

Summary
Biologia sintética permite a engenharia de proteínas com propriedades sem precedentes usando a inserção co-translational de aminoácidos não-canônicas. Aqui, apresentamos como uma variante espectralmente avermelhado de um fluoróforo GFP-tipo com propriedades espectroscópicas de fluorescência romance, denominado proteína fluorescente "ouro" (GdFP), é produzida em Escherichia coli através de incorporação de pressão seletiva (SPI).
Abstract
Proteínas fluorescentes são instrumentos fundamentais para as Ciências da vida, em particular para microscopia de fluorescência das células vivas. Enquanto o selvagem-tipo e projetou variantes da proteína verde fluorescente de Aequorea victoria (avGFP), bem como homologs de outras espécies já cobrem grandes partes do espectro óptico, uma lacuna espectral permanece na região do infravermelho próximo, para quais fluorophores baseada em avGFP não estão disponíveis. Variantes da proteína fluorescente avermelhado (FP) expandiria substancialmente o toolkit para desagregação espectral de múltiplas espécies moleculares, mas o FPs avermelhado natural derivado de corais ou anêmonas têm menor rendimento quântico de fluorescência e foto-estabilidade inferior em comparação com as variantes de avGFP. Mais manipulação e possível expansão de sistema conjugado do cromóforo para a região espectral far-red também é limitada pelo repertório de 20 aminoácidos canônicos previstos no código genético. Para superar essas limitações, biologia sintética pode alcançar ainda mais espectral vermelho de mudança através da inserção de aminoácidos não-canônicas para a Tríade de cromóforo. Descrevemos a aplicação da SPI para variantes de engenheiro avGFP com propriedades espectrais de romance. Expressão da proteína é executada em um triptofano-auxotróficos Escherichia coli cepa e completando a mídia de crescimento com precursores de indole apropriado. No interior das células, estes precursores são convertidos para os análogos de triptofano correspondente e incorporados em proteínas pela máquina ribosomal em resposta aos códons UGG. A substituição do Trp-66 na variante melhorada "ciano" do avGFP (ECFP) por um elétron-doando 4-aminotryptophan resulta em GdFP com um deslocamento de Stokes 108 nm e uma máximo de emissão fortemente avermelhado (574 nm), enquanto ser termodinamicamente mais estável do que seu antecessor ECFP. Incorporação de resíduos específicos do aminoácido não-canônicos é analisada por espectrometria de massa. As propriedades espectroscópicas de GdFP são caracterizadas por espectroscopia de fluorescência tempo-resolvido como uma das valiosas aplicações de FPs geneticamente codificado nas ciências da vida.
Introduction
Desde a descoberta da proteína verde fluorescente na Medusa Aequorea victoria (avGFP) em 19621 e a primeira expressão heteróloga em 19942 em outras células eucarióticas, tornaram-se proteínas fluorescentes da família GFP ferramentas altamente valiosas e alvos nas ciências da vida. Engenharia genética e molecular extensa incluído o ajuste de uso códon espécie-específicos, aceleração de dobramento maturação melhorada, aumento de brilho, prevenção da oligomerização e alfaiataria de propriedades espectrais e fotoquímicas incluindo a capacidade de forma reversível photoswitch3,4,5,6. GFP deve sua fluorescência do seu 4-(p- hydroxybenzylidene) imidazolidin-5-1 (HBDI) cromóforo. O último autocatalytically formado a partir da Tríade de cromóforo chamados de aminoácidos (Ser-65/Tyr-66/Gly-67 em avGFP) após a formação de uma ligação covalente adicional dentro a espinha dorsal do peptide sob a influência do oxigênio molecular7. O sistema conjugado ressonantemente estabilizado dinamicamente interage com seu ambiente molecular, permitindo a absorção na faixa do visível e fluorescência verde característica destas proteínas.
Dentro da Tríade cromóforo, a presença de um aminoácido aromático é obrigatória. No entanto, o repertório padrão de aminoácido é composto por apenas quatro resíduos aromáticos (dele, Phe, Trp e Tyr). Isso limita a mutagênese convencional abordagens para alcançar substancialmente mais avermelhado avGFP variantes em relação à FPs mais avermelhado natural como DsRed8 de Discosoma striata coralimorphs ou mKate/mNeptune9 de a anêmona Entacmaea quadricolor. Portanto, a porção far-red e infravermelha do espectro óptico acima de 600 nm é escassamente coberto por variantes GFP. Isto é, naturalmente, uma severa limitação para abordagens microscópicas de fluorescência que exigem demultiplexação espectral de várias espécies de fluoróforo ao mesmo tempo. Por exemplo, marcadores de longo comprimento de onda também são necessários para fazer uso do regime de baixa absorção dos tecidos da pele entre 700-1.000 nm em configurações para tecidos mais profundos de imagem10.
Proteínas fluorescentes, derivadas de avGFP são divididas em várias classes com base nas propriedades espectroscópicas e natureza química dos seus cromóforos11. Com sua Tríade 65-EMISSAO SERIE/Tyr/Gly-66-67, o cromóforo do selvagem-tipo existe como uma equilibrada mistura entre a forma neutra, fenólica (λmáx = 395 nm, ε = 21.000 M-1cm-1) e a forma aniônica fenolato (λmáx = 475 nm, ε = 7.100 M -1cm-1), e o espectro de emissão apresenta um único pico no 508 nm. O grupo hidroxila de EMISSAO SERIE-65 é de importância crítica, como que doa uma ligação H-a Glu-222, nas proximidades de cromóforo (distância: 3.7 Å), que promove a ionização deste carboxilato. Classe, que é caracterizada por um cromóforo fenolato aniônicos, como em EGFP (64-Phe-Leu-65/EMISSAO SERIE-Thr; λmáx = 488 nm, ε = 35.600 M-1cm-1, λem = 509 nm). Devido a substituição de Ser-65-Thr(Ala,Gly), o pico de excitação 395 nm do formulário neutro fenol é suprimida e o 470-475 nm pico do fenolato aniônico é cinco a seis vezes reforçada e deslocou a 490 nm. Classe II é composto por proteínas com um cromóforo fenólico neutra, como em safira-GFP. Aqui, a substituição de Thr-203-Ile quase completamente suprime a excitação nm 475, deixando apenas o pico em 399 nm. Desde que o cromóforo aniônico não pode ser adequadamente solvated, sua forma neutra é favorecida. Classe III compreende as variantes fluorescentes "amarelas" (EYFP; Ser-65-Gly/Val-68-leu/ser-72-ala/thr-203-Tyr; Máx ε λ = 514 nm, ε = 84, 600 M-1cm-1, λem = 527 nm) com interação π-empilhamento de uma cadeia aromática de lado e o fenolato, como trazido pelas substituições de Thr-203-His(Trp,Phe,Tyr), que levam a até 20 nm máximos de emissão avermelhado (Thr-203-Tyr). Mais substituição (Gln-69-Lys) resulta em outro 1-2 nm red shift para 529 nm, a variante mais avermelhado do avGFP conhecido11. A troca do fenol por um indol (Tyr-66-Trp) cria a classe IV, como a ECFP ciano-fluorescente (Ser-65-Thr/Tyr-66-Trp; λmax1 = 434 nm, ε = 24.800 M-1cm-1; λmax2 = 452 nm, ε = 23.600 M-1cm-1 ; Λem1 = 477 nm, λem2 = 504 nm). O alojamento de indole o volumoso provavelmente está habilitado por outras, mutações compensatórias. O maxima de excitação e emissão de ECFP cair inbetween aqueles de proteínas com cromóforos aniônicos ou neutros. Proteínas de classe V abrigam um imidazol no lugar do fenol (Tyr-66-His), por exemplo., proteínas fluorescentes azul como EBFP. Classe VI é produzido por uma troca de fenol-para-fenil favorecendo a forma neutra cromóforo exclusivamente, que consequentemente leva aos mais azulado da excitação e emissão de posições de pico (360 nm e 442 nm, respectivamente).
Mutagenesis local-dirigido clássico é especialmente adequado para a produção de variantes de cromóforo romance avGFP, pela permutação do tripeptídeo e interação resíduos no quadro dos 20 aminoácidos canônicos de 65-67. Essas possibilidades podem ser expandidas ainda mais quando as variantes não-canônicos de aminoácidos aromáticos são introduzidas durante a síntese de proteínas ribossomais12. Em princípio, há duas maneiras de conseguir isso. A primeira estratégia depende da tolerância de substrato do mecanismo de tradução do proteína, especialmente de sintetases aminoacil-tRNA (aaRSs) no sentido de análogos de aminoácidos relacionados. Para alcançar este objectivo com alta eficiência, auxotróficos estirpes de Escherichia coli expressão são empregadas que são incapazes de sintetizar o aminoácido correspondente natural. Isto permite a substituição do último adicionando apropriados aminoácidos não-canônicos (ncAAs) ou precursores respectivos para o meio de cultura. Esta estratégia, também conhecido como incorporação de pressão seletiva (SPI)13,14, permite substituições de resíduos específicos, que resultam em incorporação global do ncAA. A segunda estratégia utiliza stop códon supressor tRNAs, que são cobrados com a ncAA por engenharia aaRS enzimas. Isso resulta na ler de códons de parada em-frame e permite a incorporação de ncAA site-specific. Consequentemente, esse método de supressão de códon de parada (SCS) leva à expansão do código genético15. Através de mutagénese, um códon de parada é colocado no gene do alvo no local desejado. Em princípio, a SPI também pode ser usado para criar recombinantes peptídeos e proteínas, tendo uma única instalação do ncAA, dado que raro aminoácidos canônicos como Met ou Trp são escolhidos para substituição. Com Trp, SPI abordagens têm sido mostradas para trabalhar com uma grande variedade de análogos, incluindo 4 - F - e 5 - F - e 6-F-Trp, 7-aza-Trp, 4-OH - e 5-OH-Trp, bem como 4 - 5-NH2- Trp ou mesmo β (thienopyrrolyl) alanina derivados16 ,17,18,19,20. Assim, a SPI pode ser altamente vantajoso para a substituição de aminoácidos aromáticos de cromóforos GFP por variantes não-canônicos para explorar a possibilidade de adaptar mais espectros e deslocamento de Stokes desses FPs. Quanto a todas as modificações de sequência de proteínas, a compatibilidade com maturação de dobramento e cromóforo FP deve ser testada experimentalmente.
Neste trabalho, utilizamos a classe IV ECFP21, que carrega em vez do selvagem-tipo avGFP Tyr, um resíduo de Trp dentro sua Tríade de cromóforo. Usando SPI, este Trp-66 (e Trp-57, apenas outro Trp resíduo na ECFP) é substituído pelo 4-amino-TRP. A presença do grupo amino elétron-doação de 4-amino-Trp dentro o cromóforo favorece a estabilização de ressonância de uma transferência de prótons do estado excitado muito avermelhado (ESPT) dotada de um deslocamento de Stokes 108 nm. Esta proteína fluorescente "ouro" (GdFP) constitui a variante com o maior desvio para o vermelho da fluorescência máxima (574 nm) entre todas as proteínas derivadas de avGFP. Descrevemos o método de produção de proteína GdFP pela SPI e fornecer os protocolos para a análise obrigatória das proteínas modificadas resultantes por espectroscopia de massa. Além disso, mostramos como GdFP pode ser utilizada e analisada em abordagens de espectroscopia de fluorescência tempo-resolvido.
Protocol
1. transformação de Trp-auxotróficos Escherichia coli
- Transformar quimicamente ou electrocompetent células (50 µ l) de uma estirpe de Trp-auxotróficos Escherichia coli , por exemplo. ATTC 49980 (WP2, mutante, derivada da cepa de Escherichia coli B/R22), com 1 µ l de uma solução aquosa a 1 ng / µ l do plasmídeo de His6-ECFP pQE - 80L usando choque térmico ou eletroporação, respectivamente. Por favor consulte o JoVE ciência educação de banco de dados23,24 para obter detalhes.
Nota: O vetor de expressão pQE - 80L His6-ECFP codifica um N-terminal 6 x ECFP com sua tag21 impulsionado por um promotor T5 bacteriano com operador lac. Ele carrega mais um marcador de seleção AmpR e uma origem de replicação do colE1 (a sequência de espinha dorsal pQE - 80L vetor pode ser encontrada em: https://www.qiagen.com/mx/resources/resourcedetail?id=c3b71572-4d82-4671-a79b-96357fe926d1&lang=en & autoSuggest = true). O teórico peso molecular da proteína selvagem-tipo His6-ECFG (após a maturação de cromóforo25) é Da 28303.92. A sequência da proteína alvo traduzido é como segue (sua marca sublinhada, vetor-derivado de sequências em negrito): MRGSHHHHHHGSMVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTLTWGVQCFSRYPDHMK
QHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYISHNVYITADKQKNGIKANFKIRHNIEDGS
VQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYK. - Células transformadas em placas LB-ágar (tabela 1) suplementadas com glicose 10 g/L, 100 ampicilina µ g/mL a placa e incubar as placas a 37 ° C durante a noite.
2. expressão da proteína recombinante
-
Durante a noite cultura de Escherichia coli ATCC 49980 pQE - 80 L His6-ECFP
- Prepare-se 5 mL de meio LB (tabela 1; suplementado com glicose 10 g/L, 100 ampicilina µ g/mL) em uma estéril mL 14 cultura poliestireno tubo para crescimento aeróbio e inocular uma colônia isoladamente de uma placa de ágar usando um loop de ponta ou inoculação de pipeta estéril.
Nota: Usar colônias de células recém transformadas é recomendado. As placas com colônias bacterianas (da etapa 1.2.) podem ser armazenadas a 4 ° C por vários dias. - Incube as celulas a 37 ° C em um agitador orbital em 200-250 rpm, durante a noite.
- Prepare-se 5 mL de meio LB (tabela 1; suplementado com glicose 10 g/L, 100 ampicilina µ g/mL) em uma estéril mL 14 cultura poliestireno tubo para crescimento aeróbio e inocular uma colônia isoladamente de uma placa de ágar usando um loop de ponta ou inoculação de pipeta estéril.
-
Expressão do selvagem-tipo ECFP
- Inocular 10 mL meio fresco do LB (tabela 1; suplementado com glicose 10 g/L, 100 ampicilina µ g/mL) com 100 µ l da cultura durante a noite em um Erlenmeyer de 100 mL. Incube o frasco a 37 ° C em um agitador orbital a 200 rpm.
Nota: Opcionalmente, esta etapa pode ser executada em 10 mL NMM19 suplementado (tabela 1) com 100 µ g/L ampicilina e 0,5 mM L-triptofano (Alternativamente, indol pode ser usado). - Medir a densidade óptica em 600 nm (OD600) medir preferencialmente a cada 20 min. densidade de células, determinando a extinção em 600 nm (OD600) em um espectrofotômetro utilizando uma cubeta com um comprimento de caminho de 1 cm. sempre realizar uma referência medição usando o meio de cultura correspondente. Dilua as amostras e a misturar as amostras bem para obter um valor de medição de 0,1-0,8 e, em seguida, calcular OD600 usando o fator de diluição. Para obter detalhes, consulte anterior publicação 26.
- Ao atingir um valor de600 OD de 0,5-0,8 (aproximadamente 2-3h após a inoculação), tire amostra "antes da indução" para SDS-PAGE (electroforese do gel de polyacrylamide do sulfato dodecyl de sódio, passo 4).
- Induzir a expressão de proteínas alvo ajustando a cultura líquida de 0,5 mM IPTG (isopropil β-D-1-thiogalactopyranoside, da solução 1 M) e incube-lo a 30 ° C em um agitador orbital a 200 rpm por 4-8 h.
Nota: Ciana proteínas fluorescentes são comumente expressos em temperaturas abaixo de 37 ° C27. - Tome a amostra "após a expressão" para SDS-PAGE (passo 4.).
- Recolher as células bacterianas por centrifugação por 10min a 5.000 x g e 4 ° C.
- Descartar o sobrenadante por decantação e congelar as pelotas de célula a-20 ° C ou -80 ° C até a purificação da proteína alvo.
- Inocular 10 mL meio fresco do LB (tabela 1; suplementado com glicose 10 g/L, 100 ampicilina µ g/mL) com 100 µ l da cultura durante a noite em um Erlenmeyer de 100 mL. Incube o frasco a 37 ° C em um agitador orbital a 200 rpm.
-
SPI para a produção de GdFP
- Inocular 10 mL do meio de NMM19 (tabela 1) suplementado com 100 de µ g/mL ampicilina, 15 triptofano µM e 10 µ l de cultura durante a noite em um Erlenmeyer de 100 mL e incubar o frasco de cultura durante a noite a 30 ° C em um agitador orbital a 200 rpm.
Nota: Uma variedade de meios quimicamente definidos para o cultivo de e. coli e SPI está disponível. Além do NMM aqui usado, MOPS médio28, glicose e minerais sais médio29, Davis médio mínimo30, M9 médio mínimo31, ou GMML32 pode ser usado. - No dia seguinte, medir OD600 cada 30 min até que o valor só muda pelo menos de 0.05 mais 30 min. O valor de platô deve ser aproximadamente 1.
Nota: Os desvios de ± 0,3 unidades são aceitáveis. Dependendo da estirpe bacteriana e meio utilizado, a concentração de triptofano inicial (passo. 2.3.1) pode precisar de ajuste. - Tome a amostra "antes da indução" para SDS-PAGE (passo 4.).
- Recolher as células bacterianas por centrifugação por 10min a 5.000 x g e 4 ° C. Desprezar o sobrenadante por decantação.
- Ressuspender as células em 10 mL de meio de NMM19 com ampicilina 100 de µ g/mL para um Erlenmeyer de 100 mL e adicionar 4-amino-indol para uma concentração final de 1 mM, utilizando solução de reserva de 50 mM. Continue a incubação durante 30 min a 30 ° C em um agitador orbital a 200 rpm.
Nota: Esta etapa é recomendada devido a baixa estabilidade química de ampicilina e assegura a absorção celular de 4-amino-indol. - Induzir a expressão de proteínas alvo pela adição de IPTG para uma concentração final de 0.5 mM com estoque de 1 M e incubar a amostra durante a noite a 30 ° C, em um agitador orbital a 200 rpm.
Nota: Ciana proteínas fluorescentes são comumente expressos em temperaturas abaixo de 37 ° C27. - No dia seguinte, medir OD600.
- Tome a amostra "após a expressão" para SDS-PAGE (passo 4.).
- Recolher as células bacterianas por centrifugação por 10min a 5.000 x g e 4 ° C e descartar o sobrenadante por decantação.
- No caso desse navio não foi usado por centrifugação, transferi o centrifugado em um tubo 50 mL de poliestireno cônico usando uma espátula. Congele o centrifugado a-20 ° C ou -80 ° C até a purificação da proteína alvo.
- Inocular 10 mL do meio de NMM19 (tabela 1) suplementado com 100 de µ g/mL ampicilina, 15 triptofano µM e 10 µ l de cultura durante a noite em um Erlenmeyer de 100 mL e incubar o frasco de cultura durante a noite a 30 ° C em um agitador orbital a 200 rpm.
3. alvo de purificação de proteínas através de cromatografia de afinidade imobilizada do íon do Metal (IMAC)
-
Lise celular bacteriana
- Descongele o centrifugado no gelo por 10 a 20 min.
- Resuspenda o pellet de células em um tubo 50 mL de poliestireno cônico usando 5 mL de tampão de ligação gelada (tabela 1) no gelo.
- Adicione 20 µ l de lisozima 50mg/mL, 20 µ l de 1 mg/mL DNase I e 20 µ l de 1 mg/mL RNase r. fechar o tubo, misture suavemente por inversão de 5 vezes e mantê-lo no gelo por 30 min.
Nota: O rompimento da pilha parcial ocorre como catalisada pela lisozima. - Lisar as células por sonorização usando uma ponta de homogeneizador ultra-som usando três ciclos de 3 min em um tubo de 15 mL de poliestireno arrefecido pelo gelo de lama com 2 s de pulso, 4 s de pausa e 45% de amplitude.
Nota: Como alternativa, homogeneização de alta pressão pode ser usada, por exemplo., 20 ciclos de 14.000 libras por polegada quadrada. Se necessário, dilua usando vinculação buffer para atingir o volume mínimo do instrumento. Além disso, os reagentes de extracção de proteínas podem ser usados para rompimento da pilha. Ver tabela de materiais para exemplos. - Centrifugar a amostra por 30 min a 15.000-18.000 x g, 4 ° C.
- Transferi o sobrenadante para um tubo fresco e anote o volume de líquido.
- Filtrar a solução através de um filtro de 0,45 µm seringa utilizando uma seringa de fecho Luer plástica 5ml e um fluoreto de polivinilideno (PVDF) seringa de filtro.
- Leve a amostra "lisada" para SDS-PAGE (passo 4.).
- Resuspenda o pellet de célula detritos em ddH2O (volume igual como ex lisado).
- Leve a amostra "da pelota" para SDS-PAGE (passo 4)..
-
Purificação do IMAC
- Use um 1ml pré-embalados ou self embalado coluna IMAC FPLC (cromatografia líquida de proteína rápida) de acordo com as instruções do fabricante. Use vinculação buffer (tabela 1) para equilibração coluna, bem como para a etapa de lavagem que segue após o lisado celular foi aplicado à coluna.
- Frações de eluato coletar e piscina com GdFP, que pode ser identificado pela cor dourada luz visível.
Nota: Opcionalmente, a proteína do alvo pode ser eluída usando um gradiente linear de imidazol (0-250 mM) usando um sistema automatizado de FPLC. - Determinar a concentração de proteína usando o valor da literatura para o coeficiente de extinção em 466 nm (ɛ466 nm = 23.700 M-1 cm-1)33 com tampão de eluição como referência. Para obter detalhes sobre o procedimento, por favor consulte a anterior publicação26.
- Eluato"amostra" de SDS-PAGE e utilize 1-10 µ g de proteína por raia em caso de coloração por Coomassie.
Nota: Quantidades de amostra SDS podem variar dependendo do método de coloração e tintura sensibilidade. - Dialize uma alíquota das frações eluído contra diálise buffer ou buffer de MS usando uma membrana com um peso molecular corte (MWCO) de 5.000-10.000. Prepare a membrana de diálise, de acordo com as instruções do fabricante. Dialize uma amostra de 1 mL, três vezes contra 100 mL de tampão pelo menos 2 h. Para obter mais detalhes sobre este procedimento, consulte anterior publicação34.
- Para armazenamento, congelar amostra de proteína no buffer de diálise a-80 ° C.
Nota: Alíquotas devem ser estáveis pelo menos 6 meses.
4. preparação da amostra SDS-PAGE de extrato de células inteiras de Escherichia coli
- Transferir uma suspensão de células, equivalente a 1 mL de OD600 = 1 suspensão (ex. 500 µ l de OD600= 2) em um tubo de microcentrifugadora de 1,5 mL.
- Recolher as células por centrifugação por 10min a 5000 x g, temperatura ambiente. Desprezar o sobrenadante por pipetagem.
- Adicione 80 µ l de DDQ2O e 20 µ l de SDS 5x carregando buffer de tintura (tabela 1) para o centrifugado e misture por pipetagem.
- Desnature as células por aquecimento a 95 ° C por 5 min em um bloco de banho ou o calor da água. Posteriormente, fixe as amostras à temperatura ambiente.
- Use 10 µ l para manchadas de Coomassie SDS-PAGE de acordo com a anterior publicação35.
Nota: Quantidades de amostra SDS podem variar dependendo do método de coloração e tintura sensibilidade.
5. análise em massa de proteína intacta por cromatografia líquida de alto desempenho (HPLC) acoplado ao Electrospray ionização tempo-de-voo espectrometria de massas (LC-ESI-TOF-MS)
Nota: Buffers, configurações e gradiente HPLC podem variar dependendo da coluna de separação e o instrumento utilizado. Ver tabela de materiais para equipamento exemplar.
- Determinar a concentração de proteína de uma amostra de diálise contra buffer de MS, como descrito acima (passo 3.2.3.) usando o buffer de MS (veja a tabela dos materiais) como referência.
- Diluir a amostra de proteína a 0,1 mg/mL, usando MS buffer para um volume final de 80 µ l, misturar pipetando cuidado, transferir a solução para um tubo de mostruário de MS com inserção de vidro e fechá-la com um boné. Remova as bolhas de ar, passando rapidamente o frasco.
- Encha um segundo frasco de mostruário sem inserção de vidro (tampão em branco) com 1 mL de tampão de MS.
- Permitir que o instrumento para se aquecer. Realize a calibração de instrumento. Certifique-se que quantidades suficientes de solventes de cromatografia líquida-grau estão disponíveis (> 100 mL).
- Programar um gradiente HPLC de 20min linear de 5% para 80% de reserva um (0,1% ácido fórmico em ddH2O), combinado com o tampão B (0,1% ácido fórmico em acetonitrila).
- Começar a HPLC com um caudal de 0,3 mL/min e esperar até que a pressão de coluna é estável.
- Definir um volume de injeção de mostruário de 5 µ l para o método de LC-ESI-TOF-MS, criar uma lista de trabalho para executar em branco seguido por uma corrida de amostra e atribuir o mostruário correspondente frasco posições. Execute a lista de trabalho.
- Após a conclusão da lista de trabalho, abra o arquivo de dados de exemplo gerado. Selecione um intervalo na trama para deconvolução do íon total atual (TIC) e deconvolute o espectro de MS usando o algoritmo de deconvolução de máxima entropia.
Nota: Dependendo das condições experimentais, espécies adicionais podem ocorrer a partir de FP não amadurecidos ou íon tampão adutos.
6. fluorescência vida medições e espectros de decadência associada (DAS) de GdFP
Nota: Para a instrumentação da espectroscopia de fluorescência tempo-resolvido, consulte a Tabela de materiais para equipamento exemplar. Absorvância, bem como fluorescência de excitação e espectros de emissão de proteínas fluorescentes também podem ser gravados usando espectrofotômetros UV/Vis e fluorescência de laboratório.
-
Medição de vida de fluorescência de determinado comprimento de onda de GdFP
- Prepare-se 2 mL de uma solução de 1 µM de GdFP por diluição em tampão PBS (tabela 1), em pH 7. Preencha a solução em uma cubeta de quartzo de 1 cm.
- Instalar o ps-470 nm laser pulsado para a excitação da amostra e o filtro de passa-tempo de emissão 488 nm e ajustar o grating do tempo e comprimento de onda-correlacionados fotão único contando detector de36 (TWCSPC) para a aquisição do regime de comprimento de onda de 600 L/mm 500-700 nm.
- Adquira a emissão de fluorescência, a uma taxa de contagem de cerca de 200 x 103 fótons/s até cerca de 103 contagens são acumuladas no máximo de aquisição das curvas de decaimento de fluorescência com fotão contando software.
-
Medição da resposta instrumental função37 (IRF)
- Substitua a cubeta de amostra com uma cubeta de quartzo de 1cm repleta de sílica coloidal de 1 g/L (~ 220 m2/g) em tampão PBS pH 7.
Nota: A suspensão de sílica é preparada usando uma suspensão aquosa de 400 g/L. - Remova 488 nm de emissão de passe longo filtro e inserir cinza filtros para ajustar a taxa de contagem com o detector TWCSPC para abaixo de 100 x 103 contagens/s.
- Ajuste a grelha para a aquisição de 470 fótons nm no canal 8 do detector TWCSPC 16 canais.
- Adquira o IRF até cerca de 10 x 103 contagens são acumuladas na emissão máxima.
- 38 programa de encaixe de curvas de decaimento de fluorescência Convert e IRF para arquivos de dados ASCII com global.
- Global de conduta próprio de acordo com um modelo de uma soma de três componentes exponenciais com vidas como parâmetros vinculados.
- Trama do decaimento associado espectros (DAS) como distribuições de amplitude dos componentes individuais da decadência na dependência do comprimento de onda com software de análise de dados.
- Substitua a cubeta de amostra com uma cubeta de quartzo de 1cm repleta de sílica coloidal de 1 g/L (~ 220 m2/g) em tampão PBS pH 7.
Representative Results
Usando a técnica de incorporação de pressão seletiva, Trp-66 na tríade cromóforo do ECFP (e Trp-57, apenas outro Trp resíduo em ECFP) pode ser substituído por 4-amino-Trp, gerando assim a GdFP avermelhado com distintas Propriedades espectrais. Espectrometria de massa deve ser usada para demonstrar a desejada integração estequiométrica do aminoácido não-canônicos na proteína, com resultados mostrados na Figura 1. Depois disso, nós fornecemos dados de microscopia, espectroscopia de absorção UV-Vis, bem como o estado estacionário e tempo e comprimento de onda-resolvido por espectroscopia de fluorescência para caracterizar as propriedades do fluoróforo GdFP com foco na dependência do pH do espectros.
Para confirmar a troca dos dois resíduos Trp em ECFP por 4-amino-Trp, análise de espectrometria de massa é executada. A Figura 1 mostra um espectro de ESI-MS deconvoluted representante de GdFP. Enquanto o selvagem-tipo ECFP tem uma massa de proteína calculada da Da 28,283.9 após a maturação do cromóforo, a massa correspondente de GdFP é Da 28,313.9. O espectro de ESI-MS deconvoluted de GdFP apresenta um pico de massa principal, a 28,314.1 ± 0.1 promotor, que se desvia do valor teórico por menos de 10 ppm. Estar dentro do intervalo de precisão típico para este tipo de análise25, confirma-se a incorporação do ncAA através de SPI (valor experimental para o selvagem-tipo ECFP: Da 28,283.7).
A Figura 2 mostra a microscopia de fluorescência confocal de imagem imagens (CFIM) de células bacterianas expressando ECFP, EGFP, EYFP e GdFP em cima de ressuspensão das bactérias em tampão PBS. Todas as imagens foram adquiridas em um microscópio equipado com um UV laser e objectivo da excitação em sobre a mesma energia para cada amostra.
A figura 3A mostra uma sobreposição de imagens CFIM da bactéria e. coli expressando vários FPs, incluindo GdFP, sempre controlada com energia de excitação muito semelhantes (comprimentos de onda, como ilustrado na Figura 2). Figura 3B mostra as estruturas de cromóforo das variantes FP mostrados. Sobre o brilho da GdFP comparado a ECFP (fluorescência φ de rendimento quântico = 0,4), EGFP (φ = 0,6) e EYFP (φ = 0,6) é importante notar que, para GdFP, uma gama mais ampla de aquisição da luz da fluorescência (30 nm) foi usado em contraste com 20 nm usado para todos os outro spe cies, a fim de ajustar a intensidade das imagens para valores semelhantes. Com um ligeiramente mais baixo coeficiente de extinção e um rendimento quântico reduzido em consequência de exclusivo photophysical Propriedades, o brilho de GdFP é menor comparado com os outros FPs mostrado.
O espectro de absorção da ECFP (Figura 3) tem dois máximos característicos em 434 nm e 452 nm. Em contraste, GdFP caracteriza-se por banda larga absorção avermelhado com o máximo em 466 nm. A absorção de EGFP é mais avermelhado de 488 nm. No entanto, devido a mudança de Stokes muito maior de GdFP (108 nm) em comparação com ECFP (41 nm) e EGFP (20 nm), o espectro de emissão de GdFP é o mais avermelhado de todos os três GFP derivados investigados (Figura 3D). Enquanto a emissão de fluorescência da ECFP mostra dois máximos característicos em 475 nm e 505 nm, EGFP tem uma ampla emissão principal banda atingindo 508 nm (λmáx) com um ligeiro ombro a 540 nm. A fluorescência do GdFP aparece em cerca de 565 nm (λmáx.) (Figura 3D). Seu espectro de emissão contém uma pequena contribuição do selvagem-tipo ECFP, que também é visível como um pequeno ombro em 475 nm. Esta pequena fração ECFP é sintetizada antes da indução durante o procedimento SPI, como descrito,33.
Figura 3E mostra as alterações de pH-dependente no espectro de absorção GdFP. Para uma alteração de pH de 8 a 5, a emissão máxima desloca-se ligeiramente para o vermelho e uma ligeira ampliação da banda de absorção é observada. No entanto, a redução da amplitude absorção é apenas cerca de 10% entre pH 8 e pH 5, indicando que as propriedades do estado fundamental do cromóforo do GdFP são muito fracamente modificados pelo pH.
O tempo resolvido emissão de fluorescência, monitorado pela contagem de cada fóton é mostrado na Figura 4. As curvas de decaimento monitoradas nos canais espectrais centrados em 550 nm e 600 nm (Figura 4A) exposição um ligeiramente mais rápido decaimento de fluorescência em 600 nm, em comparação com a decadência em 550 nm. Os resultados de um ajuste global da fluorescência decaem curvas com dois resultados exponenciais componentes em dois componentes de decaimento de fluorescência espectralmente distinguíveis com constantes de tempo de 1,0 3.3 e ns ns (Figura 4 e D).
A emissão de fluorescência de GdFP fortemente depende do pH, como é típico de muitas variantes de proteína fluorescente da família GFP. Figura 4B compara a emissão de fluorescência de GdFP entre pH 5 e pH 8, que mostra claramente uma diminuição da intensidade de fluorescência em pH mais baixo, enquanto as características espectrais permanecer constantes.
Os espectros de decadência associada (DAS)39 de GdFP (Figura 4 e D) são caracterizadas por duas bandas de emissão distintos. A contribuição da lenta 3.3 componente ns é mais pronunciada na faixa de curto comprimento de onda de cerca de 550 nm (60%), com contribuição menor do componente mais rápido (40%). Em 600 nm, os dois componentes têm sobre a mesma amplitude. Após uma mudança do pH 7 (Figura 4), a pH 6 (Figura 4), as características espectrais da DAS dificilmente mudam e as constantes de tempo da rotina montagem global também são iguais (a exatidão das constantes de tempo de Souza é sobre ns ± 0,15). No entanto, que a diferença entre as amplitudes absolutas dos componentes DAS duas é claramente aparente, totalmente explica a amplitude de emissão de fluorescência reduzida mediante a mesma mudança de pH na Figura 4B.
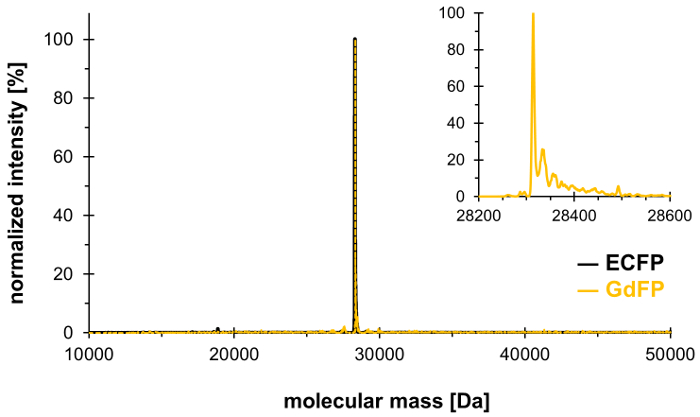
Figura 1: representante deconvoluted ESI-MS espectro de GdFP. O espectro de ESI-MS de GdFP (cor do ouro, ampliado enredo mostrado como inserir) mostra um pico principal no Da 28314.1 (calculado o valor 28313.9 Da). O espectro para o selvagem-tipo ECFP é mostrado em preto. Clique aqui para ver uma versão maior desta figura.

Figura 2: imagens de microscopia de fluorescência Confocal de populações bacterianas expressando vários FPs. As seguintes configurações de comprimento de onda foram utilizadas para aquisição de imagem: ECFP (λex = 457 nm, deteção: 461-480 nm), EGFP (λex = 488 nm, deteção: 495-515 nm), GDFP (λex = 476 nm, deteção: 560-590 nm), EYFP (λex = 514 nm, deteção: 520-530 nm). Clique aqui para ver uma versão maior desta figura.

Figura 3: Propriedades espectrais de GdFP. Imagem do (A) CFIM de uma mistura de células bacterianas expressando ECFP, EGFP e GdFP após a ressuspensão das bactérias em tampão PBS. (B) estruturas cromóforo de GdFP (com 4-amino-Trp em substituição resíduo 66), a ECFP parental (com Trp na posição 66) e EFGP (com Tyr na posição 66). (C) comparação dos espectros de absorção normalizados da GdFP, ECFP e EGFP, Considerando que em (D), o espectro de emissão de fluorescência normalizados do ECFP (excitação a 430 nm) é comparado com os espectros de emissão de fluorescência da EGFP e GdFP (ambos animado a 450 nm). (E) pH-dependência dos espectros de absorção (normalizado de absorção em 280 nm). Clique aqui para ver uma versão maior desta figura.

Figura 4: fluorescência resolvidas a tempo de GdFP. Fluorescência (A) decadência do GdFP monitorados pelo tempo e comprimento de onda-resolvido por fotão contando nos canais espectrais centrados em 550 nm e 600 nm (nm ± 12,5) após excitação com pulsos de laser 470 nm. A função de resposta instrumental (IRF) fornece informações sobre o tempo de resolução da configuração usada. (B) variação do espectro de emissão de GdFP dependente do pH (excitação em 460 nm). (C, D) Associada a decadência espectros (DAS) de GdFP a pH 7 (C) e pH 6 (D) determinado depois de deconvolução de tempo e comprimento de onda-resolvido por fluorescência decai e montagem global do decompõe-se em todos os canais por um conjunto global de duas funções exponenciais com constantes de tempo vinculado. Clique aqui para ver uma versão maior desta figura.

Figura 5: estruturas da transferência intramolecular de carga do ECFP (preto) e GdFP (ouro) cromóforos. O aumento no tamanho do sistema cromóforo pelo dador de electrões boa de um grupo amino como parte do ncAA permite a formação de estruturas mais mesomérico para conseguir a ressonância de estabilização do estado excitado. Os pontos de conexão para o cadafalso FP são mostrados como semicírculos. Clique aqui para ver uma versão maior desta figura.
| Solução-mãe | concentração, solvente | Nota | |
| 20% D-glicose | 200 g/L D-glicose em ddH2O | esterilizar por filtração através de um filtro de seringa de tamanho de poros de 0,45 µm | |
| indol | 50 mM em isopropanol | ||
| 4-amino-indol | 50 mM em etanol a 20% (20 mL de etanol em um volume final de 100 mL, enchida-se de DDQ2O) | ||
| IPTG | 1 M em ddH2O | ||
| L-triptofano | 15 mM dissolvido em ddH2O com 1 M de HCl (adicionar HCl gota a gota, sob agitação até que o pó é dissoved) | ||
| lisozima | 50 mg/mL em ddH2O | ||
| DNase eu | 1 mg/mL em ddH2O | ||
| RNase A | 1 mg/mL em ddH2O | ||
| Amp100 | Ampicilina 100 mg/mL em ddH2O | ||
| sódio-dodecylsulfate (SDS) | 200 g/L em ddH2O | ||
| sulfato de amônio ((NH4)2SO4) | 1 M em ddH2O | esterilizar em autoclave | |
| hidrogenofosfato de potássio (KH2PO4) | 1 M em ddH2O | esterilizar em autoclave | |
| fosfato di-potássio (K2HPO4) | 1 M em ddH2O | esterilizar em autoclave | |
| o sulfato de magnésio (MgSO4) | 1 M em ddH2O | esterilizar em autoclave | |
| D-glicose | 1 M em ddH2O | esterilizar por filtração através de um filtro de seringa de tamanho de poros de 0,45 µm | |
| cloreto de sódio (NaCl) | 5m em ddH2O | esterilizar em autoclave | |
| cloreto de cálcio (CaCl2) | 1 g/L | esterilizar por filtração através de um filtro de seringa de tamanho de poros de 0,45 µm | |
| cloreto de Iron(II) (FeCl2) | 1 g/L | esterilizar por filtração através de um filtro de seringa de tamanho de poros de 0,45 µm | |
| tiamina | 10 g/L | esterilizar por filtração através de um filtro de seringa de tamanho de poros de 0,45 µm | |
| Biotina | 10 g/L | esterilizar por filtração através de um filtro de seringa de tamanho de poros de 0,45 µm | |
| mistura de oligoelementos | o sulfato de cobre (CuSO4), zinco cloreto (ZnCl2), cloreto de manganês (MnCl2), molibdato de amónio (NH4)2MoO4; cada 1 mg/L em ddH2O | esterilizar por filtração através de um filtro de seringa de tamanho de poros de 0,45 µm | |
| mistura de 19 aminoácidos | 1.) dissolver 0,5 g L-fenilalanina e 0,5 g L-tirosina em 100 ml ddH2O com adição gota a gota de 1 M HCl, sob agitação até que o pó é dissolvido. | ||
| 2.) pesar 0,5 g de cada um dos restantes L-amino ácidos (exceto L-triptofano). Misture com 22 mL fo 1 M KH2PO4 e 48 mL de 1 M K2HPO4. Adicione O ddH2a cerca de 800 mL. Mexa até que a solução torna-se claro. | |||
| 3.) adicione o dissolvido L-fenilalanina e L-tirosina da etapa 1.) e ajuste o volume para 1L com DDQ2O. | |||
| 4.) esterilize a mistura de aminoácidos por filtração em vácuo com uma unidade de filtro superior da garrafa. | |||
| Buffers e mídia | Composição/preparação | ||
| SDS carregando buffer de tintura, 5x concentrado | 0,25 M Tris pH 6,8, glicerol de 50% v/v, 0,25% w/v bromofenol azul, didhiothreitol 0,5 M (DTT; alternativamente 5% β-Mercaptoetanol), 10% p/v de sódio-dodecylsulfate (SDS) | ||
| tampão de ligação | 50mm dihydrogenphosphate de sódio (NaH2PO4), 500 mM de NaCl, imidazol 10 mM, pH 8 | ||
| tampão de eluição | 50 mM dihydrogenphosphate de sódio (NaH2PO4), 500 mM de NaCl, imidazol 250 mM, pH 8 | ||
| buffer de diálise | 50 mM dihydrogenphosphate de sódio (NaH2PO4), 150 mM de NaCl, glicerol 100 mL/L, pH 8 | ||
| Buffer de MS | 10 mM Tris-HCl, pH 8 | ||
| novo meio mínimo contendo 19 L-aminoácidos excepto L-triptofano (NMM19) | Misture todas as soluções para obter as seguintes concentrações finais: 7,5 mM (NH4)2SO4, 1.7 mM NaCl, 22mm KH2PO4, 50mm K2HPO4, 1mm MgSO4, 20mm D-glicose, 50 mg/L de mix de 19 aminoácidos, 1 µ g/L de CaCl2, 1 µ g/L de FeCl2, 10 µ g/L tiamina, biotina 10 mg/L, mistura de elementos de traço de 0,01 mg/L | ||
| Meio LB | Composição: triptona 10 g/L, extrato de levedura 5 g/L, 10 g/L em NaCl, pH 7.0 em ddH2O | ||
| Preparação: | |||
| 1.) pesar fora 50 g triptona, extracto de levedura de 25 g, 5 g de NaCl em um frasco de vidro de 1 L. | |||
| 2.) adicionar ddH2O a ~ 800 mL e dissolver componentes sob agitação. | |||
| 3.) medir o pH e ajustar o pH 7 pela adição gota a gota de 1 M de HCl ou 1 NaOH de M, se necessário. Adicionar ddH2O até 1 L. | |||
| 4.) esterilize em autoclave, verificar se há perda de volume depois e adicionar ddH estéril2O para compensar se necessário. Armazenar a 4 ° C até o uso. | |||
| Placas de ágar LB | Composição: triptona 10 g/L, extrato de levedura 5 g/L, 10 g/L em NaCl, ágar-ágar 15 g/L, pH 7,0 em ddH2O | ||
| Preparação: | |||
| 1.) pesar fora 50 g triptona, extracto de levedura 25 g, 5 g de NaCl, ágar-ágar 7,5 g em um frasco de vidro de 1 L. | |||
| 2.) adicionar ddH2O até 500 mL e dissolver os componentes sob agitação. | |||
| 3.) medir o pH e ajustar o pH 7 pela adição gota a gota de 1 M de HCl ou 1 NaOH de M, se necessário. Adicionar ddH2O até 1 L. | |||
| 4.) esterilize em autoclave, verificar se há perda de volume depois e adicionar o ddH estéril2O, para compensar, se necessário. (Nota: LB ágar pode ser armazenado a 4 ° C até o uso para a preparação de placas de ágar LB. Derreta cuidadosamente solidificado ágar usando um microondas) | |||
| 5.) quando a solução é ainda morna (30-40 ° C), adicionar ampicilina para uma concentração final de 100 µ g/mL | |||
| 6.) pour cerca de 15 mL do líquido da etapa 5.) em uma estéril 10cm prato de Petri, sob condições estéreis. Quando o ágar é solidificado, placas podem ser armazenadas por 1 semana a 4 ° C até o uso. | |||
| tampão fosfato salino (PBS) | Composição: 137 mM NaCl, 2,7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 1mm CaCl2, 0,5 mM MgCl2, pH 7. Esterilize em autoclave ou filtração. | ||
Tabela 1: Solução de estoque e reserva.
Discussion
Para conseguir eficiências de incorporação de ncAA muito alta, o método SPI baseado em auxotrophy baseia-se na utilização de células metabolicamente engenharia, que não são capazes de sintetizar a contraparte natural correspondente do ncAA. Para Escherichia coli, essas estirpes estão prontamente disponíveis. Mesmo a incorporação simultânea de ncAAs múltiplas a mesma proteína é viável usando cepas multiauxotrophic. O modo de resíduos específicos de substituição e o repertório químico sendo restrito à análogos de químicos semelhantes pode ser visto como inconvenientes. Não obstante, um grande número de variantes de proteínas pode ser produzido como o aparelho de tradução bacteriana natural tolera numerosos análogos de aminoácidos. Por exemplo, mais de 50 ncAAs poderia ser incorporadas proteínas usando em vitro tradução, representando cerca de 73% de todos os códons do código genético disponível para redesignação40. Além disso, SPI também pode permitir a rotulagem multissite eficiente do alvo da proteína41. Em princípio, a metodologia SPI não está restrita a Escherichia coli, mas pode trabalhar em qualquer outro host e para cada um dos canônico de 20 aminoácidos, que cepas auxotróficos e meios de cultivo definidos estejam disponíveis. Por exemplo, dois análogos de metionina, azidohomoalanine (Aha) e homopropargylglycine (Hpg), são comercialmente disponíveis e usados para a rotulagem de proteínas e proteomes em diversos organismos. Além disso, Aha pode ser produzido intracelular e posteriormente incorporada a proteína42. Este ncAA é especialmente adequado para opaco conjugações como química clique desenvolvido pelos Tirrel e colegas de trabalho: por exemplo, na planta tecido de Arabidopsis thaliana, Bombyx mori larvas43, Drosophila células44, larval zebrafish45 bem como células de mamíferos incluindo neurônios46, as proteínas podem ser rotuladas com Aha47,48. Da mesma forma, Trp análogos foram incorporados com êxito peptídeos antimicrobianos em cepas de Trp-auxotróficos Lactococcus lactis 49. SPI é também útil para o campo de Xenobiology50,51, que explora alternativas para a composição química básica da vida. Por exemplo, baseado em trabalhos anteriores sobre Escherichia coli52 e b. subtilis53, uma estirpe de e. coli foi desenvolvida recentemente por uma estratégia evolutiva com pressão seletiva para utilizar thienopyrrole em vez de indol, resultando na substituição de todo o proteoma do triptofano por thienopyrrole-alanina no código genético de54. Geralmente, o aminoácido canônico Trp, que é codificada por um único triplet (UGG), apresenta um alvo promissor para a engenharia de proteínas devido as facetas ricas da química do indol, que oferece inúmeras variações de químicas. Recentemente e como uma alternativa para incorporação baseada no SPI, um romance plataforma SCS capaz de incorporar os análogos de Trp site-specifically em hospedeiros bacterianos e eukaryotic tem sido relatado55. Isso amplia ainda mais caixa de ferramentas na vivo engenharia de proteína baseada no ncAA, incluindo a alteração de propriedades espectrais.
Além da utilização de hosts de expressão auxotróficos, o protocolo SPI requer condições estritas de fermentação, tanto em termos de tempo de expressão do alvo e a composição do meio para alcançar alta eficiência de incorporação da ncAA e rendimento de proteína alvo 56. cultivo é realizado com o uso de meios mínimos quimicamente definidos, que contêm essencialmente além sais importantes fontes de nitrogênio (sal de amónio) e carbono (D-glucose), vitaminas e oligoelementos. Embora não seja estritamente necessário na ausência de mais auxotrophies, os restantes aminoácidos (20 -n, se n aminoácidos devem ser substituídos) são comumente adicionados para promover o crescimento bacteriano,57. Durante uma fase inicial de crescimento antes da indução da expressão da proteína-alvo, os n canônico aminoácidos a ser substituído são adicionados em limitar as concentrações. Crescimento celular continua até os alvo aminoácidos essenciais estão esgotados, como experimentalmente, indicado por uma estacionária OD600. Posteriormente, o meio de cultura é substituído pelo meio fresco que falta o aminoácido empobrecido e contém o ncAA em concentrações abundantes. Para a incorporação ribosomal de análogos de triptofano como mostrado neste protocolo, um analógico de indole é alimentado, qual intracelular torna-se convertido para a correspondente derivada de triptofano pelo triptofano sintase58. Em seguida, a expressão da proteína-alvo é induzida. Nesta fase, as células estão perto do final do crescimento logarítmico, como um equilíbrio entre o número total de células e fitness. Como a presença e a incorporação da canonical amino levaria a produção da proteína do selvagem-tipo, é fundamental para garantir que o aminoácido essencial está totalmente esgotado antes da indução. Da mesma forma, é obrigatório examinar a eficiência da incorporação da ncAA a proteína alvo, comumente por espectrometria de massa. Em caso de presença substancial do aminoácido canônico, condições de cultivo precisa ser ajustado, por exemplo, alterando a concentração da amino acid(s) essencial para a fase inicial de crescimento ou a duração do último. No caso de RAA baixa atividade em direção a ncAA, a superexpressão da enzima endógena ou expressão co de um aaRS diferentes, o que é mais ativo em direção a ncAA, pode ser conduzido59.
O aminoácido canônico Trp é dotado de três características notáveis: (i) sua abundância natural nas proteínas é baixa; (ii) suas propriedades biofísicas e químicas são exclusivas (EG., costuma ser a origem dominante da fluorescência intrínseca das proteínas e peptídeos) e (iii) contribui para uma variedade de interações bioquímicas e funções incluindo Empilhamento π, interações H-colagem e cação-π. Todas estas características são mudou radicalmente após a substituição de 4-amino-Trp → Trp em dúvida GdFP... Além, o design de uma classe de "ouro" de avGFPs é um exemplo notável para engenharia Tailor-Made autofluorescent proteínas. Com distintas Propriedades espectrais, FPs pode ser ajustado para certas janelas espectrais através de incorporação de mutagénese e ncAA. Em caso de GdFP, isso é realizado por uma simples troca química H → NH2 no frame do anel indol contido na tríade ECFP cromóforo. A Figura 5 mostra os efeitos de incorporação de ncAA dentro o cromóforo. A introdução do grupo elétron-doando provenientes de 4-amino-indol (intracelular convertido em 4-amino-Trp) permite uma variedade de estruturas mesomérico que pode explicar um estado excitado estabilizado. Espectroscopicamente, seu deslocamento de Stokes alargado e emissão de fluorescência avermelhado resultam essas propriedades distintas do sistema conjugado estendido. Como noticiado anteriormente, a transferência de carga intramolecular reforçada dentro o cromóforo GdFP é inerentemente sensível ao pH (Figura 4B) e acompanhado de uma mudança maior no momento de dipolo entre o terreno de0 S e S1 animado estado em relação à ECFP33. Como alternativos grupos elétron-doar, análogos de triptofano, possuindo um anel indol substituído com grupos hidroxi poderiam ser usados, como relatado em um estudo comparativo com o modelo da proteína barstar41.
Os espectros de absorção e fluorescência de GdFP são ampliou comparado com ECFP e EGFP (Figura 3 e D). Homogênea alargamento das bandas de absorção e fluorescência é causada geralmente por modos vibracionais no cromóforo e, além disso, por atrelar o cromóforo a presente nos anos60, proteína mais vibracional modos. O acoplamento ao ambiente local proteína é suportado por cargas localizadas sobre o cromóforo. A homogeneidade estrutural da proteína leva a variações locais do espectro vibrônico, tal acoplamento entre os espectros vibrónico o cromóforo e o resto da proteína são suportadas pela deslocalização da carga e mesomérico Estados tal como indicado no Figura 5. Este acoplamento também suporta o grande deslocamento de Stokes e necessariamente reduz o rendimento quântico de fluorescência. Em comparação com outros FPs avermelhado, GdFP nem apresenta estabilidade de proteína melhorada e uma baixa tendência para agregação33,61,62. Ele não só difere na cor de outras variantes FP, mas também exibe uma substancialmente maior estabilidade térmica e maior cooperativa dobradura33. Sua intensidade de fluorescência é preservado após aquecimento a 60 ° C, enquanto que a fluorescência ECFP é reduzida para cerca de 30% em pelo menos 90%. Em proteínas, aminoácidos aromáticos, muitas vezes, contribuem para redes de interação cadeias laterais, que geralmente têm um efeito estabilizador na estrutura terciária da proteína. avGFP abriga tal uma lado rede da cadeia, que consiste no cromóforo propriamente dito, como bem como Phe-165, His-148 e Tyr-145. Estas cadeias laterais não são apenas muito rígidas na estrutura de GdFP33, mas importante, eles formam hidrofóbicos contatos com o cromóforo. A característica mais proeminente romance identificada no GdFP que é o cromóforo aminados mais proximal a Phe-165. Essa interação é uma característica não observada em outras avGFPs conhecido. Como os dois resíduos são separados de 3.2-4.5 Å, interações de aminoácidos aromáticos podem estar também presentes. Juntamente com a estabilização de ressonância induzida por aminação do cromóforo, estes provavelmente estabiliza esta rede hidrofóbico de aminoácidos de forma cooperativa. Uma transferência intramolecular de carga mais eficaz pode ser suportada por essas interações no estado excitado em comparação com o estado fundamental do cromóforo, e é pelo menos parcialmente responsável por 108 nm Stokes turno33,62 .
No projeto racional de fluoróforo Propriedades, prevê-se um aumento no tamanho do sistema π deslocalizado para resultar em um comprimento de onda de excitação avermelhado. Esta regra é obedecida pela série de aminoácidos em 66 conduziu posição neutros cromóforos: Phe (λmáx = 355 nm) < dele (λma x= 386 nm) < Tyr (λmáx = 395 nm) < Trp (λmáx = 436 nm)63. Na natureza, esta extensão de sistema conjugado do cromóforo de ligações π foi alcançado por diferentes estratégias. Para DsRed de Discosoma striata, ele é estendido pela integração de um aminoácido adicional, assim mudando o λmáximo para 573 nm64. O cromóforo do asFP595 (λmáx = 595 nm) de Anemonia sulcata estendeu-se por um grupo imino, ampliando seu sistema π65. Desde que o cromóforo de GdFP e outros avFPs é do mesmo tamanho, um princípio diferente deve implicar um comprimento de onda de emissão na faixa do DsRed expandido e asFP595 cromóforos. A profunda mudança de Stokes de 108 nm é atribuída à estrutura distinta do cromóforo do GdFP, o que revela um novo princípio de photophysical no projeto de autofluorescent proteínas. Cálculos preliminares (conforme relatado em 62) têm mostrado que o momento dipolar de cromóforo de GdFP do estado excitado é substancialmente maior do que no estado de terra, em contraste com os respectivos valores da ECFP. Considerando que o momento dipolar de GdFP aumenta a partir D ~ 3 (Debye) no estado de0 S a ~ 15 no. S1, a mudança para o cromóforo ECFP foi bastante moderada (de 4 ~ D a ~ 6 D). Assim, a fluorescência dourada original de GdFP é causada pela transferência de carga intramolecular substancial dentro o cromóforo, que aumenta a variedade de possíveis estruturas mesomérico (ver Figura 5) que permitem a estabilização de ressonância. Isso reduz o nível de energia da qual ocorre emissão. Como consequência da mudança profunda no momento dipolar em cima da excitação, a separação de carga intramolecular é o principal motivo para as mudanças no potencial eletrostático do ambiente cromóforo. A proteína matriz circundante, por sua vez, ajusta-se às mudanças na distribuição carga após a excitação do cromóforo. O subsequente relaxamento estrutural reduz o nível de energia do cromóforo do animado, que desloca o espectro de fluorescência para o vermelho devido a seu caráter de transferência de carga. Pela mesma razão, em consequência da grande deslocamento de Stokes e taxas aprimoradas dos processos radiationless, o rendimento quântico de fluorescência da GdFP é reduzida em comparação a ECFP33.
O rendimento quântico de alta e um pequeno deslocamento de Stokes da ECFP e EGFP são normalmente atribuídas a um ambiente de proteína rígida do cromóforo, que reduz os graus de liberdade e, consequentemente, conversão interna para favorecer o relaxamento radiativo do estado excitado 66. por conseguinte, o design molecular de cromóforos mais rigidamente incorporados com acoplamento reduzido para a matriz de proteínas restantes pode servir como um guia para produzir mais avermelhado derivados GFP com rendimento quântico de fluorescência de alta. Portanto, para novas abordagens para produzir proteínas autofluorescent avermelhado de engenharia, o alargamento do sistema de elétrons π e uma estrutura rígida cromóforo com fraco acoplamento para o ambiente de proteína é altamente desejável. Tais modificações também poderiam ser introduzidas diretamente no GFP-baseado cromóforos ou pela colocação de ncAAs desejado nas proximidades de cromóforo.
Disclosures
Os autores declaram que eles têm não tem interesses financeiro concorrente.
Acknowledgments
Este trabalho foi financiado pela alemão Research Foundation (Cluster de excelência "unificar conceitos em catálise) T.F. e N.B. e pelo Ministério Federal da educação e da ciência (BMBF programa"HSP 2020", TU-WIMIplus projeto SynTUBio) de F.-J.S.
Materials
| Name | Company | Catalog Number | Comments |
| Chemicals | |||
| 4-aminoindole | Sigma-Aldrich | 525022 | |
| acetonitrile | VWR | HiPerSolv CHROMANORM ULTRA for LC-MS, 83642 | LC-MS grade required |
| agar-agar | Carl Roth | 5210 | |
| ammonium molybdate ((NH4)2MoO4) | Sigma-Aldrich | 277908 | |
| ammonium sulfate ((NH4)2SO4) | Sigma-Aldrich | A4418 | |
| ampicillin sodium salt | Carl Roth | K029 | |
| biotin | Sigma-Aldrich | B4501 | |
| bromophenol blue | Sigma-Aldrich | B0126 | |
| calcium chloride (CaCl2) | Sigma-Aldrich | C5670 | |
| colloidal silica | Sigma-Aldrich | Ludox HS-40, 420816 | |
| Coomassie Brillant Blue R 250 | Carl Roth | 3862 | |
| copper sulfate (CuSO4) | Carl Roth | CP86.1 | |
| D-glucose | Carl Roth | 6780 | |
| di-sodium hydrogen phosphate (Na2HPO4) | Carl Roth | X987 | |
| di-potassium hydrogen phosphate (K2HPO4) | Carl Roth | P749.1 | |
| 1,4-dithiothreitol (DTT) | Carl Roth | 6908 | |
| DNase I | Sigma-Aldrich | D5025 | |
| ethanol | Carl Roth | 9065.1 | |
| formic acid | VWR | HiPerSolv CHROMANORM for LC-MS, 84865 | LC-MS grade required |
| glycerol | Carl Roth | 3783 | |
| imidazole | Carl Roth | X998 | |
| indole | Sigma-Aldrich | I3408 | |
| iron(II) chloride (FeCl2) | Sigma-Aldrich | 380024 | |
| isopropanol | Carl Roth | AE73.1 | |
| isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma-Aldrich | I6758 | |
| lysozyme | Sigma-Aldrich | L6876 | |
| magnesium chloride (MgCl2) | Carl Roth | KK36.1 | |
| magnesium sulfate (MgSO4) | Carl Roth | 8283.2 | |
| manganese chloride (MnCl2) | Sigma-Aldrich | 63535 | |
| β-mercaptoethanol | Carl Roth | 4227.3 | |
| potassium chloride (KCl) | Carl Roth | 6781.3 | |
| potassium dihydrogen phosphate (KH2PO4) | Sigma-Aldrich | P5655 | |
| RNase A | Carl Roth | 7156 | |
| sodium chloride (NaCl) | Carl Roth | P029 | |
| sodium dihydrogen phosphate (NaH2PO4) | Carl Roth | T879 | |
| sodium dodecyl sulphate (NaC12H25SO4) | Carl Roth | 0183 | |
| thiamine | Sigma-Aldrich | T4625 | |
| Tris(hydroxymethyl)-aminomethane (Tris) | Carl Roth | 5429 | |
| Tris hydrochloride (Tris-HCl) | Sigma-Aldrich | 857645 | |
| tryptone | Carl Roth | 8952 | |
| yeast extract | Carl Roth | 2363 | |
| zinc chloride (ZnCl2) | Sigma-Aldrich | 229997 | |
| Name | Company | Catalog Number | Comments |
| amino acids | |||
| L-alanine | Sigma-Aldrich | A7627 | |
| L-arginine | Sigma-Aldrich | A5006 | |
| L-asparagine | Sigma-Aldrich | A8381 | |
| L-aspartic acid | Sigma-Aldrich | A0884 | |
| L-cysteine | Sigma-Aldrich | C7352 | |
| L-glutamic acid | Sigma-Aldrich | G2128 | |
| L-glutamine | Sigma-Aldrich | G3126 | |
| L-glycine | Sigma-Aldrich | G7126 | |
| L-histidine | Sigma-Aldrich | H8000 | |
| L-isoleucine | Sigma-Aldrich | I2752 | |
| L-leucine | Sigma-Aldrich | L8000 | |
| L-lysine | Sigma-Aldrich | L5501 | |
| L-methionine | Sigma-Aldrich | M9625 | |
| L-proline | Sigma-Aldrich | P0380 | |
| L-phenylalanine | Sigma-Aldrich | P2126 | |
| L-serine | Sigma-Aldrich | S4500 | |
| L-threonine | Sigma-Aldrich | T8625 | |
| L-tryptophan | Sigma-Aldrich | T0254 | |
| L-tyrosine | Sigma-Aldrich | T3754 | |
| L-valine | Sigma-Aldrich | V0500 | |
| Name | Company | Catalog Number | Comments |
| Lab materials | |||
| 0.45 µm syringe filter with PVDF membrane | Carl Roth | CCY1.1 | |
| 1.5 mL microcentrifuge tubes | Eppendorf | 30120086 | |
| conical polystyrene (Falcon) tubes, 50 mL | Fisher Scientific | 14-432-22 | |
| Luer-Lock syringe 5 mL | Carl Roth | EP96.1 | |
| dialysis membrane, Molecular Weight Cut-Off (MWCO) 5,000 | Spectrum Medical Industries | Spectra/Por MWCO 5000 dialysis membrane, 133198 | |
| Immobilized Metal ion Affinity Chromatography (IMAC) column 1 mL, Ni-NTA | Macherey Nagel | Protino series, 745410.5 | |
| petri dishes (polystyrene, sterile) | Carl Roth | TA19 | |
| pQE-80L plasmid vector | Qiagen | no longer available | replaced by N-terminus pQE Vector set Cat No./ID: 32915 |
| protein extraction reagent BugBuster | EMB Millipore | 70921-4 | |
| round-bottom polystyrene tubes, 14 mL | Fisher Scientific | Corning Falcon, 14-959-1B | |
| Trp-auxotrophic E. coli strain | ATCC | ATCC 49980 | Bridges BA et al., Chem Biol Interact., 1972, 5(2):77-84; see main text for alternatives |
| Name | Company | Catalog Number | Comments |
| Mass Spectrometry equipment | |||
| mass spectrometer for LC-ESI-TOF-MS | Agilent | Agilent 6530 Accurate-Mass QTOF | coupled with Infinity LC system |
| mass spectrometry data analysis software | Agilent | MassHunter Qualitative Analysis software v. B.06.00 | |
| High-Performance Liquid Chromatography (HPLC) column for LC-ESI-TOF-MS | Sigma-Aldrich | Supelco Discovery BIO Wide Pore C5 HPLC column, 3 µm particle size, 10 cm x 2.1 mm | |
| HPLC autosampler vials 1.5 mL | Sigma-Aldrich | Supelco 854165 | with conical 0.1 mL glass inserts, screw caps and septa |
| Name | Company | Catalog Number | Comments |
| General equipment | |||
| benchtop centrifuge for 1.5 mL Eppendorf tubes | Eppendorf | 5427 R | |
| cooling centrifuge for 50 mL Falcon tubes | Eppendorf | 5810 R | |
| high pressure microfluidizer for bacterial cell disruption | Microfluidics | LM series with “Z” type chamber | |
| peristaltic pump for LC | GE Healthcare | P-1 | |
| Fast Protein Liquid Chromatography (FPLC) system | GE Healthcare | ÄKTA pure 25 L | |
| orbital shaker for bacterial cultivation | Infors HT | Minitron | |
| UV/Vis spectrophotometer | Biochrom | ULTROSPEC 2100 | |
| ultrasonic homogenizer for bacterial cell disruption | Omnilab | Bandelin SONOPULS HD 3200, 5650182 | with MS72 sonifier tip |
| Name | Company | Catalog Number | Comments |
| Fluorescence spectroscopy equipment | |||
| ps-pulsed laser 470 nm | Picoquant GmbH | PDL-470 | |
| time- and wavelength-correlated single photon counting (TWSPC) acquisition software | Picoquant GmbH | SymPhoTime 64 | |
| time- and wavelength-correlated single photon counting (TWSPC) detector | Picoquant GmbH | PML-16C | 16 spectral channels, to be selected by grating settings |
| single photon counting software | Picoquant GmbH | SPCM 9.75 | |
| global fitting software | Picoquant GmbH | SPC2Glo(R) | |
| fluorescence decay data analysis software | Picoquant GmbH | FluoFit program | |
| data analysis software | OriginLab Inc. | Origin 9.2 | |
| neutral density filter set | Schott | NG1 to NG11 | (400 - 650 nm, transmission 50 %, 20%, 10 %, 5 %) |
| 488 nm long-pass emission filter | AHF Analysentechnik | AHF-488 | |
| quartz cuvette | Thorlabs GmbH | CV10Q1400 | 1 cm pathlength |
References
- Shimomura, O., Johnson, F. H., Saiga, Y. Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan, Aequorea. J Cell Compar Physl. 59 (3), 223-239 (1962).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263 (5148), 802-805 (1994).
- Andresen, M., et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. P Natl Acad Sci USA. 102 (37), 13070-13074 (2005).
- Andresen, M., et al. Structural basis for reversible photoswitching in Dronpa. P Natl Acad Sci USA. 104 (32), 13005-13009 (2007).
- Brakemann, T., et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat Biotechnol. 29 (10), 942-947 (2011).
- Kremers, G. -J., Gilbert, S. G., Cranfill, P. J., Davidson, M. W., Piston, D. W. Fluorescent proteins at a glance. J Cell Sci. 124 (Pt 2), 157-160 (2011).
- Shimomura, O. Structure of the chromophore of aequorea 0. shimomura green fluorescent protein. FEBS Lett. 104 (2), 220-222 (1979).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 22 (12), 1567-1572 (2004).
- Shcherbo, D., et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 4 (9), 741-746 (2007).
- Shcherbakova, D. M., Subach, O. M., Verkhusha, V. V. Red fluorescent proteins: advanced imaging applications and future design. Angew Chem Int Edit. 51 (43), 10724-10738 (2012).
- Stepanenko, O. V., Verkhusha, V. V., Kuznetsova, I. M., Uversky, V. N., Turoverov, K. K. Fluorescent proteins as biomarkers and biosensors: throwing color lights on molecular and cellular processes. Curr Protein Pept Sc. 9 (4), 338-369 (2008).
- Wang, L., Xie, J., Deniz, A. A., Schultz, P. G. Unnatural amino acid mutagenesis of green fluorescent protein. J Org Chem. 68 (1), 174-176 (2003).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Sharma, N., Furter, R., Kast, P., Tirrell, D. A. Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett. 467 (1), 37-40 (2000).
- Liu, C. C., Schultz, P. G. Adding new chemistries to the genetic code. Annu Rev Biochem. 79, 413-444 (2010).
- Twine, S. M., Murphy, L., Phillips, R. S., Callis, P., Cash, M. T., Szabo, A. G. The Photophysical Properties of 6-Azaindole. J Phys Chem B. 107 (2), 637-645 (2003).
- Lepthien, S., Hoesl, M. G., Merkel, L., Budisa, N. Azatryptophans endow proteins with intrinsic blue fluorescence. P Natl Acad Sci USA. 105 (42), 16095-16100 (2008).
- Budisa, N., et al. Probing the role of tryptophans in Aequorea victoria green fluorescent proteins with an expanded genetic code. Biol Chem. 385 (2), 191-202 (2004).
- Ross, J. B., et al. Spectral enhancement of proteins: biological incorporation and fluorescence characterization of 5-hydroxytryptophan in bacteriophage lambda cI repressor. P Natl Acad Sci USA. 89 (24), 12023-12027 (1992).
- Soumillion, P., Jespers, L., Vervoort, J., Fastrez, J. Biosynthetic incorporation of 7-azatryptophan into the phage lambda lysozyme: Estimation of tryptophan accessibility, effect on enzymatic activity and protein stability. Protein Eng Des Sel. 8 (5), 451-456 (1995).
- Heim, R., Tsien, R. Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 6 (2), 178-182 (1996).
- Bridges, B. A., Mottershead, R. P., Rothwell, M. A., Green, M. H. L. Repair-deficient bacterial strains suitable for mutagenicity screening: tests with the fungicide captain. Chem Biol Interact. 5 (2), 77-84 (1972).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: The Heat Shock Method. J Vis Exp. , (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Bacterial Transformation: Electroporation. J Vis Exp. , (2017).
- Grigorenko, B. L., Krylov, A. I., Nemukhin, A. V. Molecular modeling clarifies the mechanism of chromophore maturation in the green fluorescent protein. J Am Chem Soc. , (2017).
- JoVE Science Education Database. General Laboratory Techniques. Introduction to the Spectrophotometer. J Vis Exp. , (2017).
- Goedhart, J., et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun. 3, 751 (2012).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F. Culture medium for enterobacteria. J Bacteriol. 119 (3), 736-747 (1974).
- Hörnsten, E. G. On culturing Escherichia coli on a mineral salts medium during anaerobic conditions. Bioprocess Eng. 12 (3), 157-162 (1995).
- Davis, B. D. The Isolation of Biochemically Deficient Mutants of Bacteria by Means of Penicillin. P Natl Acad Sci USA. 35 (1), 1-10 (1949).
- Sambrook, J., Russell, D. W. Molecular Cloning: A Laboratory Manual. , Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY, USA. (2001).
- Wang, Y. -S., et al. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives. Mol Biosyst. 7 (3), 714-717 (2011).
- Bae, J. H., et al. Expansion of the genetic code enables design of a novel "gold" class of green fluorescent proteins. J Mol Biol. 328 (5), 1071-1081 (2003).
- JoVE Science Education Database. Dialysis: Diffusion Based Separation. J Vis Exp. , Cambridge, MA. (2017).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Separating Protein with SDS-PAGE. J Vis Exp. , (2017).
- Petrásek, Z., et al. Excitation energy transfer from phycobiliprotein to chlorophyll d in intact cells of Acaryochloris marina studied by time- and wavelength-resolved fluorescence spectroscopy. Photoch Photobio Sci. 4 (12), 1016-1022 (2005).
- Kolber, Z. S., Barkley, M. D. Comparison of approaches to the instrumental response function in fluorescence decay measurements. Anal Biochem. 152 (1), 6-21 (1986).
- Pelet, S., Previte, M. J. R., Laiho, L. H., So, P. T. C. A fast global fitting algorithm for fluorescence lifetime imaging microscopy based on image segmentation. Biophys J. 87 (4), 2807-2817 (2004).
- Loefroth, J. E. Time-resolved emission spectra, decay-associated spectra, and species-associated spectra. J Phys Chem. 90 (6), 1160-1168 (1986).
- Hartman, M. C. T., Josephson, K., Lin, C. -W., Szostak, J. W. An expanded set of amino acid analogs for the ribosomal translation of unnatural peptides. PLoS One. 2 (10), e972 (2007).
- Budisa, N., et al. Global replacement of tryptophan with aminotryptophans generates non-invasive protein-based optical pH sensors. Angew Chem Int Edit. 41 (21), 4066-4069 (2002).
- Ma, Y., Biava, H., Contestabile, R., Budisa, N., di Salvo, M. L. Coupling bioorthogonal chemistries with artificial metabolism: intracellular biosynthesis of azidohomoalanine and its incorporation into recombinant proteins. Molecules. 19 (1), 1004-1022 (2014).
- Teramoto, H., Kojima, K. Incorporation of Methionine Analogues Into Bombyx mori Silk Fibroin for Click Modifications. Macromol Biosci. 15 (5), 719-727 (2015).
- Deal, R. B., Henikoff, J. G., Henikoff, S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 328 (5982), 1161-1164 (2010).
- Hinz, F. I., Dieterich, D. C., Tirrell, D. A., Schuman, E. M. Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci. 3 (1), 40-49 (2012).
- Dieterich, D. C., et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 13 (7), 897-905 (2010).
- Dieterich, D. C., Link, A. J., Graumann, J., Tirrell, D. A., Schuman, E. M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). P Natl Acad Sci USA. 103 (25), 9482-9487 (2006).
- Glenn, W. S., et al. Bioorthogonal Noncanonical Amino Acid Tagging (BONCAT) Enables Time-Resolved Analysis of Protein Synthesis in Native Plant Tissue. Plant Physiol. 173 (3), 1543-1553 (2017).
- Zhou, L., et al. Incorporation of tryptophan analogues into the lantibiotic nisin. Amino Acids. 48 (5), 1309-1318 (2016).
- Acevedo-Rocha, C. G., Budisa, N. Xenomicrobiology: a roadmap for genetic code engineering. Microb Biotechnol. 9 (5), 666-676 (2016).
- Agostini, F., Völler, J. -S., Koksch, B., Acevedo-Rocha, C. G., Kubyshkin, V., Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew Chem Int Edit. 56 (33), 9680-9703 (2017).
- Bacher, J. M., Ellington, A. D. Selection and characterization of Escherichia coli variants capable of growth on an otherwise toxic tryptophan analogue. J Bacteriol. 183 (18), 5414-5425 (2001).
- Wong, J. T. Membership mutation of the genetic code: loss of fitness by tryptophan. Pc Natl Acad Sci USA. 80 (20), 6303-6306 (1983).
- Hoesl, M. G., et al. Chemical Evolution of a Bacterial Proteome. Angew Chem Int Edit. 54 (34), 10030-10034 (2015).
- Italia, J. S., et al. An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nat Chem Biol. 13 (4), 446-450 (2017).
- Völler, J. -S., Thi To, T. M., Biava, H., Koksch, B., Budisa, N. Global substitution of hemeproteins with noncanonical amino acids in Escherichia coli with intact cofactor maturation machinery. Enzyme Microb Tech. 106, 55-59 (2017).
- Budisa, N., Steipe, B., Demange, P., Eckerskorn, C., Kellermann, J., Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem. 230 (2), 788-796 (1995).
- Völler, J. -S., Budisa, N. Coupling genetic code expansion and metabolic engineering for synthetic cells. Curr Opin Biotech. 48, 1-7 (2017).
- Johnson, J. A., Lu, Y. Y., Van Deventer, J. A., Tirrell, D. A. Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Curr Opin Chem Biol. 14 (6), 774-780 (2010).
- Somsen, O. J., van Grondelle, R., van Amerongen, H. Spectral broadening of interacting pigments: polarized absorption by photosynthetic proteins. Biophys J. 71 (4), 1934-1951 (1996).
- Kurschus, F. C., Pal, P. P., Bäumler, P., Jenne, D. E., Wiltschi, B., Budisa, N. Gold fluorescent annexin A5 as a novel apoptosis detection tool. Cytom Part A. 75 (7), 626-633 (2009).
- Lepthien, S., Wiltschi, B., Bolic, B., Budisa, N. In vivo engineering of proteins with nitrogen-containing tryptophan analogs. Appl Microbiol Biot. 73 (4), 740-754 (2006).
- Wachter, R. M., Elsliger, M. -A., Kallio, K., Hanson, G. T., Remington, S. J. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure. 6 (10), 1267-1277 (1998).
- Verkhusha, V. V., Lukyanov, K. A. The molecular properties and applications of Anthozoa fluorescent proteins and chromoproteins. Nat Biotechnol. 22 (3), 289-296 (2004).
- Martynov, V. I., Savitsky, A. P., Martynova, N. Y., Savitsky, P. A., Lukyanov, K. A., Lukyanov, S. A. Alternative cyclization in GFP-like proteins family. The formation and structure of the chromophore of a purple chromoprotein from Anemonia sulcata. J Biol Chem. 276 (24), 21012-21016 (2001).
- Piatkevich, K. D., Malashkevich, V. N., Morozova, K. S., Nemkovich, N. A., Almo, S. C., Verkhusha, V. V. Extended Stokes shift in fluorescent proteins: chromophore-protein interactions in a near-infrared TagRFP675 variant. Sci Rep. 3 (1), 1847 (2013).
