

Summary
Biologie synthétique permet l’ingénierie des protéines avec des propriétés sans précédent à l’aide de l’insertion précurseur des aminoacides non canonique. Ici, nous avons présenté comment une variante spectralement décalée vers le rouge d’un fluorophore GFP-type avec des propriétés spectroscopiques de fluorescence roman, appelée protéine fluorescente « or » (GdFP), est produite par e. coli par incorporation de pression sélective (SPI).
Abstract
Protéines fluorescentes sont des outils fondamentaux pour les sciences de la vie, en particulier pour la microscopie de fluorescence des cellules vivantes. Bien que sauvage et conçus des variantes de la protéine fluorescente verte de Aequorea victoria (avGFP), mais aussi des homologues chez d’autres espèces couvrent déjà les grandes parties du spectre, un décalage spectral demeure dans la région du proche infrarouge, pour les fluorophores axée sur les avGFP ne sont pas disponibles. Variantes de la protéine fluorescente décalée vers le rouge (FP) augmenterait sensiblement le toolkit for déconvolution spectrale des espèces moléculaires multiples, mais les FPs décalée vers le rouge naturel dérivé de coraux ou anémones de mer ont bas rendement quantique de fluorescence et photo-stabilité inférieure par rapport aux variantes d’avGFP. Autre manipulation et élargissement possible du système conjugué de chromophore vers la région spectrale rouge sombre est également limitée par le répertoire des 20 acides aminés canoniques prescrites par le code génétique. Pour surmonter ces limites, la biologie synthétique peut atteindre davantage spectrale rouge changeante via l’insertion d’aminoacides non canonique dans la triade du chromophore. Nous décrivons l’application de SPI à l’ingénieur avGFP variantes aux nouvelles propriétés spectrales. Expression de la protéine est réalisée dans un tryptophane-auxotrophes Escherichia coli souche et en complétant des milieux de culture avec les précurseurs de l’indole approprié. L’intérieur des cellules, ces précurseurs sont convertis au analogues tryptophane correspondants et incorporés dans les protéines par la machinerie ribosomale en réponse aux codons UGG. Le remplacement du Trp-66 dans la variante « cyan » amélioré d’avGFP (ECFP) par un donneur d’électron 4-aminotryptophan traduit par GdFP mettant en vedette un déplacement de Stokes 108 nm et une émission fortement décalée vers le rouge maximale (574 nm), tout en étant thermodynamiquement plus stable que son prédécesseur ECFP. Incorporation des résidus spécifiques de l’acide aminé non canonique est analysée par spectrométrie de masse. Les propriétés spectroscopiques des GdFP sont caractérisées par spectroscopie de fluorescence résolue en temps comme l’une des applications utiles de génétiquement encodé IPS en sciences de la vie.
Introduction
Depuis la découverte de la protéine fluorescente verte dans la méduse Aequorea victoria (avGFP) en 19621 et la première expression hétérologue dans 19942 dans d’autres cellules eucaryotes, sont devenus des protéines fluorescentes de la famille GFP des outils très précieux et cibles dans les sciences de la vie. Une vaste génie génétique moléculaire et inclus l’ajustement de l’utilisation de codon spécifique à l’espèce, l’accélération de pliage, maturation améliorée, une luminosité, prévention d’oligomérisation et adaptation des propriétés spectrales et photochimiques y compris la capacité de façon réversible photoswitch3,4,5,6. GFP doit sa fluorescence de sa 4-(p- hydroxybenzylidène) imidazolidin-5-one (HBDI) chromophore. Ce dernier est autocatalytique formé de la triade de chromophore ce qu’on appelle des acides aminés (Ser-65/Tyr-66/Gly-67 à avGFP) après la formation d’une liaison covalente supplémentaire au sein de l’épine dorsale du peptide sous l’influence de l’oxygène moléculaire7. La résonance stabilisé système conjugué interagit dynamiquement avec l’environnement moléculaire, ce qui permet l’absorption dans le domaine du visible et fluorescence verte caractéristique de ces protéines.
Au sein de la triade de chromophore, la présence d’un acide aminé aromatique est obligatoire. Cependant, le répertoire des acides aminés standard comprend seulement quatre résidus aromatiques (His, Phe, Trp et Tyr). Cela limite les approches classiques de mutagenèse pour atteindre sensiblement plus décalée vers le rouge avGFP de variantes par rapport à la FPF naturelle plus décalée vers le rouge par exemple DsRed8 de coralimorphs Duncanopsammia striata ou mKate/mNeptune9 de l’anémone de mer Entacmaea quadricolor. Par conséquent, la partie rouge sombre et du proche infrarouge du spectre optique au-dessus de 600 nm est faiblement couvert par des variantes GFP. Il s’agit, bien sûr, une limitation sévère des approches microscopiques de fluorescence nécessitant la DEMULTIPLEXAGE spectrale de plusieurs espèces de fluorophore en même temps. Par exemple, des marqueurs de longueur d’onde sont également nécessaires pour faire utiliser du régime faible absorption du tissu cutané entre 700-1 000 nm dans paramètres pour10d’imagerie des tissus profonds.
Des protéines fluorescentes provenant d’avGFP sont divisés en plusieurs classes basées sur les propriétés spectroscopiques et de la nature chimique de leurs chromophores11. Avec sa triade Ser-65/Tyr-66/Gly-67, le chromophore sauvage existe sous la forme un mélange équilibré entre la forme neutre et phénolique (λmax = 395 nm, ε = 21 000 M-1cm-1) et la forme anionique phénolate (λmax = 475 nm, ε = 7 100 M -1cm-1), et le spectre d’émission présente un seul pic à 508 nm. Le groupement hydroxyle de Ser-65 est d’une importance cruciale, car il donne une liaisons hydrogènes à Glu-222 dans les environs de chromophore (distance : 3.7 Å), qui favorise l’ionisation de ce carboxylate. Classe I est caractérisée par un chromophore phénolate anioniques, comme EGFP (Phe-64-Leu/Ser-65-Thr ; λmax = 488 nm, ε = 35 600 M-1cm-1, λem = 509 nm). En raison de la substitution de Ser-65-Thr(Ala,Gly), le pic d’excitation 395 nm de la forme neutre de phénol est supprimée et le 470-475 nm sommet du phénolate anionique est cinq à six fois amélioré et décalé à 490 nm. Classe II compose de protéines avec un chromophore phénolique neutre, comme dans saphir-GFP. Ici, la substitution de Thr-203-Ile supprime presque totalement l’excitation nm 475, laissant seulement le pic à 399 nm. Étant donné que le chromophore anionique ne peut pas être correctement solvatés, sa forme neutre est favorisée. Classe III comprend les variantes fluorescents « jaunes » (EYFP ; SER-65-Gly/Val-68-Leu/ser-72-Ala/Thr-203-Tyr ; Lesmax ε λ = 514 nm, ε = 84, 600 M-1cm-1, λem = 527 nm) avec un empilement π l’interaction d’une chaîne latérale aromatique et le phénolate, comme provoquée par les substitutions de Thr-203-His(Trp,Phe,Tyr), qui conduisent à jusqu'à 20 nm maxima d’émission décalée vers le rouge (Thr-203-Tyr). Autres (Gln-69-Lys), donnent un autre décalage vers le rouge 1 à 2 nm à 529 nm, la plus décalée vers le rouge avGFP variante connue11. L’échange du phénol pour un indole (Tyr-66-Trp) crée la classe IV, comme dans le ECFP fluorescente cyan (Ser-65-Thr/Tyr-66-Trp ; λmax1 = 434 nm, ε = 24 800 M-1cm-1; λmax2 = 452 nm, ε = 23 600 M-1cm-1 ; Λem1 = 477 nm, λem2 = 504 nm). L’hébergement de l’indole encombrant est probablement activé par d’autres, mutations compensatoires. Les maxima d’excitation et d’émission de ECFP automne inbetween ceux des protéines avec des chromophores neutres ou anioniques. Protéines de classe V abritent un imidazole en lieu et place du phénol (Tyr-66-His), par exemple., protéines fluorescentes bleu comme EBFP. Classe VI est produit par un échange de phénol-à-phényl favorisant la forme neutre chromophore exclusivement, qui entraîne par conséquent les plus bleu-décalé d’excitation et d’émission positions des pics (360 nm et 442 nm, respectivement).
Mutagenèse classique est particulièrement adapté pour la production de variantes de chromophore avGFP roman, par la permutation du tripeptide et résidus qui interagissent dans le cadre des 20 acides aminés canoniques 65-67. Ces possibilités peuvent être étendues lorsque les variantes non canoniques des acides aminés aromatiques sont introduits lors de la synthèse de protéine ribosomique12. En principe, il y a deux façons d’y parvenir. La première stratégie repose sur la tolérance de substrat de la machinerie de traduction protéique, surtout d’aminoacyl-tRNA synthétases (aaRSs) vers analogues d’acides aminés liés. Pour y parvenir avec une grande efficacité, auxotrophe souches d’expression e. coli sont employés qui sont incapables de synthétiser l’acide aminé naturel correspondant. Cela permet le remplacement de ce dernier en ajoutant adapté des acides aminés non canoniques (ANAC) ou précurseurs celle-ci au milieu de culture. Cette stratégie, appelée aussi Incorporation de pression sélective (SPI)13,14, permet aux résidus propres remplacements, entraînant une incorporation globale de la ncAA. La deuxième stratégie utilise codon stop ARNt suppresseurs qui est chargés de la ncAA par ingénierie enzymes RAA. Cela se traduit par le phénomène de codons stop dans le cadre et permet l’incorporation de ncAA spécifique. Par conséquent, cette méthode de suppression de codon stop (SCS) conduit à l’expansion du code génétique15. Par mutagénèse, un codon d’arrêt est placé dans le gène cible à l’emplacement désiré. En principe, SPI permet également de créer recombinants peptides et protéines portant une installation unique de ncAA, étant donné que rares acides aminés canoniques comme Met ou Trp sont choisis pour la substitution. Avec Trp, auraient dû être divulgués SPI approches pour travailler avec une grande variété d’analogues dont 4 - F - 5 - F - et 6-F-Trp, Trp-aza-7, 4-OH - et 5-OH-Trp, ainsi que 4 - et 5-NH2- Trp ou même β (thienopyrrolyl) alanine dérivés16 ,17,18,19,20. Ainsi, les SPI pourrait être très avantageux pour le remplacement des acides aminés aromatiques des chromophores de la GFP par les variantes non canoniques d’étudier la possibilité d’adapter davantage les spectres et déplacement de Stokes de ces FPs. En ce qui concerne toutes les modifications de séquences protéiques, la compatibilité avec la maturation de pliage et de chromophore FP doit être testée expérimentalement.
Dans ce travail, nous utilisons la classe IV ECFP21, qui porte au lieu de l’avGFP sauvage Tyr, un résidu Trp au sein de la triade du chromophore. À l’aide de SPI, ce Trp-66 (et Trp-57, le seul autre résidu de Trp dans ECFP) sont substitué par 4-amino-TRP. La présence du groupe amino électrodonneurs du 4-amino-PRT dans le chromophore favorise la stabilisation de résonance d’un transfert de proton état excité bien décalée vers le rouge (ESPT) doté d’un déplacement de Stokes 108 nm. Cette protéine fluorescente « or » (GdFP) constitue la variante avec le plus grand décalage vers le rouge de la fluorescence maximale (574 nm) parmi toutes les protéines dérivées d’avGFP. Nous décrivons la méthode de production de protéines GdFP de SPI et fournir les protocoles pour l’analyse obligatoire des résultantes protéines modifiées par spectrométrie de masse. En outre, nous montrons comment les GdFP peut être utilisé et analysé dans les approches de spectroscopie de fluorescence résolue en temps.
Protocol
1. transformation de Trp-auxotrophes e. coli
- Transformer chimiquement ou electrocompetent (50 µL) de cellules d’une souche auxotrophe-Trp e. coli , par exemple. CIFA 49980 (WP2, mutant, dérivée de la souche e. coli B/R22), avec 1 µL d’une solution aqueuse de 1 ng/µL du plasmide d’expérience - 80 L His6-ECFP utilisant un choc thermique ou électroporation, respectivement. Veuillez vous référer à la JoVE Science Education de base de données23,24 pour plus de détails.
Remarque : Le vecteur d’expression d’expérience - 80L His6-ECFP encode une N-terminale 6 x ECFP His-tag21 pilotée par un promoteur bactérien de T5 avec opérateur lac. Elle porte également un marqueur de sélection AmpR et une origine colE1 de réplication (la séquence d’épine dorsale de vecteur d’expérience - 80 L se trouvent sur : https://www.qiagen.com/mx/resources/resourcedetail?id=c3b71572-4d82-4671-a79b-96357fe926d1&lang=en & autoSuggest = true). Le poids moléculaire théorique de la protéine de type sauvage His6-ECFG (après maturation de chromophore25) est Da 28303.92. La séquence des protéines traduites cible est comme suit (His-tag a souligné, séquence dérivée d’un vecteur en caractères gras) : MRGSHHHHHHGSMVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTLTWGVQCFSRYPDHMK
QHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYISHNVYITADKQKNGIKANFKIRHNIEDGS
VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYK. - Plaque des cellules transformées sur milieu gélosé-LB (tableau 1) additionnés de 10 g/L de glucose, 100 ampicilline µg/mL et incuber les plaques à 37 ° C pendant la nuit.
2. recombinant Protein Expression
-
Une culture d’e. coli ATCC 49980 d’expérience - 80 L His6-ECFP
- Préparer 5 mL de milieu LB (tableau 1; additionné de 10 g/L de glucose, 100 ampicilline µg/mL) dans un stérile 14 mL polystyrène culture tube croissance aérobie et ensemencer avec un séparément de la colonie d’un plat d’agar en utilisant une boucle d’extrémité ou inoculation pipette stérile.
Remarque : À l’aide de colonies de cellules fraîchement transformées est recommandé. Les plaques avec des colonies bactériennes (à l’étape 1.2.) peuvent être stockés à 4 ° C pendant plusieurs jours. - Incuber les cellules à 37 ° C dans un agitateur orbital à 200-250 tr/min pendant la nuit.
- Préparer 5 mL de milieu LB (tableau 1; additionné de 10 g/L de glucose, 100 ampicilline µg/mL) dans un stérile 14 mL polystyrène culture tube croissance aérobie et ensemencer avec un séparément de la colonie d’un plat d’agar en utilisant une boucle d’extrémité ou inoculation pipette stérile.
-
Expression de type sauvage ECFP
- Ensemencer un milieu LB frais 10 mL (tableau 1; additionné de 10 g/L de glucose, 100 ampicilline µg/mL) avec 100µl de la culture au jour le jour dans un erlenmeyer de 100 mL. Incuber le ballon à 37 ° C dans un agitateur orbital à 200 tr/min.
Remarque : En option, cette étape peut être réalisée en moyenne 10 mL NMM19 (tableau 1) additionné de 100 µg/L ampicilline et à 0,5 mM de L-tryptophane (alternativement, indole peut être utilisé). - Mesurer la densité optique à 600 nm (OD600) chaque 20 min. préférentiellement mesurer densité de cellules en déterminant l’extinction à 600 nm (OD600) dans un spectrophotomètre en utilisant une cuvette avec une longueur de chemin d’accès de 1 cm. toujours effectuer une référence mesure à l’aide du milieu de culture correspondant. Diluer les échantillons et mélanger les échantillons bien pour obtenir une valeur de mesure de 0,1 à 0,8, alors calculer OD600 en utilisant le facteur de dilution. Pour plus de détails, veuillez vous référer à la précédente publication 26.
- En arrivant à une valeur de600 OD de 0,5 à 0,8 (environ 2-3 h après l’inoculation), prélever « avant l’induction » pour SDS-PAGE (électrophorèse sur gel de polyacrylamide dodécyl sulfate, étape 4).
- Induire l’expression de la protéine cible en ajustant la culture liquide à 0,5 mM IPTG (isopropyl β-D-1-thiogalactopyranoside, de solution mère de 1 M) et il incuber à 30 ° C dans un agitateur orbital à 200 tr/min pendant 4 à 8 heures.
NOTE : Cyan protéines fluorescentes sont couramment exprimées aux températures inférieures à 37 ° C,27. - Prélever « après l’expression » pour SDS-PAGE (étape 4.).
- Récolter les cellules bactériennes par centrifugation pendant 10 min à 5 000 x g et 4 ° C.
- Jeter le surnageant par décantation et geler les granules cellulaires à-20 ° C ou à-80 ° C jusqu'à ce que la purification des protéines cibles.
- Ensemencer un milieu LB frais 10 mL (tableau 1; additionné de 10 g/L de glucose, 100 ampicilline µg/mL) avec 100µl de la culture au jour le jour dans un erlenmeyer de 100 mL. Incuber le ballon à 37 ° C dans un agitateur orbital à 200 tr/min.
-
SPI de production GdFP
- Ensemencer 10 mL de milieu NMM19 (tableau 1) additionné de 100 µg/mL ampicilline, à 15 tryptophane µM et 10 µL de culture pendant la nuit dans un erlenmeyer de 100 mL et incuber le flacon de culture du jour au lendemain à 30 ° C dans un agitateur orbital à 200 tr/min.
Remarque : Une variété de milieux chimiquement définis pour la culture d’e. coli et SPI est disponible. En plus de NMM utilisé dans les présentes, vadrouilles moyenne28, glucose et minéraux sels moyenne29, Davis minimale moyenne30, M9 minimale moyenne31ou GMML32 peut être utilisé. - Le lendemain, mesurer OD600 toutes les 30 minutes jusqu'à ce que la valeur change uniquement de moins de 0,05 pendant 30 min. La valeur de plateau doit être approximativement de 1.
NOTE : Les écarts de ± 0,3 unités sont acceptables. Selon la souche bactérienne et le support utilisé, la concentration initiale de tryptophane (étape. 2.3.1) peut avoir besoin d’ajustement. - Prélever « avant l’induction » pour SDS-PAGE (étape 4.).
- Récolter les cellules bactériennes par centrifugation pendant 10 min à 5 000 x g et 4 ° C. Jeter le surnageant par décantation.
- Remettre en suspension les cellules dans 10 mL de milieu de NMM19 avec de l’ampicilline 100 µg/mL dans une fiole Erlenmeyer de 100 mL et ajouter 4-amino-acide indole à une concentration finale de 1 mM à l’aide de la solution mère de 50 mM. Poursuivre l’incubation pendant 30 min à 30 ° C dans un agitateur orbital à 200 tr/min.
Remarque : Cette étape est recommandée en raison de la faible stabilité chimique d’ampicilline et assure une absorption cellulaire du 4-amino-acide indole. - Induire l’expression de la protéine cible en ajoutant IPTG à une concentration finale de 0,5 mM à l’aide de stock de 1 M et incuber l’échantillon pendant la nuit à 30 ° C dans un agitateur orbital à 200 tr/min.
NOTE : Cyan protéines fluorescentes sont couramment exprimées aux températures inférieures à 37 ° C,27. - Le lendemain, mesurer OD600.
- Prélever « après l’expression » pour SDS-PAGE (étape 4.).
- Récolter les cellules bactériennes par centrifugation pendant 10 min à 5 000 x g et 4 ° C et éliminer le surnageant par décantation.
- Dans le cas où un tel navire n’était pas utilisé pour la centrifugation, transférer le culot cellulaire dans un tube conique en polystyrène de 50 mL à l’aide d’une spatule. Congeler le culot cellulaire à-20 ° C ou à-80 ° C jusqu'à ce que la purification des protéines cibles.
- Ensemencer 10 mL de milieu NMM19 (tableau 1) additionné de 100 µg/mL ampicilline, à 15 tryptophane µM et 10 µL de culture pendant la nuit dans un erlenmeyer de 100 mL et incuber le flacon de culture du jour au lendemain à 30 ° C dans un agitateur orbital à 200 tr/min.
3. cible Protein Purification par chromatographie d’affinité immobilisée Ion métallique (IMAC)
-
Lyse des cellules bactériennes
- Décongeler le culot cellulaire sur la glace pendant 10 à 20 min.
- Resuspendre le culot dans un tube conique en polystyrène de 50 mL à l’aide de 5 mL de tampon de liaison glacee (tableau 1) sur la glace.
- Ajouter 20 µL du lysozyme 50 mg/mL, 20 µL de 1 mg/mL DNase I et 20 µL de 1 mg/mL RNase A. fermer le tube, mélanger doucement en retournant le 5 fois et garder sur glace pendant 30 min.
NOTE : Désorganisation cellulaire partielle se produit comme catalysée par la lysozyme. - Lyse des cellules par sonication à l’aide d’une pointe de homogénisateur ultrasons à l’aide de trois cycles de 3 min dans un tube de 15 mL polystyrène refroidi par glacées avec 2 s d’impulsion, de 4 s d’amplitude de pause et de 45 %.
Remarque : Vous pouvez également l’homogénéisation à haute pression peut être utilisée, par exemple., 20 cycles à 14 000 lb/po2. Si nécessaire, diluer à l’aide de tampon de liaison pour atteindre le volume minimal d’instrument. En outre, réactifs d’extraction de protéine peuvent être utilisés pour la rupture de la cellule. Voir le tableau des matériaux pour obtenir des exemples. - Centrifuger l’échantillon pendant 30 min à 15 000-18 000 x g, 4 ° C.
- Transvaser le surnageant dans un nouveau tube et noter le volume de liquid.
- Filtrer la solution sur un filtre de 0,45 : seringue à l’aide d’une seringue de verrouillage Luer en plastique 5 mL et un filtre de seringue Polyfluorure de vinylidène (PVDF).
- Prélever « lysat » pour SDS-PAGE (étape 4.).
- Resuspendre le culot de débris cellulaire en FD2O (volume égal comme l’ancien lysat).
- Prenez échantillon « pellet » pour SDS-PAGE (étape 4.).
-
Purification de l’IMAC
- Utiliser un 1 mL préemballées ou emballé Self colonne IMAC FPLC (chromatographie liquide de protéine rapide) selon les instructions du fabricant. Utiliser le tampon de liaison (tableau 1) pour l’équilibration de colonne ainsi que pour l’étape de lavage qui suit après que le lysat cellulaire a été appliquée à la colonne.
- À frais virés et piscine éluat fractions avec des GdFP qui peuvent être identifiés par la couleur dorée lumière visible.
Remarque : En option, la protéine-cible peut être éluée utilisant un gradient linéaire imidazole (0-250 mM) à l’aide d’un système automatisé des FPLC. - Déterminer la concentration de la protéine à l’aide de la valeur de la littérature pour le coefficient d’extinction à 466 nm (ɛ466 nm = 23 700 M-1 cm-1)33 avec tampon d’élution comme référence. Pour plus d’informations sur la procédure, veuillez vous référer à la précédente publication26.
- Prendre l’échantillon « éluat » pour SDS-PAGE et utiliser de 1 à 10 µg de protéines par voie en cas de coloration au bleu de Coomassie.
NOTE : Quantités d’échantillon SDS peuvent varier selon la méthode de coloration et de la sensibilité de la teinture. - Dialyser une partie aliquote des fractions éluat contre tampon de dialyse ou MS tampon à l’aide d’une membrane dont le poids moléculaire coupure (MWCO) 5 000-10 000. Préparer la membrane de dialyse conformément aux instructions du fabricant. Dialyser un échantillon de 1 mL, trois fois contre 100 mL de tampon pendant au moins 2 h. Pour plus d’informations sur cette procédure, veuillez vous référer à la précédente publication34.
- Pour le stockage, geler l’échantillon de protéine dans le tampon de la dialyse à-80 ° C.
Remarque : Les aliquotes doivent être stables depuis au moins 6 mois.
4. SDS-PAGE échantillon préparation d’extrait de cellules entières d’e. coli
- Transférer une suspension de cellules équivalente à 1 mL de OD600 = 1 suspension (e.g. 500 µL de OD600= 2) dans un tube de microtubes de 1,5 mL.
- Récolter les cellules par centrifugation pendant 10 min à 5000 x g, température ambiante. Jeter le surnageant de pipetage.
- Ajouter 80 µL de ddH2O et 20 µL de SDS x 5 chargement de tampon de colorant (tableau 1) au culot et mélanger en pipettant également.
- Dénaturer les cellules par chauffage à 95 ° C pendant 5 min dans un bloc de baignoire ou de la chaleur de l’eau. Par la suite, laisser refroidir les échantillons à la température ambiante.
- Utiliser 10 µL pour coloration bleu de Coomassie SDS-PAGE selon la précédente publication35.
NOTE : Quantités d’échantillon SDS peuvent varier selon la méthode de coloration et de la sensibilité de la teinture.
5. analyse de masse des protéines intactes par chromatographie liquide à haute performance (HPLC), couplé à Electrospray Ionization Time-of-flight Mass Spectrometry (LC-ESI-TOF-MS)
NOTE : Tampons, paramètres et gradient de HPLC peuvent varier selon l’instrument utilisé et la colonne de séparation. Voir le tableau des matériaux pour l’équipement exemplaire.
- Déterminer la concentration de protéines d’un échantillon dialysée contre MS tampon comme décrit ci-dessus (étape 3.2.3.) en utilisant MS tampon (voir table des matières) comme référence.
- Diluer l’échantillon de protéine à 0,1 mg/mL à l’aide de tampons de MS pour un volume final de 80 µL, mélanger en pipettant également, prudent, transvaser la solution dans un flacon pour échantillonneur automatique de MS avec insert en verre et fermer avec un bouchon. Enlever les bulles d’air en effleurant le flacon.
- Remplir un deuxième auto-échantillonneur sans verre insert (tampon vide) avec 1 mL de tampon de MS.
- Laissez l’instrument pour se réchauffer. Effectuer le calibrage de l’instrument. S’assurer que des quantités suffisantes de solvants de chromatographie en phase liquide de qualité sont disponibles (> 100 mL).
- Programmer un gradient HPLC linéaire de 20 min de 5 % à 80 % tampon un (0,1 % acide formique dans ddH2O), combiné avec le tampon B (0,1 % de l’acide formique dans l’acétonitrile).
- Commencer par CLHP à un débit de 0,3 mL/mn et attendre que la pression de la colonne est stable.
- Définir un volume d’injection échantillonneur automatique de 5 µL pour la méthode LC-ESI-TOF-MS, créer une liste des tâches pour un témoin suivie d’un essai de l’échantillon et assignez l’échantillonneur automatique correspondant flacon postes. Exécuter la liste des tâches.
- Après la fin de la liste des tâches, ouvrez le fichier de données exemple généré. Sélectionnez une plage dans l’intrigue de (TIC) courant ionique total de déconvolution et deconvolute le spectre MS à l’aide de l’algorithme de déconvolution d’entropie maximale.
Remarque : Selon les conditions expérimentales, espèces supplémentaires peuvent s’appliquer de FP n’ou tampon ion adduits.
6. fluorescence Lifetime Mensurations et les spectres associés à désintégration (DAS) de GdFP
Remarque : Pour l’instrumentation de la spectroscopie de fluorescence résolue en temps, veuillez vous référer à la Table des matières pour l’équipement exemplaire. Absorbance comme excitation de fluorescence et spectres d’émission des protéines fluorescentes peuvent également être enregistrées à l’aide de spectrophotomètres UV/visible et de fluorescence de laboratoire.
-
Mesure de durée de vie de fluorescence résolue en longueur d’onde de GdFP
- Préparer 2 mL de 1 solution µM de GdFP de dilution dans le tampon PBS (tableau 1) à un pH de 7. Verser la solution dans une cuvette de quartz de 1 cm.
- Installer laser 470 nm ps-pulsé pour l’excitation de l’échantillon et le filtre de passe longue émission 488 nm et ajuster la grille du photon unique temps - et la longueur d’onde-corrélées comptant36 (TWCSPC) détecteur pour l’acquisition du régime d’onde de 600 L/mm 500 à 700 nm.
- Acquérir l’émission de fluorescence à un taux de comptage de photons d’environ 200 x 10 3/s jusqu'à environ 103 chefs d’accusation sont accumulent dans le maximum de l’acquisition des courbes de décomposition de fluorescence avec photon unique logiciel de comptage.
-
Mesure de la réponse instrumentale fonction37 (IRF)
- Remplacer la cuvette de l’échantillon avec une cuvette de quartz de 1 cm remplie de silice colloïdale de 1 g/L (~ 220 m2/g) dans un tampon PBS à pH 7.
NOTE : La suspension de la silice est établie à l’aide d’une suspension aqueuse de 400 g/L. - Supprimer les 488 nm-filtre passe-longue émission et insert gris filtres pour ajuster le taux de comptage sur le détecteur de TWCSPC au-dessous de 100 x 103 chefs d’accusation/s.
- Ajuster la grille pour l’acquisition de 470 photons nm au canal 8 du détecteur TWCSPC 16 canaux.
- Acquérir la Fri jusqu'à environ 10 x 103 chefs d’accusation sont accumulent dans l’émission maximale.
- Courbes de décomposition de fluorescence Convert et IRF aux fichiers de données ASCII avec global raccord38 programme.
- Comportement Global s’adapter selon un modèle d’une somme de trois composantes exponentielles avec des durées de vie en tant que paramètres liés.
- Désintégration de terrain associés spectres (DAS) comme distributions d’amplitude des composants individuels de désintégration en dépendance de la longueur d’onde avec logiciel d’analyse de données.
- Remplacer la cuvette de l’échantillon avec une cuvette de quartz de 1 cm remplie de silice colloïdale de 1 g/L (~ 220 m2/g) dans un tampon PBS à pH 7.
Representative Results
En utilisant la technique d’incorporation de la pression sélective, Trp-66 dans la triade chromophore ECFP (et Trp-57, le seul autre résidu de Trp dans ECFP) peut être remplacé par 4-amino-Trp, générant ainsi la GdFP décalée vers le rouge avec des propriétés spectrales distinctes. Spectrométrie de masse doit servir à démontrer l’intégration stoechiométrique désirée de l’acide aminé non canonique dans la protéine, avec les résultats présentés dans la Figure 1. Par la suite, nous fournir les données de microscopie, spectroscopie d’absorption UV-Vis ainsi que la spectroscopie de fluorescence stationnaire et résolue en temps et en longueur d’onde pour caractériser les propriétés du fluorophore GdFP en mettant l’accent sur l’influence du pH sur la spectres.
Pour confirmer l’échange des deux résidus Trp dans ECFP par 4-amino-Trp, analyse par spectrométrie de masse est effectuée. La figure 1 montre un éventail représentatif de ESI-MS déconvoluée de GdFP. ECFP sauvage a une masse de protéine calculé de 28,283.9 Da après la maturation du chromophore, la masse correspondante de GdFP est Da 28,313.9. Le spectre de ESI-MS déconvoluée du GdFP présente un pic de masse principal à 28,314.1 ± 0,1 Da, ce qui s’écarte de la valeur théorique de moins de 10 ppm. Être dans la fourchette de précision typique pour ce type d’analyse25, cela confirme l’incorporation de la ncAA via SPI (valeur expérimentale pour ECFP sauvage : Da 28,283.7).
La figure 2 montre les imagerie microscopie confocal fluorescence images (CFIM) de cellules bactériennes exprimant EGFP, EYFP, ECFP et GdFP à la remise en suspension des bactéries dans le tampon PBS. Toutes les images ont été acquises sur un microscope équipé d’une UV objectif et laser d’excitation à propos la même énergie pour chaque échantillon.
Figure 3 a montre une superposition d’images CFIM de bactérie e. coli exprimant différents FPs y compris GdFP, toujours surveillé avec énergie d’excitation très similaire (longueurs d’onde comme dans la Figure 2). Figure 3 b montre les structures chromophore des variantes FP montrés. En ce qui concerne la luminosité de GdFP par rapport à ECFP (fluorescence quantique rendement φ = 0,4), EGFP (φ = 0,6) et EYFP (φ = 0,6) il est important de noter que, pour GdFP, une gamme plus large d’acquisition de la lumière de fluorescence (30 nm) a été utilisé contrairement à 20 nm utilisé pour tous les autre spe CIES, afin d’ajuster l’intensité des images à des valeurs semblables. Avec un coefficient d’extinction légèrement plus faible et un rendement quantique réduite en raison des propriétés photophysiques unique, la luminosité de la GdFP est plus faible comparativement aux autres FPs montré.
Le spectre d’absorption de ECFP (Figure 3) a deux maxima caractéristiques à 434 nm et 452 nm. En revanche, les GdFP se caractérise par une bande d’absorption large décalée vers le rouge avec le maximum à 466 nm. L’absorption de EGFP est plus décalée vers le rouge à 488 nm. Cependant, en raison du déplacement de Stokes beaucoup plus grand de GdFP (108 nm) par rapport à ECFP (41 nm) et EGFP (20 nm), le spectre d’émission de GdFP est la plus décalée vers le rouge de tous les trois GFP dérivés étudiés ici (Figure 3D). Alors que l’émission de fluorescence de ECFP montre deux maxima caractéristiques à 475 nm et 505 nm, EGFP a une émission principale large bande avec un pic à 508 nm (λmax) avec une légère épaule à 540 nm. La fluorescence de la GdFP apparaît à environ 565 nm (λmax.) (Figure 3D). Son spectre d’émission contient une petite contribution de ECFP sauvage qui est également visible comme un petit épaulement à 475 nm. Cette petite fraction ECFP est synthétisée avant l’induction au cours de la procédure SPI, comme décrit33.
3E figure montre les changements de pH-dépendante dans le spectre d’absorption de GdFP. Pour un changement de pH de 8 à 5, l’émission maximale se déplace légèrement vers le rouge et un léger élargissement de la bande d’absorption est observé. Toutefois, la réduction de l’amplitude de l’absorption est seulement environ 10 % entre le pH de 8 et 5, ce qui indique que les propriétés de l’état fondamental du chromophore GdFP sont très faiblement modifié par pH.
Le temps résolu émission de fluorescence contrôlée par comptage de photon unique est illustrée à la Figure 4. Les courbes de décroissance surveillés dans les canaux spectraux centrées à 550 nm et 600 nm (Figure 4 a) présentent une légèrement plus rapide décroissance de fluorescence à 600 nm par rapport à la désintégration à 550 nm. Les résultats d’un ajustement global de la fluorescence de désintégration des courbes avec deux résultats de composantes exponentielles en deux composantes de désintégration de fluorescence spectralement différente avec des constantes de temps de 1,0 ns et 3.3 ns (Figure 4 et D).
L’émission de fluorescence de GdFP dépend fortement de pH, comme il est typique de nombreuses variantes de la protéine fluorescente de la famille GFP. Figure 4 b compare l’émission de fluorescence de GdFP entre pH 5 et pH 8, ce qui montre clairement une diminution de l’intensité de fluorescence à pH plus bas, alors que les caractéristiques spectrales restent constants.
Les spectres associés à décroissance (DAS)39 de GdFP (Figure 4 et D) sont caractérisés par deux bandes d’émission distincts. La contribution de la lente 3.3 composant ns est plus prononcée dans la gamme de longueur d’onde courte environ 550 nm (60 %) avec une contribution mineure de la composante plus rapide (40 %). À 600 nm, les deux composantes ont sur la même amplitude. Après un changement de pH 7 (Figure 4) à pH 6 (Figure 4), les caractéristiques spectrales de la DAS guère le changement et les constantes de temps de la routine de raccord global sont les mêmes (la précision des constantes de temps DAS est d’environ ± 0,15 ns). Cependant, la différence dans l’amplitude absolue des deux composants DAS est clairement apparente, qui tient pleinement compte de l’amplitude d’émission de fluorescence réduite sur le même changement de pH dans la Figure 4 b.
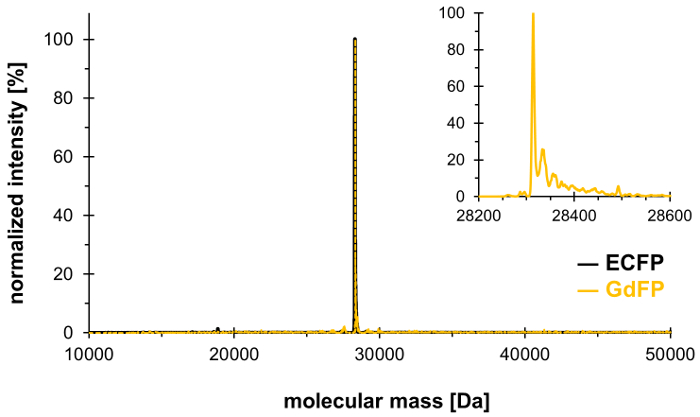
Figure 1 : spectre de ESI-MS déconvoluée représentatif de GdFP. Le spectre de l’ESI-MS de GdFP (couleur or, agrandie intrigue montré comme encart) montre un pic principal à 28314.1 Da (valeur 28313.9 Da). Le spectre de type sauvage ECFP est représenté en noir. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 2 : images de Microscopie Confocal fluorescence provenant de populations bactériennes exprimant différents FPs. Les paramètres suivants de la longueur d’onde ont été utilisés pour l’acquisition d’images : ECFP (λex = 457 nm, détection : 461-480 nm), EGFP (λex = 488 nm, détection : 495-515 nm), GDFP (λex = 476 nm, détection : 560-590 nm), EYFP (λex = 514 nm, détection : 520-530 nm). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 3 : propriétés spectrales de GdFP. (A), CFIM image d’un mélange de cellules bactériennes exprimant EGFP, ECFP et GdFP après la remise en suspension des bactéries dans le tampon PBS. (B) structures de Chromophore du GdFP (avec 4-amino-Trp à la place de résidus 66), ECFP parentale (avec Trp postées sur 66) et EFGP (avec Tyr postées sur 66). (C) Comparaison des spectres d’absorption normalisés de GdFP, ECFP et EGFP, considérant que (D), le spectre d’émission de fluorescence normalisée de ECFP (excitation à 430 nm) est contre les spectres d’émission de fluorescence de EGFP et GdFP (à la fois excitée à 450 nm). (E)-influence du pH sur les spectres d’absorption (normalisé d’absorption à 280 nm). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 4 : Time-resolved fluorescence de GdFP. Fluorescence (A) décomposition de GdFP surveillé par et longueur d’onde-résolution temporelle monophotonique comptage dans les canaux spectraux centrées à 550 nm et 600 nm (± 12,5 nm) après excitation avec 470 nm laser pulsé. La fonction de réponse instrumentale (IRF) fournit des informations sur la résolution au moment de la configuration utilisée. (B) Variation du spectre d’émission de GdFP dépend du pH (excitation à 460 nm). (C, D) Carie associées aux spectres (DAS) de GdFP à pH 7 (C) et pH 6 (D), déterminé après que déconvolution de fluorescence résolue en temps et en longueur d’onde se désintègre et montage global des désintégrations dans tous les canaux par un ensemble global de deux fonctions exponentielles avec constantes de temps lié. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 5 : Structures du transfert de charge intramoléculaire de ECFP (noir) et GdFP (or) chromophores. L’augmentation de taille du système chromophore par le donneur d’électrons bonne d’un groupement aminé dans le cadre de la ncAA permet la formation de structures plus mésomère pour atteindre la stabilisation par résonance de l’état excité. Les points de connexion à l’échafaud FP apparaissent comme des demi-cercles. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
| Solution mère | concentration, solvant | Remarque | |
| 20 % D-glucose | 200 g/L D-glucose en FD2O | stériliser par filtration à travers un filtre de seringue de taille de pore 0,45 µm | |
| indole | 50 mM dans l’isopropanol | ||
| 4-amino-acide indole | 50 mM de 20 % d’éthanol (20 mL d’éthanol dans un volume final de 100 mL rempli de ddH2O) | ||
| IPTG | 1 M de ddH2O | ||
| L-tryptophane | 15 mM dissous dans ddH2O avec 1 M HCl (HCl, ajouter quelques gouttes en remuant jusqu'à ce que la poudre est dissoved) | ||
| lysozyme | 50 mg/mL dans ddH2O | ||
| DNase I | 1 mg/mL en FD2O | ||
| RNase A | 1 mg/mL en FD2O | ||
| Amp100 | ampicilline 100 mg/mL en FD2O | ||
| dodécylsulfate de sodium (SDS) | 200 g/L en FD2O | ||
| sulfate d’ammonium ((NH4)2SO4) | 1 M de ddH2O | stériliser à l’autoclave | |
| phosphate monopotassique (KH2PO4) | 1 M de ddH2O | stériliser à l’autoclave | |
| hydrogénophosphate de di-potassium (K2HPO4) | 1 M de ddH2O | stériliser à l’autoclave | |
| sulfate de magnésium (MgSO4) | 1 M de ddH2O | stériliser à l’autoclave | |
| D-glucose | 1 M de ddH2O | stériliser par filtration à travers un filtre de seringue de taille de pore 0,45 µm | |
| chlorure de sodium (NaCl) | DdH2O de 5 M | stériliser à l’autoclave | |
| chlorure de calcium (CaCl2) | 1 g/L | stériliser par filtration à travers un filtre de seringue de taille de pore 0,45 µm | |
| chlorure de fer (II) (FeCl2) | 1 g/L | stériliser par filtration à travers un filtre de seringue de taille de pore 0,45 µm | |
| thiamine | 10 g/L | stériliser par filtration à travers un filtre de seringue de taille de pore 0,45 µm | |
| biotine | 10 g/L | stériliser par filtration à travers un filtre de seringue de taille de pore 0,45 µm | |
| mélange d’oligo-éléments | sulfate de cuivre CuSO4, zinc chlorure (ZnCl2), chlorure de manganèse (MnCl2), molybdate d’ammonium (NH4)2MoO4; chaque 1 mg/L dans les ddH2O | stériliser par filtration à travers un filtre de seringue de taille de pore 0,45 µm | |
| 19 acides aminés mix | 1.) dissoudre 0,5 g de L-phénylalanine et 0,5 g L-tyrosine en 100 ml FD2O goutte à goutte additionnée de 1 M de HCl en remuant jusqu'à ce que la poudre se dissout. | ||
| 2.) peser 0,5 g de chacun des acides aminé restantes (sauf le L-tryptophane). Mélanger avec fo 22 mL 1 M KH2PO4 et 48 mL de 1 M K2HPO4. Ajouter ddH2O 800 ml environ. Remuer jusqu'à ce que la solution devienne claire. | |||
| 3.) ajouter le dissous L-phénylalanine, L-tyrosine de l’étape 1.) et régler le volume à 1 L avec FD2O. | |||
| 4.) stériliser le mélange d’acides aminés par une filtration sous vide avec une unité de filtration haut de bouteille. | |||
| Tampons et medias | Composition/préparation | ||
| SDS tampon de colorant de chargement, 5 x concentré | 0,25 M Tris pH 6,8, glycérol à 50 % v/v, 0,25 % w/v bromophénol bleu, 0,5 M didhiothreitol (DTT ; sinon 5 % de β-mercaptoéthanol), 10 % p/v-dodécylsulfate de sodium (SDS) | ||
| tampon de liaison | dihydrogenphosphate de sodium de 50 mM (NaH2PO4), 500 mM NaCl, imidazole de 10 mM, pH 8 | ||
| tampon d’élution | dihydrogenphosphate de sodium de 50 mM (NaH2PO4), 500 mM NaCl, imidazole de 250 mM, pH 8 | ||
| tampon de dialyse | dihydrogenphosphate de sodium de 50 mM (NaH2PO4), 150 mM NaCl, glycérol 100 mL/L, pH 8 | ||
| Tampon de MS | 10 mM Tris-HCl, pH 8 | ||
| nouveau milieu minimal contenant 19 acides aminés L que le L-tryptophane (NMM19) | Mélanger les solutions-mères pour obtenir les concentrations finales suivantes : 7,5 mM (NH4)2SO4, 1,7 mM NaCl, 22 mM KH2PO4, 50 mM K2HPO4, 1 mM MgSO4, 20 mM D-glucose, 50 mg/L de 19 acides aminés mix, 1 µg/L CaCl2, 1 µg/L FeCl2, 10 thiamine µg/L, biotine de 10 mg/L, mélange d’oligo-éléments 0,01 mg/L | ||
| Milieu LB | Composition : tryptone 10 g/L, extrait de levure 5 g/L, 10 g/L de NaCl, pH 7.0 à FD2O | ||
| Préparation : | |||
| 1.) peser 50 g tryptone, extrait de 25 g de levure, 5 g de NaCl dans un flacon de verre de 1 L. | |||
| 2.) Add ddH2O à ~ 800 mL et dissoudre composants sous agitation. | |||
| 3.) mesurer le pH et ajuster le pH 7 par addition de goutte de 1 M HCl ou 1 NaOH M, si nécessaire. Ajouter ddH2O jusqu'à 1 L. | |||
| 4.) stériliser à l’autoclave, vérifier par la suite pour la perte de volume et ajouter ddH stérile2O d’indemniser si nécessaire. Conserver à 4 ° C jusqu'à l’utilisation. | |||
| Boîtes de gélose LB | Composition : tryptone 10 g/L, extrait de levure 5 g/L, 10 g/L de NaCl, agar-agar 15 g/L, pH 7.0 à FD2O | ||
| Préparation : | |||
| 1.) peser 50 g tryptone, extrait de 25 g de levure, 5 g de NaCl, agar-agar 7,5 g dans une bouteille de verre de 1 L. | |||
| 2.) Ajouter ddH2O jusqu'à 500 mL et dissoudre des composants sous agitation. | |||
| 3.) mesurer le pH et ajuster le pH 7 par addition de goutte de 1 M HCl ou 1 NaOH M, si nécessaire. Ajouter ddH2O jusqu'à 1 L. | |||
| 4.) stériliser à l’autoclave, vérifier par la suite pour la perte de volume et ajouter des ddH stérile2O pour compenser, si nécessaire. (Note : LB agar peut être stocké à 4 ° C jusqu'à l’utilisation pour la préparation des boîtes de gélose LB. Soigneusement fonte solidifiée gélose à l’aide d’un four à micro-ondes) | |||
| 5.) lorsque la solution est encore tiède (30-40 ° C), ajouter l’ampicilline à une concentration finale de 100 µg/mL | |||
| 6.) pour environ 15 mL du liquide de l’étape 5.) dans un stérile 10 cm boîte de Pétri dans des conditions stériles. Lorsque l’agar est solidifié, plaques peuvent être conservées pendant 1 semaine à 4 ° C jusqu'à l’utilisation. | |||
| solution saline tamponnée au phosphate (PBS) | Composition : 137 mM NaCl, KCl, 2,7 mM 10 mM Na2HPO4, 1,8 mM KH2PO4, 1 mM CaCl2, 0,5 mM MgCl2, pH 7. Stériliser à l’autoclave ou filtration. | ||
Tableau 1 : Solution mère et tampon.
Discussion
Pour atteindre des rendements très élevés d’incorporation ncAA, la méthode SPI auxotrophie repose sur l’utilisation des cellules-hôtes métaboliquement machiné, qui ne sont pas capables de synthétiser le corollaire naturel correspondante de la ncAA. Pour e. coli, ces souches sont disponibles. Même l’intégration simultanée de multiples ANAC dans la même protéine est réalisable à l’aide de souches multiauxotrophic. Le mode de résidus spécifiques de remplacement et le répertoire chimique étant limitée aux analogues chimiques similaires peut considérer comme des inconvénients. Néanmoins, un grand nombre de variantes de la protéine peut être produit comme les appareils de traduction bactérienne naturelle tolèrent de nombreux analogues d’acides aminés. Par exemple, plus de 50 ANAC pourrait comprendre des protéines à l’aide de la traduction in vitro , comptent pour environ 73 % de tous les codons du code génétique d’être disponibles pour réattribution,40. Par ailleurs, SPI peut aussi permettre un marquage efficace multisite de la protéine de cible41. En principe, la méthodologie SPI ne se limite pas à e. coli, mais peut fonctionner dans n’importe quel autre hôte et, pour chacun des 20 acides aminés canoniques, sous réserve que les souches auxotrophes et supports de culture définis sont disponibles. Par exemple, deux analogues de méthionine, azidohomoalanine (Aha) et homopropargylglycine (Hpg), sont commercialement disponibles et utilisés pour le marquage des protéines et protéomes dans divers organismes. En outre, les Aha peut être produite intracellulairement et par la suite incorporé dans la protéine42. Ce ncAA est particulièrement adapté aux conjugaisons de bioorthogonal comme chimie click développé par Tirrel et collègues de travail : par exemple, plantez le tissu de l' Arabidopsis thaliana, Bombyx mori larves43, Drosophila cellules44, larves de poisson zèbre45 ainsi que les cellules mammifères, y compris les neurones46, les protéines peuvent être étiquetés Aha47,,48. De même, Trp analogues ont été intégrées avec succès des peptides antimicrobiens en Trp-auxotrophes Lactococcus lactis souches49. SPI est également utile pour le champ de xénobiologie50,51, qui explore des solutions de rechange à la base composition chimique de la vie. Par exemple, basé sur des travaux antérieurs sur e. coli52 et53de la b. subtilis, une souche d’e. coli a été développée récemment par une stratégie évolutive avec une pression sélective d’utiliser thienopyrrole au lieu de indole, résultant en substitution à l’échelle du protéome du tryptophane par thienopyrrole-alanine dans le code génétique54. Généralement, l’aminoacide canonique Trp, codée par un seul triplet (UGG), présente une cible prometteuse pour l’ingénierie des protéines en raison les riches facettes de chimie indole, qui propose de nombreuses variations chimiques. Récemment et comme une alternative à l’incorporation axée sur le SPI, un roman SCS plateforme capable d’intégrer Trp analogues relativement chez les hôtes de bactéries et des eucaryotes a été rapporté55. Cela élargit encore la boîte à outils d’ingénierie en vivo axée sur le ncAA de protéines, y compris la modification des propriétés spectrales.
Hormis l’utilisation d’hôtes expression auxotrophes, le protocole SPI nécessite des conditions de fermentation stricte, tant en termes de calendrier d’expression cible et la composition du milieu afin d’atteindre une haute efficacité d’incorporation de ncAA et rendement de protéine cible 56. la culture est pratiquée au moyen de médias minimal chimiquement défini, qui contiennent essentiellement en dehors des principaux sels les sources d’azote (sel d’ammonium) et de carbone (D-glucose), de vitamines et d’oligo-éléments. Bien que pas strictement nécessaire en l’absence d’autres auxotrophies, les autres acides aminés (20 -n, si n aminoacides sont remplacés) sont généralement ajoutés pour promouvoir la croissance bactérienne57. Pendant une phase initiale de croissance avant l’induction de l’expression de la protéine cible, les acides aminés canonique de n à remplacer sont ajoutés pour limiter les concentrations. Croissance cellulaire continue jusqu'à ce que les acides aminés essentiels ciblées sont épuisés, indiqué comme expérimentalement par un stationnaire OD600. Par la suite, le milieu de culture est remplacé par un milieu frais qui manque de l’acide aminé appauvri et contient de la ncAA en concentrations abondantes. Pour l’incorporation ribosomique des analogues du tryptophane comme indiqué dans le présent protocole, un indole analogique est chargée, qui devient intracellulairement converti à la correspondante dérivée de tryptophane par tryptophane synthase58. Ensuite, l’expression de la protéine cible est induite. À ce stade, les cellules sont proches de la fin de la croissance logarithmique, comme un équilibre entre le nombre total de cellules et de remise en forme. Comme la présence et l’incorporation de la canonique amino entraînerait la production de protéines de type sauvage, il est essentiel de s’assurer que l’acide aminé essentiel est entièrement épuisée avant l’induction. De même, il est obligatoire de vérifier l’efficacité de l’incorporation de ncAA dans la protéine cible généralement par spectrométrie de masse. En cas de présence importante de l’acide aminé canonique, la culture des conditions nécessaire d’ajuster, par exemple, en modifiant la concentration de l’ordre acides aminés essentiels pour la phase initiale de croissance ou de la durée de ce dernier. Dans le cas de RAA faible activité vers la ncAA, la surexpression de l’enzyme endogène ou la co-expression d’un RAA différent, ce qui est plus active vers la ncAA, peut être menée59.
L’acide aminé canonique Trp est doté de trois caractéristiques remarquables : (i) son abondance naturelle en protéines est faible ; (ii) ses propriétés biophysiques et chimiques sont uniques (p. ex.., c’est généralement l’origine dominante de la fluorescence intrinsèque des protéines et des peptides) et (iii) il contribue à une variété d’interactions biochimiques et fonctions y compris Un empilement π, interactions hydrogènes et cation-π. Toutes ces caractéristiques sont radicalement changés après substitution 4-amino-Trp Trp → doute GdFP. au-delà, la conception d’une classe de « or » d’avGFPs est un exemple remarquable pour l’ingénierie des protéines auto-fluorescente sur mesure. Avec des propriétés spectrales distinctes, FPs peut être réglé vers certaines fenêtres spectrales par incorporation de mutagénèse et de la ncAA. En cas de GdFP, ceci est accompli par un simple échange chimique H → NH2 dans le cadre de l’anneau indole contenue dans la triade de chromophore ECFP. La figure 5 affiche les effets de l’incorporation de ncAA dans le chromophore. L’introduction du groupe électrodonneur provenant du 4-amino-acide indole (intracellulairement converti en 4-amino-Trp) permet une variété de structures mésomère qui peut expliquer un état excité stabilisé. Par spectroscopie, son déplacement de Stokes élargie et l’émission de fluorescence décalée vers le rouge résultent de ces propriétés distinctes du système conjugué prolongée. Comme indiqué précédemment, le transfert de charge intramoléculaire améliorée dans le chromophore GdFP est par nature sensible au pH (Figure 4 b) et accompagné d’un plus grand changement de moment dipolaire entre le sol de0 S et S1 état excité par rapport à ECFP33. Comme les autres groupes électrodonneurs, analogues de tryptophane portant un anneau indole substitué par des groupes hydroxyle pourraient être utilisés, tel que rapporté dans une étude comparative avec le modèle protéine barstar41.
Les spectres d’absorption et de fluorescence de GdFP sont élargies par rapport à ECFP et EGFP (Figure 3 et D). Un élargissement homogène des bandes d’absorption et de fluorescence est généralement causée par des modes de vibration dans le chromophore et, en outre, par couplage du chromophore aux autres modes vibrationnels présents dans les protéines60. Le couplage avec l’environnement local de protéine est pris en charge par les charges localisées sur le chromophore. Comme l’hétérogénéité structurelle de la protéine entraîne des variations locales du spectre vibronique, ce couplage entre les spectres de vibration du chromophore et le reste de la protéine sont pris en charge par la délocalisation de la charge et mésomère États figurant dans la Figure 5. Ce couplage prend également en charge le déplacement de Stokes grand et nécessairement réduit le rendement quantique de fluorescence. Par rapport aux autres FPs décalée vers le rouge, GdFP expositions même stabilité améliorée des protéines et une faible tendance pour agrégation33,,du6162. Il ne diffère en couleur à partir d’autres variantes de la FP mais présente aussi une thermostabilité une augmentation substantielle et coopératif amélioré pliage33. Son intensité de fluorescence est au moins 90 % conservé par chauffage à 60 ° C, alors que la fluorescence ECFP est réduite à environ 30 %. En protéines, acides aminés aromatiques contribuent souvent à des réseaux d’interaction des chaînes latérales, qui ont généralement un effet stabilisateur sur la structure tertiaire de la protéine. avGFP héberge un tel côté chaîne réseau, qui se compose du chromophore lui-même, aussi bien que Phe-165, His-148 et Tyr-145. Ces chaînes latérales ne sont pas assez rigides dans la structure de GdFP33, mais surtout, ils forment des contacts hydrophobes avec le chromophore. La plus importante nouveauté identifiée dans GdFP que le chromophore aminé est plus proximal à Phe-165. Cette interaction est une fonctionnalité ne pas observée dans les autres avGFPs connus. Comme les deux résidus de 3.2 à 4.5 Å apart, interactions aminé aromatique peuvent être également présentes. Ainsi que la stabilisation par résonance induite par l’amination du chromophore, ces probablement stabilise ce réseau hydrophobe des acides aminés de manière coopérative. Un transfert de charge intramoléculaire plus efficace peut être prise en charge de ces interactions dans l’état excité par rapport à l’état fondamental du chromophore, et elle représente au moins en partie, les 108 nm Stokes MAJ33,62 .
Dans une conception rationnelle des propriétés fluorophore, une augmentation de la taille du système π délocalisé prévoit d’aboutir à une longueur d’onde d’excitation décalée vers le rouge. Cette règle est respectée par la série des acides aminés en entraînant 66 position neutres chromophores : Phe (λmax = 355 nm) < son (λma x= 386 nm) < Tyr (λmax = 395 nm) < Trp (λmax = 436 nm)63. Dans la nature, cette extension du système conjugué du chromophore de liaisons π a été réalisée par différentes stratégies. Pour DsRed d’Acropora striata, elle est prolongée par l’intégration d’un acide aminé supplémentaire, passant ainsi λmax à 573 nm64. Le chromophore d’asFP595 (λmax = 595 nm) depuis Anemonia sulcata a été étendu par un groupe imino, élargissant son système π65. Le chromophore de GdFP et autres avFPs étant de la même taille, un principe différent doit entraîner une longueur d’onde d’émission de l’ordre de la DsRed élargi et asFP595 chromophores. Le déplacement de Stokes profond de 108 nm est attribuée à la structure distincte du chromophore GdFP, qui révèle un nouveau principe de photophysique dans la conception des protéines auto-fluorescente. Calculs préliminaires (comme signalé dans 62) ont montré que le moment dipolaire du chromophore état excité de GdFP est nettement plus important que dans l’état fondamental, par contraste avec les valeurs respectives de ECFP. Considérant que le moment dipolaire du GdFP augmente de 3 ~ D (Debye) dans l’état de0 S à ~ 15 S1, le changement pour le chromophore ECFP a été plutôt modéré (de D ~ 4 à ~ 6 D). Ainsi, la fluorescence or unique de GdFP est provoquée par transfert de charge intramoléculaire substantiel dans le chromophore, ce qui augmente la diversité des structures mésomère possibles (voir Figure 5) permettant la stabilisation par résonance. Cela réduit le niveau d’énergie dont l’émission se produit. En raison du profond changement le moment dipolaire à l’excitation, la séparation de charge intramoléculaire est la principale raison de l’évolution du potentiel électrostatique de l’environnement du chromophore. La matrice de protéine environnante, à son tour, s’ajuste aux changements dans la distribution de charge après excitation chromophore. La relaxation structurale ultérieure abaisse le niveau d’énergie du chromophore excité, qui déplace le spectre de fluorescence vers le rouge en raison de son caractère de transfert de charge. Pour la même raison, conséquence des gros déplacement de Stokes et taux améliorés des processus sans radiation, le rendement quantique de fluorescence de GdFP est réduite par rapport à ECFP33.
Le rendement quantique élevée et un petit déplacement de Stokes de ECFP et EGFP sont généralement attribuées à un environnement de protéine rigide du chromophore, ce qui réduit les degrés de liberté et, par conséquent, la conversion interne pour favoriser la relaxation radiative de l’état excité 66. par conséquent, la conception moléculaire des plus rigidement incorporées chromophores avec couplage réduit à la matrice de protéines restantes peut-être servir de guide pour produire plus loin décalée vers le rouge GFP dérivés avec rendement quantique de fluorescence élevée. Pour davantage d’ingénierie approches pour produire des protéines auto-fluorescente décalée vers le rouge, l’élargissement du système d’électrons π et une structure rigide chromophore avec couplage à l’environnement de la protéine faible est donc hautement souhaitable. Adaptations pourraient également être introduites directement sur chromophores GFP-basé ou de placement de l’ANAC désiré dans le voisinage du chromophore.
Disclosures
Les auteurs déclarent qu’ils n’ont aucun intérêt financier concurrentes.
Acknowledgments
Ce travail a été soutenu par la Fondation de recherche allemande (Cluster d’Excellence « Unifying Concepts en catalyse) à T.F. et n.-b. et par le ministère fédéral de l’éducation et des sciences (BMBF programme « HSP 2020", TU-WIMIplus projet SynTUBio) à F.-J.S.
Materials
| Name | Company | Catalog Number | Comments |
| Chemicals | |||
| 4-aminoindole | Sigma-Aldrich | 525022 | |
| acetonitrile | VWR | HiPerSolv CHROMANORM ULTRA for LC-MS, 83642 | LC-MS grade required |
| agar-agar | Carl Roth | 5210 | |
| ammonium molybdate ((NH4)2MoO4) | Sigma-Aldrich | 277908 | |
| ammonium sulfate ((NH4)2SO4) | Sigma-Aldrich | A4418 | |
| ampicillin sodium salt | Carl Roth | K029 | |
| biotin | Sigma-Aldrich | B4501 | |
| bromophenol blue | Sigma-Aldrich | B0126 | |
| calcium chloride (CaCl2) | Sigma-Aldrich | C5670 | |
| colloidal silica | Sigma-Aldrich | Ludox HS-40, 420816 | |
| Coomassie Brillant Blue R 250 | Carl Roth | 3862 | |
| copper sulfate (CuSO4) | Carl Roth | CP86.1 | |
| D-glucose | Carl Roth | 6780 | |
| di-sodium hydrogen phosphate (Na2HPO4) | Carl Roth | X987 | |
| di-potassium hydrogen phosphate (K2HPO4) | Carl Roth | P749.1 | |
| 1,4-dithiothreitol (DTT) | Carl Roth | 6908 | |
| DNase I | Sigma-Aldrich | D5025 | |
| ethanol | Carl Roth | 9065.1 | |
| formic acid | VWR | HiPerSolv CHROMANORM for LC-MS, 84865 | LC-MS grade required |
| glycerol | Carl Roth | 3783 | |
| imidazole | Carl Roth | X998 | |
| indole | Sigma-Aldrich | I3408 | |
| iron(II) chloride (FeCl2) | Sigma-Aldrich | 380024 | |
| isopropanol | Carl Roth | AE73.1 | |
| isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma-Aldrich | I6758 | |
| lysozyme | Sigma-Aldrich | L6876 | |
| magnesium chloride (MgCl2) | Carl Roth | KK36.1 | |
| magnesium sulfate (MgSO4) | Carl Roth | 8283.2 | |
| manganese chloride (MnCl2) | Sigma-Aldrich | 63535 | |
| β-mercaptoethanol | Carl Roth | 4227.3 | |
| potassium chloride (KCl) | Carl Roth | 6781.3 | |
| potassium dihydrogen phosphate (KH2PO4) | Sigma-Aldrich | P5655 | |
| RNase A | Carl Roth | 7156 | |
| sodium chloride (NaCl) | Carl Roth | P029 | |
| sodium dihydrogen phosphate (NaH2PO4) | Carl Roth | T879 | |
| sodium dodecyl sulphate (NaC12H25SO4) | Carl Roth | 0183 | |
| thiamine | Sigma-Aldrich | T4625 | |
| Tris(hydroxymethyl)-aminomethane (Tris) | Carl Roth | 5429 | |
| Tris hydrochloride (Tris-HCl) | Sigma-Aldrich | 857645 | |
| tryptone | Carl Roth | 8952 | |
| yeast extract | Carl Roth | 2363 | |
| zinc chloride (ZnCl2) | Sigma-Aldrich | 229997 | |
| Name | Company | Catalog Number | Comments |
| amino acids | |||
| L-alanine | Sigma-Aldrich | A7627 | |
| L-arginine | Sigma-Aldrich | A5006 | |
| L-asparagine | Sigma-Aldrich | A8381 | |
| L-aspartic acid | Sigma-Aldrich | A0884 | |
| L-cysteine | Sigma-Aldrich | C7352 | |
| L-glutamic acid | Sigma-Aldrich | G2128 | |
| L-glutamine | Sigma-Aldrich | G3126 | |
| L-glycine | Sigma-Aldrich | G7126 | |
| L-histidine | Sigma-Aldrich | H8000 | |
| L-isoleucine | Sigma-Aldrich | I2752 | |
| L-leucine | Sigma-Aldrich | L8000 | |
| L-lysine | Sigma-Aldrich | L5501 | |
| L-methionine | Sigma-Aldrich | M9625 | |
| L-proline | Sigma-Aldrich | P0380 | |
| L-phenylalanine | Sigma-Aldrich | P2126 | |
| L-serine | Sigma-Aldrich | S4500 | |
| L-threonine | Sigma-Aldrich | T8625 | |
| L-tryptophan | Sigma-Aldrich | T0254 | |
| L-tyrosine | Sigma-Aldrich | T3754 | |
| L-valine | Sigma-Aldrich | V0500 | |
| Name | Company | Catalog Number | Comments |
| Lab materials | |||
| 0.45 µm syringe filter with PVDF membrane | Carl Roth | CCY1.1 | |
| 1.5 mL microcentrifuge tubes | Eppendorf | 30120086 | |
| conical polystyrene (Falcon) tubes, 50 mL | Fisher Scientific | 14-432-22 | |
| Luer-Lock syringe 5 mL | Carl Roth | EP96.1 | |
| dialysis membrane, Molecular Weight Cut-Off (MWCO) 5,000 | Spectrum Medical Industries | Spectra/Por MWCO 5000 dialysis membrane, 133198 | |
| Immobilized Metal ion Affinity Chromatography (IMAC) column 1 mL, Ni-NTA | Macherey Nagel | Protino series, 745410.5 | |
| petri dishes (polystyrene, sterile) | Carl Roth | TA19 | |
| pQE-80L plasmid vector | Qiagen | no longer available | replaced by N-terminus pQE Vector set Cat No./ID: 32915 |
| protein extraction reagent BugBuster | EMB Millipore | 70921-4 | |
| round-bottom polystyrene tubes, 14 mL | Fisher Scientific | Corning Falcon, 14-959-1B | |
| Trp-auxotrophic E. coli strain | ATCC | ATCC 49980 | Bridges BA et al., Chem Biol Interact., 1972, 5(2):77-84; see main text for alternatives |
| Name | Company | Catalog Number | Comments |
| Mass Spectrometry equipment | |||
| mass spectrometer for LC-ESI-TOF-MS | Agilent | Agilent 6530 Accurate-Mass QTOF | coupled with Infinity LC system |
| mass spectrometry data analysis software | Agilent | MassHunter Qualitative Analysis software v. B.06.00 | |
| High-Performance Liquid Chromatography (HPLC) column for LC-ESI-TOF-MS | Sigma-Aldrich | Supelco Discovery BIO Wide Pore C5 HPLC column, 3 µm particle size, 10 cm x 2.1 mm | |
| HPLC autosampler vials 1.5 mL | Sigma-Aldrich | Supelco 854165 | with conical 0.1 mL glass inserts, screw caps and septa |
| Name | Company | Catalog Number | Comments |
| General equipment | |||
| benchtop centrifuge for 1.5 mL Eppendorf tubes | Eppendorf | 5427 R | |
| cooling centrifuge for 50 mL Falcon tubes | Eppendorf | 5810 R | |
| high pressure microfluidizer for bacterial cell disruption | Microfluidics | LM series with “Z” type chamber | |
| peristaltic pump for LC | GE Healthcare | P-1 | |
| Fast Protein Liquid Chromatography (FPLC) system | GE Healthcare | ÄKTA pure 25 L | |
| orbital shaker for bacterial cultivation | Infors HT | Minitron | |
| UV/Vis spectrophotometer | Biochrom | ULTROSPEC 2100 | |
| ultrasonic homogenizer for bacterial cell disruption | Omnilab | Bandelin SONOPULS HD 3200, 5650182 | with MS72 sonifier tip |
| Name | Company | Catalog Number | Comments |
| Fluorescence spectroscopy equipment | |||
| ps-pulsed laser 470 nm | Picoquant GmbH | PDL-470 | |
| time- and wavelength-correlated single photon counting (TWSPC) acquisition software | Picoquant GmbH | SymPhoTime 64 | |
| time- and wavelength-correlated single photon counting (TWSPC) detector | Picoquant GmbH | PML-16C | 16 spectral channels, to be selected by grating settings |
| single photon counting software | Picoquant GmbH | SPCM 9.75 | |
| global fitting software | Picoquant GmbH | SPC2Glo(R) | |
| fluorescence decay data analysis software | Picoquant GmbH | FluoFit program | |
| data analysis software | OriginLab Inc. | Origin 9.2 | |
| neutral density filter set | Schott | NG1 to NG11 | (400 - 650 nm, transmission 50 %, 20%, 10 %, 5 %) |
| 488 nm long-pass emission filter | AHF Analysentechnik | AHF-488 | |
| quartz cuvette | Thorlabs GmbH | CV10Q1400 | 1 cm pathlength |
References
- Shimomura, O., Johnson, F. H., Saiga, Y. Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan, Aequorea. J Cell Compar Physl. 59 (3), 223-239 (1962).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263 (5148), 802-805 (1994).
- Andresen, M., et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. P Natl Acad Sci USA. 102 (37), 13070-13074 (2005).
- Andresen, M., et al. Structural basis for reversible photoswitching in Dronpa. P Natl Acad Sci USA. 104 (32), 13005-13009 (2007).
- Brakemann, T., et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat Biotechnol. 29 (10), 942-947 (2011).
- Kremers, G. -J., Gilbert, S. G., Cranfill, P. J., Davidson, M. W., Piston, D. W. Fluorescent proteins at a glance. J Cell Sci. 124 (Pt 2), 157-160 (2011).
- Shimomura, O. Structure of the chromophore of aequorea 0. shimomura green fluorescent protein. FEBS Lett. 104 (2), 220-222 (1979).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 22 (12), 1567-1572 (2004).
- Shcherbo, D., et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 4 (9), 741-746 (2007).
- Shcherbakova, D. M., Subach, O. M., Verkhusha, V. V. Red fluorescent proteins: advanced imaging applications and future design. Angew Chem Int Edit. 51 (43), 10724-10738 (2012).
- Stepanenko, O. V., Verkhusha, V. V., Kuznetsova, I. M., Uversky, V. N., Turoverov, K. K. Fluorescent proteins as biomarkers and biosensors: throwing color lights on molecular and cellular processes. Curr Protein Pept Sc. 9 (4), 338-369 (2008).
- Wang, L., Xie, J., Deniz, A. A., Schultz, P. G. Unnatural amino acid mutagenesis of green fluorescent protein. J Org Chem. 68 (1), 174-176 (2003).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Sharma, N., Furter, R., Kast, P., Tirrell, D. A. Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett. 467 (1), 37-40 (2000).
- Liu, C. C., Schultz, P. G. Adding new chemistries to the genetic code. Annu Rev Biochem. 79, 413-444 (2010).
- Twine, S. M., Murphy, L., Phillips, R. S., Callis, P., Cash, M. T., Szabo, A. G. The Photophysical Properties of 6-Azaindole. J Phys Chem B. 107 (2), 637-645 (2003).
- Lepthien, S., Hoesl, M. G., Merkel, L., Budisa, N. Azatryptophans endow proteins with intrinsic blue fluorescence. P Natl Acad Sci USA. 105 (42), 16095-16100 (2008).
- Budisa, N., et al. Probing the role of tryptophans in Aequorea victoria green fluorescent proteins with an expanded genetic code. Biol Chem. 385 (2), 191-202 (2004).
- Ross, J. B., et al. Spectral enhancement of proteins: biological incorporation and fluorescence characterization of 5-hydroxytryptophan in bacteriophage lambda cI repressor. P Natl Acad Sci USA. 89 (24), 12023-12027 (1992).
- Soumillion, P., Jespers, L., Vervoort, J., Fastrez, J. Biosynthetic incorporation of 7-azatryptophan into the phage lambda lysozyme: Estimation of tryptophan accessibility, effect on enzymatic activity and protein stability. Protein Eng Des Sel. 8 (5), 451-456 (1995).
- Heim, R., Tsien, R. Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 6 (2), 178-182 (1996).
- Bridges, B. A., Mottershead, R. P., Rothwell, M. A., Green, M. H. L. Repair-deficient bacterial strains suitable for mutagenicity screening: tests with the fungicide captain. Chem Biol Interact. 5 (2), 77-84 (1972).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: The Heat Shock Method. J Vis Exp. , (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: Electroporation. J Vis Exp. , (2017).
- Grigorenko, B. L., Krylov, A. I., Nemukhin, A. V. Molecular modeling clarifies the mechanism of chromophore maturation in the green fluorescent protein. J Am Chem Soc. , (2017).
- JoVE Science Education Database. General Laboratory Techniques. Introduction to the Spectrophotometer. J Vis Exp. , (2017).
- Goedhart, J., et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun. 3, 751 (2012).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F. Culture medium for enterobacteria. J Bacteriol. 119 (3), 736-747 (1974).
- Hörnsten, E. G. On culturing Escherichia coli on a mineral salts medium during anaerobic conditions. Bioprocess Eng. 12 (3), 157-162 (1995).
- Davis, B. D. The Isolation of Biochemically Deficient Mutants of Bacteria by Means of Penicillin. P Natl Acad Sci USA. 35 (1), 1-10 (1949).
- Sambrook, J., Russell, D. W. Molecular Cloning: A Laboratory Manual. , Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY, USA. (2001).
- Wang, Y. -S., et al. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives. Mol Biosyst. 7 (3), 714-717 (2011).
- Bae, J. H., et al. Expansion of the genetic code enables design of a novel "gold" class of green fluorescent proteins. J Mol Biol. 328 (5), 1071-1081 (2003).
- JoVE Science Education Database. Dialysis: Diffusion Based Separation. J Vis Exp. , Cambridge, MA. (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Separating Protein with SDS-PAGE. J Vis Exp. , (2017).
- Petrásek, Z., et al. Excitation energy transfer from phycobiliprotein to chlorophyll d in intact cells of Acaryochloris marina studied by time- and wavelength-resolved fluorescence spectroscopy. Photoch Photobio Sci. 4 (12), 1016-1022 (2005).
- Kolber, Z. S., Barkley, M. D. Comparison of approaches to the instrumental response function in fluorescence decay measurements. Anal Biochem. 152 (1), 6-21 (1986).
- Pelet, S., Previte, M. J. R., Laiho, L. H., So, P. T. C. A fast global fitting algorithm for fluorescence lifetime imaging microscopy based on image segmentation. Biophys J. 87 (4), 2807-2817 (2004).
- Loefroth, J. E. Time-resolved emission spectra, decay-associated spectra, and species-associated spectra. J Phys Chem. 90 (6), 1160-1168 (1986).
- Hartman, M. C. T., Josephson, K., Lin, C. -W., Szostak, J. W. An expanded set of amino acid analogs for the ribosomal translation of unnatural peptides. PLoS One. 2 (10), e972 (2007).
- Budisa, N., et al. Global replacement of tryptophan with aminotryptophans generates non-invasive protein-based optical pH sensors. Angew Chem Int Edit. 41 (21), 4066-4069 (2002).
- Ma, Y., Biava, H., Contestabile, R., Budisa, N., di Salvo, M. L. Coupling bioorthogonal chemistries with artificial metabolism: intracellular biosynthesis of azidohomoalanine and its incorporation into recombinant proteins. Molecules. 19 (1), 1004-1022 (2014).
- Teramoto, H., Kojima, K. Incorporation of Methionine Analogues Into Bombyx mori Silk Fibroin for Click Modifications. Macromol Biosci. 15 (5), 719-727 (2015).
- Deal, R. B., Henikoff, J. G., Henikoff, S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 328 (5982), 1161-1164 (2010).
- Hinz, F. I., Dieterich, D. C., Tirrell, D. A., Schuman, E. M. Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci. 3 (1), 40-49 (2012).
- Dieterich, D. C., et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 13 (7), 897-905 (2010).
- Dieterich, D. C., Link, A. J., Graumann, J., Tirrell, D. A., Schuman, E. M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). P Natl Acad Sci USA. 103 (25), 9482-9487 (2006).
- Glenn, W. S., et al. Bioorthogonal Noncanonical Amino Acid Tagging (BONCAT) Enables Time-Resolved Analysis of Protein Synthesis in Native Plant Tissue. Plant Physiol. 173 (3), 1543-1553 (2017).
- Zhou, L., et al. Incorporation of tryptophan analogues into the lantibiotic nisin. Amino Acids. 48 (5), 1309-1318 (2016).
- Acevedo-Rocha, C. G., Budisa, N. Xenomicrobiology: a roadmap for genetic code engineering. Microb Biotechnol. 9 (5), 666-676 (2016).
- Agostini, F., Völler, J. -S., Koksch, B., Acevedo-Rocha, C. G., Kubyshkin, V., Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew Chem Int Edit. 56 (33), 9680-9703 (2017).
- Bacher, J. M., Ellington, A. D. Selection and characterization of Escherichia coli variants capable of growth on an otherwise toxic tryptophan analogue. J Bacteriol. 183 (18), 5414-5425 (2001).
- Wong, J. T. Membership mutation of the genetic code: loss of fitness by tryptophan. Pc Natl Acad Sci USA. 80 (20), 6303-6306 (1983).
- Hoesl, M. G., et al. Chemical Evolution of a Bacterial Proteome. Angew Chem Int Edit. 54 (34), 10030-10034 (2015).
- Italia, J. S., et al. An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nat Chem Biol. 13 (4), 446-450 (2017).
- Völler, J. -S., Thi To, T. M., Biava, H., Koksch, B., Budisa, N. Global substitution of hemeproteins with noncanonical amino acids in Escherichia coli with intact cofactor maturation machinery. Enzyme Microb Tech. 106, 55-59 (2017).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Völler, J. -S., Budisa, N. Coupling genetic code expansion and metabolic engineering for synthetic cells. Curr Opin Biotech. 48, 1-7 (2017).
- Johnson, J. A., Lu, Y. Y., Van Deventer, J. A., Tirrell, D. A. Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Curr Opin Chem Biol. 14 (6), 774-780 (2010).
- Somsen, O. J., van Grondelle, R., van Amerongen, H. Spectral broadening of interacting pigments: polarized absorption by photosynthetic proteins. Biophys J. 71 (4), 1934-1951 (1996).
- Kurschus, F. C., Pal, P. P., Bäumler, P., Jenne, D. E., Wiltschi, B., Budisa, N. Gold fluorescent annexin A5 as a novel apoptosis detection tool. Cytom Part A. 75 (7), 626-633 (2009).
- Lepthien, S., Wiltschi, B., Bolic, B., Budisa, N. In vivo engineering of proteins with nitrogen-containing tryptophan analogs. Appl Microbiol Biot. 73 (4), 740-754 (2006).
- Wachter, R. M., Elsliger, M. -A., Kallio, K., Hanson, G. T., Remington, S. J. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure. 6 (10), 1267-1277 (1998).
- Verkhusha, V. V., Lukyanov, K. A. The molecular properties and applications of Anthozoa fluorescent proteins and chromoproteins. Nat Biotechnol. 22 (3), 289-296 (2004).
- Martynov, V. I., Savitsky, A. P., Martynova, N. Y., Savitsky, P. A., Lukyanov, K. A., Lukyanov, S. A. Alternative cyclization in GFP-like proteins family. The formation and structure of the chromophore of a purple chromoprotein from Anemonia sulcata. J Biol Chem. 276 (24), 21012-21016 (2001).
- Piatkevich, K. D., Malashkevich, V. N., Morozova, K. S., Nemkovich, N. A., Almo, S. C., Verkhusha, V. V. Extended Stokes shift in fluorescent proteins: chromophore-protein interactions in a near-infrared TagRFP675 variant. Sci Rep. 3 (1), 1847 (2013).
