

Summary
Hier berichten wir, der Synthese und Kristallisation von 3,5-Lutidine N -Oxid zu entwässern, durch ein einfaches Protokoll, das unterscheidet sich von der klassischen Synthese von Pyridin- N-oxid. Dieses Protokoll nutzt verschiedene Ausgangsmaterial und beinhaltet weniger Reaktionszeit auf eine neue SOLVATISIERTE supramolekulare Struktur ergeben, der unter langsame Verdunstung kristallisiert.
Abstract
Die Synthese von 3,5-Lutidine N -Oxid zu entwässern, 1, wurde in der Synthese von 2-amino-Pyridin-3,5-dicarboxylic Säure erreicht. Ochiai zuerst die Methodik für nicht ersetzt Pyridines im Jahre 1957 in einem 12 h-Prozess, aber kein Röntgen geeignet Kristalle stammen. Die substituierte Ring verwendet, in der hier vorgestellten Methodik beeinflusst deutlich die Zugabe von Wasser-Moleküle in die asymmetrische Einheit, die eine unterschiedliche nukleophilen Stärke 1verleiht. Die x-ray geeignet Kristall zusammengesetzte 1 war möglich, da die Stabilisierung der negativen Ladung in der Sauerstoff durch die Anwesenheit von zwei Wassermolekülen wo Spenden die Wasserstoffatome positiven Ladung in den Ring; Diese Wassermoleküle auch dazu dienen, eine supramolekulare Interaktion zu konstruieren. Die hydratisierten Moleküle können für das alkalische System möglich, die erreicht wird durch Einstellen des pH-Werts um 10. Wichtig ist, die doppelte Methyl ersetzt, Ring und einer Reaktionszeit von 5 h, macht es eine vielseitigere Methode und mit breiteren chemische Anwendungen für zukünftige Ring Einfügungen.
Introduction
Heute haben Wissenschaftler auf der ganzen Welt Ressourcen in die Entwicklung neuer Synthesewege für die Funktionalisierung von aromatischen Gruppen, investiert die Hinzufügung Reaktionen1,2für niedrige Reaktivität Front bekannt sind, 3. Pyridin, wo ein Stickstoffatom ein Kohlenstoffatom ersetzt, stellt eine ähnliche chemische Reaktivität auf Analog, die ausschließlich aus Kohlenstoff-Atomen3Ringe komponiert, und er erfährt in der Regel eine Substitution Mechanismus als Ergänzung. N-Oxide zeichnen sich durch die Anwesenheit einer Spender-Bindung zwischen Stickstoff und Sauerstoff durch die Überlappung der nonbonding Elektronenpaar am Stickstoff mit einer leeren Orbital auf dem Sauerstoff-Atom-3gebildet. Vor allem, Pyridin N-Oxide sind Lewis-Basen, weil ihre N-O Glyko-kann als ein Elektron Spender handeln, und sie möglicherweise mit Lewis-Säuren bilden die entsprechenden Lewis-Säure-Basen-Paare verbinden. Diese Eigenschaft hat eine wesentliche chemische Folge, weil es kann die Nucleophilicity von Lewis-Säuren gegenüber potenziellen Electrophiles erhöhen und somit, unter Bedingungen zu reagieren ermöglichen, wo würde normalerweise die Reaktion nicht auftreten. Wahrscheinlich ist die häufigste Verwendung solcher Verbindungen in verschiedenen Oxidationsreaktionen, wo sie als Oxidationsmittel4handeln. Pyridin- N-Oxide und vieler ihrer Ring funktionalisiert Derivate sind wiederkehrende Moleküle von biologisch aktiven und pharmakologische Wirkstoffe5und eine klare räumliche Verteilung durch verschiedene spektroskopische Werkzeuge wurde eingerichtet für einige von ihnen6,7. In der Forschung über die Pyridin Ring verschiedene Gruppen zuweisen haben Wissenschaftler testeten verschiedene Methoden um eine einfache und konventionelle Methode zu produzieren, da Isoxazolines eine katalytische Menge an Base wie DBU erfordert in siedendem Xylol zu bilden 6- ersetzt-2-Aminopyridine N-Oxide8,9. Eine Vielzahl von Pyridin-Derivate wurden umgewandelt in ihre entsprechenden N-Oxide in der Gegenwart eine katalytische Menge an Mangan tetrakis(2,6-diclorophenyl) Porphyrin und Ammonium-Acetat CH2Cl2/CH3 CN8,10. Andere Pyridines sind auf ihre Oxide mit H2O2 in Anwesenheit von katalytische Mengen von Methyltrioxorhenium8,11, oder durch die Zugabe von überschüssigen Dimethyldioxirane in CH2Cl2 oxidiert. bei 0 ° C, wodurch die entsprechenden N-Oxide8,12,13,14. BIZ (Trimethylsilyl) Wasserstoffperoxid in Gegenwart von Trioxorhenium in CH2Cl2 wird für die Synthese von Pyridin- N-Oxide8,11. Die Synthese von Aminopyridine N-Oxide mit Acylation mit Carosche Säure (Peroxomonosulfuric Säure) auch wurde berichtet,8. Dennoch, die Methodik hier berichtet, und die Teil der Methodik von Ochiai1, berichtet verwendet liefert sehr gute Ergebnisse mit dem Einsatz von billiger und zugänglich Reagenzien, H2O2 und Eisessig. Diese Praxis ist besser geeignet für den Einsatz in großem Maßstab Vorbereitungen, die auf tertiäre Amine wirken, es produziert gute Erträge in einer Reaktion, die nur 30 % Wasserstoffperoxid erfordert und Eisessig in eine Temperatur zwischen 70-80 ° C und es nutzt einen Reinigungsprozess in den meisten Synthese Laboratorien wie Destillation, ohne den Einsatz von Katalysatoren oder teurer Reagenzien1verfügbar ist. Die Literatur berichtet, dass andere Methoden häufig auch Fristen von 10-24 h und Temperaturen über 100 ° C 4,8 beinhalten, und der Ertrag von gut ausgebildeten Kristallen für Röntgen-Analysen wird selten berichtet.
Reaktiv, werden verschiedene N -oxid-Derivate verwendet, um angemessen die Lutidine Ring auf einer nukleophilen oder elektrophiler Weise aktivieren. Der nukleophilen oder elektrophiler Faktor betrifft die Substituenten. Mit dem Pyridin Ring wird die Aberkennung der Elektron-Gruppen ist der Hauptfaktor der nukleophilen charakteristischen1. Die kostenlose N-oxid Verbindungen sind selten isoliert als geeignete Kristalle für röntgenstrahlanalyse durch delokalisierte Ladung in den aromatischen Ring. Die Solvatation Faktor ist jedoch entscheidend für die negative Dichte der Sauerstoff15zu stabilisieren.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Reaktion
- In einer Dampfhaube eine geöffnete Runde 100 mL-Flasche mit 0,5 Mol (29,8 mL) Eisessig geben Sie und 0.051 Mol (5,82 mL) 3,5-Dimethylpyridine und 5 mL H2O2 (35 %). Halten Sie die Reaktion der Mischung unter ständigem Rühren magnetische, eine innere Temperatur von 80 ° C für 5 h.
- Kühlen Sie nach der Einwirkzeit die Küvette auf 24 ° C mit Eis (Do nicht aussetzen der Essigsäure auf das Eis Gase) und stecken Sie es auf eine hohe Vakuumdestillation Einheit für 90-120 min, überschüssige Essigsäure zu entfernen.
Achtung: Verwenden Sie nicht heißes Material. Warten Sie, bis das Glas eine überschaubare Temperatur erreicht hat. Dies vermeidet auch Dämpfe in die Top der Destillation-Einheit. - Fügen Sie destilliertes Wasser (10 mL) zweimal, um sicherzustellen, dass die entfernt von jeder Spur von Essigsäure und die Mischung zu konzentrieren viel wie möglich.
(2) Basizität Anpassung und Extraktion
- Isolierte zähflüssige und transparentere Produkt in Bi-distillated Wasser auflösen und ein Potentiometer zur Einstellung des pH-Wertes auf 10 mit pure solid Na2CO3verwenden.
- Legen Sie die Lösung in einem 250 mL-Trennung-Trichter vorsichtig und entpacken Sie es 5 Mal mit 250 mL Kchl3 , um den Ertrag zu verbessern. Wiederherstellen der organische Schicht und trocknen Sie es SO über solide Na24 30 Minuten maximal, die das Produkt enthält. Falls erforderlich, wieder extrahieren der wässrigen Phase mit der gewünschten Menge an Kchl3.
Achtung: Kchl3 können Schläfrigkeit und Benommenheit verursachen; behandeln Sie mit Sorgfalt und in einer Dampfhaube. - Entfernen Sie das Lösungsmittel unter vermindertem Druck mit einem hohen Vakuum-Destillation bis zur Bildung des ein sehr hygroskopisch klar Beige, kristallines Pulver (70 %).
3. die Kristallisation
- 4,3 g kristallines Pulver in 50 mL kalten hohe durchgeführten Flüssigkeitschromatographie (HPLC) Grade Diethylether auflösen. Vakuum-Filter die Lösung für jede Spur von soliden Ausgangsmaterial oder sogar Staub zu entfernen. Das Filtrat in ein Glas Petrischale Gießen, so dass es zu langsam verdunsten bei 4 ° C in einem Labor-Kühlschrank.
- Stellen Sie sicher, dass nach zwei Tagen, klare farblose Kristalle gewonnen werden. Dann Messen Sie den Schmelzpunkt, die im Bereich von 310-311 K. sein sollte
4. Analyse der 3,5-Lutidine N -oxid gelief
- Entfernen Sie die Kristalle, die prismatische Form und farblos, durch Dekantieren von der Küvette Wänden für weitere röntgenstrahlanalyse gebildet werden. Wenn nicht sofort verwendet, halten Sie die Kristalle in Diethylether, Kristall Flüssigkeitszufuhr zu vermeiden.
- Auflösen von 0,010 g 3,5-Lutidine N -oxid entwässern in 0,4 mL CDCl3 , NMR H1 und C-13 -Analyse, beweisen die Wirksamkeit des Verfahrens durchzuführen.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Das Protokoll ist im Wesentlichen eine Erweiterung des Ochiai Technik1. Allerdings gelten niedrigere Temperatur und weniger Zeit. Diese einfache Methode lässt sich eine vielseitige Liganden zu erhalten, eine ersetzte Pyridin- N-oxid Ableitung. Um die Bildung von 1zu bestätigen, werden NMR 1H und 13C Analyse bevorzugt, um die Wirksamkeit des Verfahrens prüfen.
Die chemische Verschiebung zeigt die Bildung von 1. Das Signal bei 2,28 ppm (Teile pro million) entspricht sechs gleichwertige Wasserstoffatome von zwei Methylgruppen in den 3 und 5 Positionen, die das magnetische Feld in weniger Anteil als die dauerhafte magnetische wahrnehmen. Es gibt zwei Arten von septolen: eine gehört das Proton in der C-Position bei 7,9, welche verdoppelt die Größe des anderen Signals am 6.9, die das Proton in der Position a Abbildung 1 gehört die chemischen Verschiebungen provoziert durch die Anwesenheit von Sauerstoff Atom Bonde zeigt Das Stickstoffatom des Pyridin Ring d. Das Sauerstoffatom ist Electro-zurückziehen und die engere Wasserstoffatome an das Sauerstoffatom (c und a) Show Verschiebung zu einer höheren Frequenz als die für die Methyl-Wasserstoffatome (b).
Der gleiche Prozess wird für die NMR- 13C-Spektrum, Abbildung 2, gezeichnet wo die Signale für die engere Kohlebürsten das Sauerstoffatom (c und eine) zeigen Frequenzabstand zwischen den Signalen des Δc = 1.300 Hz und Δein = 200 Hz. Wieder einmal zeigen die Methyl-Kohlebürsten keine Änderung. IR-Spektrum kann verwendet werden, um den Erfolg der Methode als auch sehen.
Das ORTEP Diagramm, Abbildung 4zeigt das Vorhandensein von zwei Molekülen Wasser rund um die asymmetrische Molekül. Diese Moleküle werden geglaubt, um die N-O-Bindung zu stabilisieren. In ähnlichen Fällen, es wurde beschrieben für Pyridin- N-Oxid und damit verbundenen aromatischen Oxide. Gibt es eine erhebliche Stabilisierung π-Typ O→N Rücken-Spende, spiegelt sich in einer berechneten Anleihe Reihenfolge höher als 1 und eine Reihe von einsamen Elektronenpaaren auf das O-Atom weniger als 36.
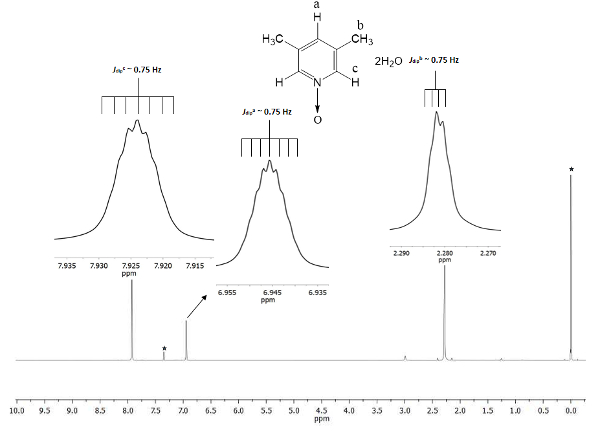
Abbildung 1 . TMS verwiesen CDCl3 500 MHz NMR 1H Spektrum von 1. Die Integrationen und chemischen Verschiebungen der drei Signale stimmen mit drei verschiedenen Arten von Wasserstoff-Atomen im Lutine N-oxid. Bitte klicken Sie hier für eine größere Version dieser Figur.
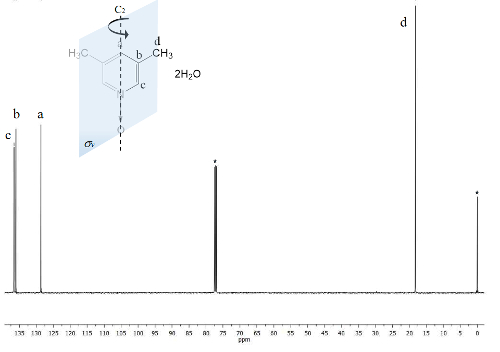
Abbildung 2 . TMS verwiesen CDCl3 100 MHz NMR 13C Spektrum von 1. Drei Signale werden für die fünf aromatische Kohlenstoffe und eines für die beiden Methylgruppen beobachtet. Bitte klicken Sie hier für eine größere Version dieser Figur.
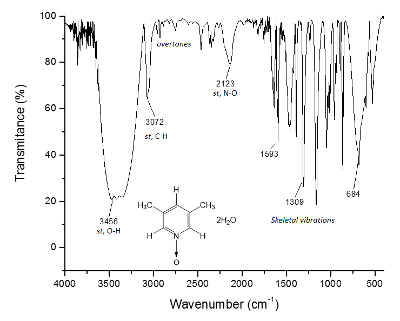
Abbildung 3 . IR-Spektrum von 1. Die O-H-Bindungen vor 3.300 cm-1, sind verantwortlich für die supramolekulare Strukturbildung und die Kristallbildung. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 4 . ORTEP Diagramm 1 wo zwei Moleküle von H2O Formen Wasserstoffbrückenbindungen mit Lutidine Sauerstoff überbrücken, fahren ihre Wasserstoff-Atomen in Richtung das Sauerstoffatom. Diese Zahl wurde aus Merino García Et Al. modifiziert 12 Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 5 . Bilder von geeigneten Röntgenbeugung Kristalle 1 in Diethylether (oben) und beim open Air (unten). Einer dieser Kristalle wurde bestätigt in der Röntgen-Diffraktometer und zeigte einen Beugung Pfad von x-ray, die übersetzt und verfeinert durch spezielle computergestützte Programme in einer molekularen und kristalline Struktur24,25, 26,27,28. Bitte klicken Sie hier für eine größere Version dieser Figur.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Das Protokoll hier vorgestellten ist eine konventionelle Methode, um ein Sauerstoffatom an das Stickstoffatom des 3,5-Lutidine als eine Methode der Funktionalisierung der Substrate zu verknüpfen. Diese Technik ist auch etablierte Röntgen geeignet dehydriert Kristalle (Abbildung 5, fotografiert mit einer Sony DSC-HX300 Cyber-Shot-Kamera) zu liefern. Soweit wir betroffen sind, haben nicht viele Berichte die Produktion solcher Kristalle16beschrieben. Viele Verbindungen wachsen ideal Kristalle für röntgenstrahlanalyse, wenn sie von verschiedenen Metallen17,18,19,20chelated sind. Sobald das kristalline Pulver gebildet wird, ist es wichtig, sie von ihrer Mutter Liköre mit einem Kitasato Kolben und einen Büchner-Trichter zu extrahieren. Mit Gummischläuchen, Kitasato-Kolben mit einer Vakuumleitung verbunden ist und oben drauf wird Buchner-Trichter mit einem Filterpapier gelegt. Sobald das Vakuum aktiviert wurde, wird das Filterpapier mit einer kleinen Menge des Lösungsmittels angefeuchtet, das Produkt kristallisiert. Dadurch wird verhindert, dass das kristalline Pulver rieselt in den Buchner Trichter durch den Vakuumeffekt. Nach der Sicherung des Filterpapiers, ist die Lösung mit dem kristallinen Pulver geschüttelt, um sicherzustellen, dass das kristalline Pulver wird gefiltert, und keiner im unteren Teil der Flasche bleibt. Die Lösung ist schnell über die Buchner Trichter gegossen. Das kristalline Pulver erhalten bleibt für ca. 10 min auf dem Filterpapier und dann das Vakuum ist ausgeschaltet und kristalline Pulver ist losgelöst vom Papier und gespeichert in einer Milchglas-Fläschchen, beschriftet mit seinem Code und bis zur weiteren Analyse bei 4 ° C aufbewahrt. Die Filterrohr Flüssigkeit wird in ein Glas Petrischale, wobei es langsam verdunsten bei 4 ° C zur Verbesserung der angemessene Kristallbildung für röntgenstrahlanalyse gegossen.
Es ist wichtig zu bemerken, dass dieses Protokoll verwendet Lösungsmittel und Materialien, die leicht erreichbar und befinden sich in der Regel Forschungslabor. Die pH-Einstellung durch die Zugabe von Na2CO3 und das konsequente magnetische rühren sind entscheidend für den Ertrag des Endproduktes. Allerdings ist es wichtig, in allen Prozessschritten, vor allem in der extraktionsstufe besonders vorsichtig achten wo keine Spur des Ausgangsmaterials zu leisten, die Bildung von kristallines Pulver und anschließend Kristalle vorhanden sein muss. Somit kann diese Extraktion/Reinigungsschritt durch NMR oder IR-Spektroskopie, die Qualität des Produktes sicherzustellen überwacht werden.
Um die Reproduzierbarkeit dieses Protokolls sicherzustellen, ist NMR ein ausgezeichnetes Werkzeug. Auch feine Details sind sichtbar im Spektrum. Alle Signale werden als Intarsien in Abbildung 1dargestellt. Diese Einschübe zeigen deutlich eine Teilung, nämlich die Vielfalt aller Signale. Zum Beispiel die Protonen-b (JDipb ~ 0,75 Hz) zeigen vier Gipfel auf dem Kamm des Signals mit einer Trennung unter ihnen mulmig Konstante (ΔSpitze-Spitze) ~ 0,0075 ppm. 0,0075 ppm kann Energie mit Hilfe der folgenden Gleichung21 umgewandelt werden
 Gleichung 1
Gleichung 1

Die Transformation wird empfohlen, weil die Signale, die Entfaltung von dipolar räumliche Interaktion unter den Kernen drei Wasserstoffatome der Methylgruppe kommen, und obwohl sie weiter als 4 Single-Anleihen mit Protonen C und a, sie sind in der Lage zu erkennen sind die zweipoligen magnetischen Impuls Interaktionen22. Darüber hinaus ermöglicht die kostenlose Sigma Bindung Drehung in die Methylgruppe die super Hyperfein Proton-Proton-Interaktion zu sehen in der Mannigfaltigkeit des Signals sein. Das septolen von Protonen ein und c bei 6,9 und 7,9 ppm bzw. stammen von der gleichen zweipoligen Naturphänomen. In diesen Fällen Protonen ein und c kann die Protonen in der Methyl-Gruppe für die gleiche dynamische Rotation unterscheiden. Zuletzt, wie erwartet, die berechnete JDip für a, b und c haben kaum den gleichen Wert, ~ 0,75 Hz. Diese Mengen der Interaktionen bestätigen die Wasserstoff Kerne räumliche Anordnung während der magnetischen Anisotropie.
Auf der anderen Seite macht die C2v Symmetrie von 1 entspricht Kohlebürsten23. 13C Spektrum, Abbildung 2zeigt das typische Signal für Methylgruppen an den aromatischen Ringen befestigt Kohlebürsten d bei 18 ppm. Darüber hinaus ein Signal bei 129 ppm ist sichtbar in dieser Region aufgrund der weniger wissenschaftlich Element beeinflusst Kohlenstoff ein. Bei hohen Frequenzen werden das Signal für die exponierten Kohlebürsten Kerne auf das magnetische Feld um 137 ppm22vorgestellt.
Die vorgestellte Methodik ist sehr nützlich für die Synthese von Pyridin-N-Oxide, bietet gute Erträge, innerhalb eines angemessenen Zeitraums mit weichen Reaktionsbedingungen und billig und leicht zugängliche Reagenzien, die keine zusätzliche Katalysatoren benötigen. Diese Bedingungen können für die wissenschaftliche und pädagogische Gemeinschaft verwendet werden, um eine breite Palette von Pyridines N-Oxide als Vorläufer für andere Moleküle von Interesse zu erhalten. Die geeignete Methode bietet die Möglichkeit, grundlegende experimentelle und konzeptionelle Werkzeuge in pädagogischen Laboratorien für Studenten, beweist eine gelungene Synthese von Verbindungen und das Glück zu sehen, die Bildung von Kristallen zu erwerben. Es ist jedoch wichtig zu betonen, dass wie eine chemische Reaktion, es notwendig ist, alle Vorkehrungen zu treffen, da in der Regel verwendeten Reagenzien gefährlich sind.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Alle Autoren erklären keinen Interessenskonflikt.
Acknowledgments
Die vorliegende Arbeit wurde unterstützt von Vicerrectoría de Investigación y Estudios de Posgrado von BUAP und Verbreitung der Wissenschaft Projekte Nr. REOY-NAT14, 15, 16-G. HEAŞ-NAT17. RMG Dank CONACyT (Mexiko) für Stipendium 417887.
Materials
| Name | Company | Catalog Number | Comments |
| 3,5-lutidine | Sigma-Aldrich | L4206-500ML | |
| Glacial acetic acid | Fermont | 3015 | |
| Hidrogen peroxide (35%) | Sigma-Aldrich | 349887-500ML | |
| Na2CO3 anhydrous | Productos Químicos Monterrey | 1792 | |
| Na2SO4 anhydrous | Alfa reactivos | 25051-C | |
| CHCl3 | Fermont | 6205 | |
| Ethyl eter | Mercury Chemist | QME0309 | |
| Distilled water | Comercializadora Química Poblana | not-existent |
References
- Ochiai, E. Recent Japanese work on the chemistry of pyridine 1-oxide and related compounds. J. Org. Chem. 18 (5), 534-551 (1953).
- Solomons, T. W. G. Organic Chemistry 2nd Edition. , John Wiley & Sons. 1110 (1976).
- Albini, A., Pietra, S. Heterocyclic N-Oxides. , CRC Press. ISBN: 0849345529 328 (1991).
- Koukal, P., Ulc, J., Necas, D., Kotora, Heterocyclic N.-Oxides. Topics in Heterocyclic Chemistry. 53, 29-58 (2017).
- Wen-Man, Z., Jian-Jun, D., Xu, J., Jun, X., Huan-Jian, X. Visible-Light-Induced C2 alkylation of pyridine N.-oxides. J. Org. Chem. 82 (4), 2059-2066 (2017).
- Merino García, M. R., Ríos-Merino, F. J., Bernès, S., Reyes-Ortega, Y. Crystal structure of 3,5-dimethylpyridine N-oxide dihydrate. Acta Cryst. 72 (12), 1687-1690 (2016).
- Sarma, R., Karmakar, A., Baruah, J. B. N-Oxides in Metal-Containing Multicomponent Molecular Complexes. Inorg. Chem. 47 (3), 763-765 (2008).
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. ARKIVOC. 2001, 242-268 (2001).
- Chucholowski, A. W., Uhlendorf, S. Base catalyzed rearrangement of 5-cyanomethyl-2-isoxazolines; novel pathway for the formation of 2-aminopyridine N-oxides. Tetrahedron Lett. 31 (14), 1949-1952 (1990).
- Thellend, A., Battioni, P., Sanderson, W., Mansuy, D. Oxidation of N-Heterocycles by H2O2 Catalyzed by a Mn-Porphyrin: An Easy Access to N-Oxides Under Mild Conditions. Synthesis. 1997 (12), 1387-1388 (1997).
- Copéret, C., Adolfson, H., Tinh-Alfredo, V. K. h, Yudin, A. K., Sharpless, K. B. A simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 63 (5), 1740-1741 (1998).
- Ferrer, M., Sánchez-Baeza, F., Messeguer, A. On the preparation of amine N-oxides by using dioxiranes. Tetrahedron. 53 (46), 15877-15888 (1997).
- Adam, W., Briviba, K., Duschek, F., Golsch, D., Kiefer, W., Sies, H. Formation of singlet oxygen in the deoxygenation of heteroarene N-oxides by dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1995 (18), 1831-1832 (1995).
- Murray, R. W., Singh, M. A Facile One-Step Synthesis of C-Arylnitrones Using Dimethyldioxirane. J.Org.Chem. 55 (9), 2954-2957 (1990).
- Kim, S. W., Um, T., Shin, S. Brønsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides. Chem. Commun. 53 (18), 2733-2736 (2017).
- Campeau, L., Rousseaux, R., Fagnou, K. A solution to the 2-pyridyl organometallic cross-coupling problem: regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 127 (51), 18020-18021 (2005).
- Gang, L., et al. Metal-free methylation of a pyridine N-oxide C-H bond by using peroxides. Org. Biomol. Chem. 13 (46), 11184-11188 (2015).
- May, D., Nyman,, Hampden-Smith, M. J., Duesler, E. N. Synthesis, characterization, and reactivity of group 12 metal thiocarboxylates M(SOCR)2Lut2[M) Cd, Zn; R ) CH3, C(CH3)3; Lut ) 3,5-Dimethylpyridine (Lutidine)]. Inorg. Chem. 36 (10), 2218-2224 (1997).
- Cho, S. H., Hwang, S. J., Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 130 (29), 9254-9256 (2008).
- Ide, Y., et al. Spin-crossover between high-spin (S = 5/2) and low-spin (S = 1/2) states in six-coordinate iron(III) porphyrin complexes having two pyridine-N. oxide derivatives. Dalton Trans. 46 (1), 242-249 (2017).
- Drago, R. S. Physical Methods in Chemistry. , Saunders College Publishing USA. 750 (1977).
- Cervantes-Mejía, V., et al. Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies. American Journal of Analytical Chemistry. 5 (16), 1090-1101 (2014).
- Huheey, J. E., Keiter, E. A., Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th Edition. , Oxford University Press. Mexico. ISBN: 9706131620 1023 (1997).
- Rigaku, CrysAlisPRO. , (2013).
- Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. A short history of SHELX. Acta Cryst. 64 (1), 112-122 (2008).
- Macrae, C. F., et al. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 41 (2), 466-470 (2008).
- ChemBioDraw Ultra 13. , PerkinElmer. (2013).
