnsten, E. G. On culturing Escherichia coli on a mineral salts medium during anaerobic conditions. Bioprocess Eng. 12 (3), 157-162 (1995).
- Davis, B. D. The Isolation of Biochemically Deficient Mutants of Bacteria by Means of Penicillin. P Natl Acad Sci USA. 35 (1), 1-10 (1949).
- Sambrook, J., Russell, D. W. Molecular Cloning: A Laboratory Manual. , Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY, USA. (2001).
- Wang, Y. -S., et al. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives. Mol Biosyst. 7 (3), 714-717 (2011).
- Bae, J. H., et al. Expansion of the genetic code enables design of a novel "gold" class of green fluorescent proteins. J Mol Biol. 328 (5), 1071-1081 (2003).
- JoVE Science Education Database. Dialysis: Diffusion Based Separation. J Vis Exp. , Cambridge, MA. (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Separating Protein with SDS-PAGE. J Vis Exp. , (2017).
- Petrásek, Z., et al. Excitation energy transfer from phycobiliprotein to chlorophyll d in intact cells of Acaryochloris marina studied by time- and wavelength-resolved fluorescence spectroscopy. Photoch Photobio Sci. 4 (12), 1016-1022 (2005).
- Kolber, Z. S., Barkley, M. D. Comparison of approaches to the instrumental response function in fluorescence decay measurements. Anal Biochem. 152 (1), 6-21 (1986).
- Pelet, S., Previte, M. J. R., Laiho, L. H., So, P. T. C. A fast global fitting algorithm for fluorescence lifetime imaging microscopy based on image segmentation. Biophys J. 87 (4), 2807-2817 (2004).
- Loefroth, J. E. Time-resolved emission spectra, decay-associated spectra, and species-associated spectra. J Phys Chem. 90 (6), 1160-1168 (1986).
- Hartman, M. C. T., Josephson, K., Lin, C. -W., Szostak, J. W. An expanded set of amino acid analogs for the ribosomal translation of unnatural peptides. PLoS One. 2 (10), e972 (2007).
- Budisa, N., et al. Global replacement of tryptophan with aminotryptophans generates non-invasive protein-based optical pH sensors. Angew Chem Int Edit. 41 (21), 4066-4069 (2002).
- Ma, Y., Biava, H., Contestabile, R., Budisa, N., di Salvo, M. L. Coupling bioorthogonal chemistries with artificial metabolism: intracellular biosynthesis of azidohomoalanine and its incorporation into recombinant proteins. Molecules. 19 (1), 1004-1022 (2014).
- Teramoto, H., Kojima, K. Incorporation of Methionine Analogues Into Bombyx mori Silk Fibroin for Click Modifications. Macromol Biosci. 15 (5), 719-727 (2015).
- Deal, R. B., Henikoff, J. G., Henikoff, S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 328 (5982), 1161-1164 (2010).
- Hinz, F. I., Dieterich, D. C., Tirrell, D. A., Schuman, E. M. Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci. 3 (1), 40-49 (2012).
- Dieterich, D. C., et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 13 (7), 897-905 (2010).
- Dieterich, D. C., Link, A. J., Graumann, J., Tirrell, D. A., Schuman, E. M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). P Natl Acad Sci USA. 103 (25), 9482-9487 (2006).
- Glenn, W. S., et al. Bioorthogonal Noncanonical Amino Acid Tagging (BONCAT) Enables Time-Resolved Analysis of Protein Synthesis in Native Plant Tissue. Plant Physiol. 173 (3), 1543-1553 (2017).
- Zhou, L., et al. Incorporation of tryptophan analogues into the lantibiotic nisin. Amino Acids. 48 (5), 1309-1318 (2016).
- Acevedo-Rocha, C. G., Budisa, N. Xenomicrobiology: a roadmap for genetic code engineering. Microb Biotechnol. 9 (5), 666-676 (2016).
- Agostini, F., Völler, J. -S., Koksch, B., Acevedo-Rocha, C. G., Kubyshkin, V., Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew Chem Int Edit. 56 (33), 9680-9703 (2017).
- Bacher, J. M., Ellington, A. D. Selection and characterization of Escherichia coli variants capable of growth on an otherwise toxic tryptophan analogue. J Bacteriol. 183 (18), 5414-5425 (2001).
- Wong, J. T. Membership mutation of the genetic code: loss of fitness by tryptophan. Pc Natl Acad Sci USA. 80 (20), 6303-6306 (1983).
- Hoesl, M. G., et al. Chemical Evolution of a Bacterial Proteome. Angew Chem Int Edit. 54 (34), 10030-10034 (2015).
- Italia, J. S., et al. An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nat Chem Biol. 13 (4), 446-450 (2017).
- Völler, J. -S., Thi To, T. M., Biava, H., Koksch, B., Budisa, N. Global substitution of hemeproteins with noncanonical amino acids in Escherichia coli with intact cofactor maturation machinery. Enzyme Microb Tech. 106, 55-59 (2017).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Völler, J. -S., Budisa, N. Coupling genetic code expansion and metabolic engineering for synthetic cells. Curr Opin Biotech. 48, 1-7 (2017).
- Johnson, J. A., Lu, Y. Y., Van Deventer, J. A., Tirrell, D. A. Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Curr Opin Chem Biol. 14 (6), 774-780 (2010).
- Somsen, O. J., van Grondelle, R., van Amerongen, H. Spectral broadening of interacting pigments: polarized absorption by photosynthetic proteins. Biophys J. 71 (4), 1934-1951 (1996).
- Kurschus, F. C., Pal, P. P., Bäumler, P., Jenne, D. E., Wiltschi, B., Budisa, N. Gold fluorescent annexin A5 as a novel apoptosis detection tool. Cytom Part A. 75 (7), 626-633 (2009).
- Lepthien, S., Wiltschi, B., Bolic, B., Budisa, N. In vivo engineering of proteins with nitrogen-containing tryptophan analogs. Appl Microbiol Biot. 73 (4), 740-754 (2006).
- Wachter, R. M., Elsliger, M. -A., Kallio, K., Hanson, G. T., Remington, S. J. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure. 6 (10), 1267-1277 (1998).
- Verkhusha, V. V., Lukyanov, K. A. The molecular properties and applications of Anthozoa fluorescent proteins and chromoproteins. Nat Biotechnol. 22 (3), 289-296 (2004).
- Martynov, V. I., Savitsky, A. P., Martynova, N. Y., Savitsky, P. A., Lukyanov, K. A., Lukyanov, S. A. Alternative cyclization in GFP-like proteins family. The formation and structure of the chromophore of a purple chromoprotein from Anemonia sulcata. J Biol Chem. 276 (24), 21012-21016 (2001).
- Piatkevich, K. D., Malashkevich, V. N., Morozova, K. S., Nemkovich, N. A., Almo, S. C., Verkhusha, V. V. Extended Stokes shift in fluorescent proteins: chromophore-protein interactions in a near-infrared TagRFP675 variant. Sci Rep. 3 (1), 1847 (2013).
