

Summary
Herein, we report the synthesis and crystallization of 3,5-lutidine N-oxide dehydrate by a simple protocol that differs from the classical synthesis of pyridine N-oxide. This protocol utilizes different starting material and involves less reaction time to yield a new solvated supramolecular structure, which crystallizes under slow evaporation.
Abstract
The synthesis of 3,5-lutidine N-oxide dehydrate, 1, was achieved in the synthesis route of 2-amino-pyridine-3,5-dicarboxylic acid. Ochiai first used the methodology for non-substituted pyridines in 1957 in a 12 h process, but no X-ray suitable crystals were obtained. The substituted ring used in the methodology presented here clearly influenced the addition of water molecules into the asymmetric unit, which confers a different nucleophilic strength in 1. The X-ray suitable crystal compound 1 was possible due to the stabilization of the negative charge in the oxygen by the presence of two water molecules where the hydrogen atoms donate positive charge into the ring; such water molecules serve well to construct a supramolecular interaction. The hydrated molecules may be possible for the alkaline system that is reached by adjusting the pH to 10. Importantly, the double methyl substituted ring and a reaction time of 5 h, makes it a more versatile method and with wider chemical applications for future ring insertions.
Introduction
Nowadays, scientists around the globe have been investing resources into the development of new synthetic routes for the functionalization of aromatic groups, which are known for low reactivity front to addition reactions1,2,3. Pyridine, where a nitrogen atom substitutes a carbon atom, presents a similar chemical reactivity to analogue rings composed solely of carbon atoms3, and it usually undergoes a substitution mechanism rather than addition. N-oxides are distinctive by the presence of a donor bond between nitrogen and oxygen formed by the overlap of the nonbonding electron pair on the nitrogen with an empty orbital on the oxygen atom3. Particularly, pyridine N-oxides are Lewis bases, because their N-O moiety may act as an electron donor, and they may combine with Lewis acids forming the corresponding Lewis acid-base pairs. This property has an essential chemical consequence, because it can increase the nucleophilicity of the Lewis acids towards potential electrophiles and thus allow them to react under conditions where normally the reaction would not occur. Probably the most frequent use of such compounds is in various oxidation reactions where they act as oxidants4. Pyridine N-oxides and many of their ring-functionalized derivatives are recurrent molecules of biologically active and pharmacological agents5, and a clear spatial distribution by different spectroscopic tools has been established for some of them6,7. In research on attaching different groups to the pyridine ring, scientists have tested various methodologies to produce an easy and conventional method, since isoxazolines requires a catalytic amount of base such as DBU in boiling xylene to form 6-substituted-2-aminopyridine N-oxides8,9. A variety of pyridine derivatives were converted into their corresponding N-oxides in the presence of a catalytic amount of manganese tetrakis(2,6-diclorophenyl)porphyrin and ammonium acetate in CH2Cl2/CH3CN8,10. Other pyridines are oxidized to their oxides using H2O2 in the presence of catalytic amounts of methyltrioxorhenium8,11, or by the addition of excess dimethyldioxirane in CH2Cl2 at 0 °C, which leads to the corresponding N-oxides8,12,13,14. Bis(trimethylsilyl)peroxide in the presence of trioxorhenium in CH2Cl2 has been used for the synthesis of pyridine N-oxides8,11. The synthesis of aminopyridine N-oxides involving acylation using Caro's acid (peroxomonosulfuric acid) has also been reported8. Nevertheless, the methodology reported here, and which uses part of the methodology reported by Ochiai1, provides very good results with the use of cheaper and accessible reagents, H2O2 and glacial acetic acid. This practice is more suitable for use in large scale preparations that act on tertiary amines, it produces good yields in a reaction that only requires 30% hydrogen peroxide and glacial acetic acid in a temperature between 70-80 °C, and it uses a purification process that is available in most synthesis laboratories like distillation, without the use of catalyst or more expensive reagents1. The literature reports that other methodologies also frequently involve time frames from 10-24 h and temperatures above 100 °C 4,8, and the yield of well-formed crystals for X-ray analyses is rarely reported.
Reactively, various N-oxide derivates are used to adequately activate the lutidine ring, in either a nucleophilic or electrophilic way. The nucleophilic or electrophilic factor is affected by the substituents. With the pyridine ring being the electron-withdrawing groups, the main factor is the nucleophilic characteristic1. The free N-oxide compounds are rarely isolated as suitable crystals for X-ray analysis due to the delocalized charge in the aromatic ring. However, the solvation factor is critical to stabilize the negative density of the oxygen15.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Reaction
- Place in a fume hood an opened round 100 mL flask with 0.5 mol (29.8 mL) glacial acetic acid and add 0.051 mol (5.82 mL) of 3,5-dimethylpyridine and 5 mL of H2O2 (35%). Keep the mixture reaction under constant magnetic stirring, at an inner temperature of 80 °C for 5 h.
- After the reaction time, cool the flask to 24 °C with ice (do not expose the acetic acid gases to the ice), and plug it to a high vacuum distillation unit for 90-120 min to remove excess acetic acid.
Caution: Do not use hot material. Wait until the glassware reaches a manageable temperature. This will also avoid vapors entering the top of the distillation unit. - Add distilled water (10 mL) twice to ensure the removal of any trace of acetic acid and to concentrate the mixture much as possible.
2. Basicity Adjustment and Extraction
- Dissolve in bi-distillated water the isolated viscous and transparent product and use a potentiometer to adjust the pH to 10 with pure solid Na2CO3.
- Place carefully the solution in a 250 mL separation funnel and extract it 5 times with 250 mL of CHCl3 to improve the yield. Recover the organic layer and dry it over solid Na2SO4 for 30 min maximum, which will contain the product. If necessary, re-extract the aqueous phase with the desired amount of CHCl3.
Caution: CHCl3 may cause drowsiness and dizziness; handle with care and inside a fume hood. - Remove the solvent under reduced pressure with a high vacuum distillation unit, until the formation of a very hygroscopic clear beige crystalline powder (70%).
3. Crystallization Process
- Dissolve 4.3 g of the crystalline powder in 50 mL of cold high performed liquid chromatography (HPLC) grade diethyl ether. Vacuum filter the solution to remove any trace of solid starting material or even dust. Pour the filtrate into a glass Petri dish, leaving it to slow evaporate at 4 °C in a laboratory fridge.
- Ensure that after two days, clear colorless crystals are obtained. Then measure the melting point, which should be in the range of 310-311 K.
4. Analysis of 3,5-Lutidine N-oxide Dehydrate
- Remove the crystals that are formed, of prismatic shape and colorless, by decantation from the flask's walls for further X-ray analysis. If not immediately used, keep the crystals in diethyl ether to avoid crystal hydration.
- Dissolve 0.010 g of 3,5-lutidine N-oxide dehydrate in 0.4 mL of CDCl3 to perform NMR H1 and C13 analysis to prove the effectiveness of the procedure.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
The protocol is essentially an extension of Ochiai's technique1. However, lower temperature and less time are applied. This simple method can be used to obtain a versatile ligand, which is a substituted pyridine N-oxide derivate. To confirm the formation of 1, NMR 1H and 13C analysis are preferred to test the effectiveness of the procedure.
The chemical shift demonstrates the formation of 1. The signal at 2.28 ppm (parts per million) corresponds to the six equivalent hydrogens of the two methyl groups in the 3 and 5 positions, which perceive the magnetic field in less proportion than the permanent magnetic. There are two sets of septuplets: one belongs to the proton in the c position at 7.9, which doubles the size of the other signal at 6.9 that belongs to the proton in the position a. Figure 1 shows the chemical shifts provoked by the presence of the oxygen atom bonded to the nitrogen atom of the pyridine ring. The oxygen atom is electro-withdrawing and the closer hydrogen atoms to the oxygen atom (c and a) show displacement to a higher frequency than that for the methyl hydrogens (b).
The same process is plotted for the NMR 13C spectrum, Figure 2, where the signals for the closer carbons to the oxygen atom (c and a) show frequency separation between their signals of Δc = 1,300 Hz and Δa = 200 Hz. Once again, the methyl carbons do not show any change. The IR spectrum can be used to see the success of the method as well.
The ORTEP diagram, Figure 4, demonstrates the presence of two molecules of water surrounding the asymmetric molecule. These molecules are believed to stabilize the N-O bond. In similar cases, it has been described for pyridine N-oxide and related aromatic oxides. There is a significant stabilizing π-type O→N back-donation, reflected in a calculated bond order higher than 1 and a number of electron lone pairs on the O atom lower than 36.
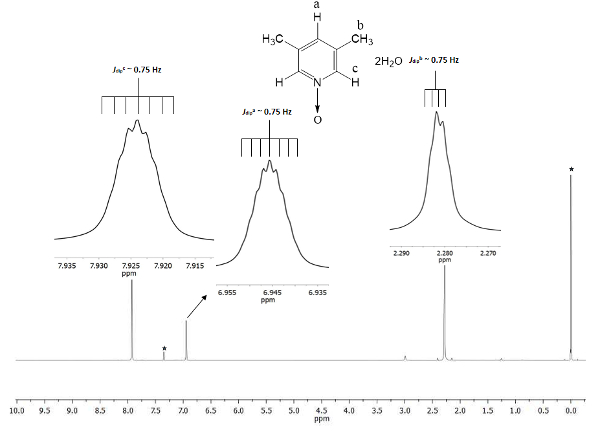
Figure 1. TMS referenced CDCl3 500 MHz NMR 1H spectrum of 1. The integrations and chemical shifts of the three signals agree with three different types of hydrogen atoms present in lutine N-oxide. Please click here to view a larger version of this figure.
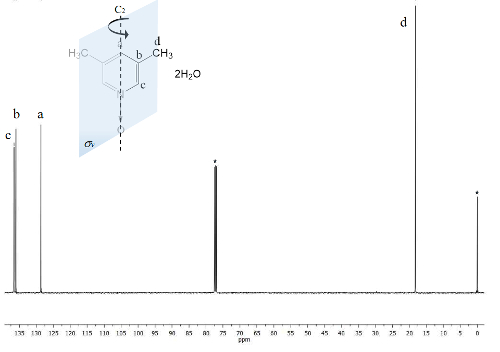
Figure 2. TMS referenced CDCl3 100 MHz NMR 13C spectrum of 1. Three signals are observed for the five aromatic carbons and one for the two methyl groups. Please click here to view a larger version of this figure.
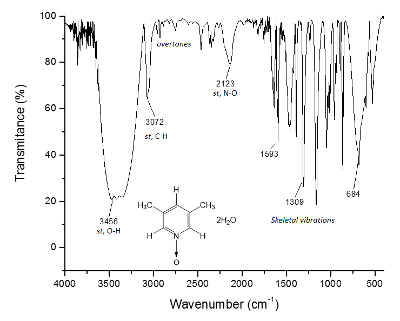
Figure 3. IR spectrum of 1. The O-H bonds, above of 3,300 cm-1, are responsible for the supramolecular structure formation and the crystal formation. Please click here to view a larger version of this figure.

Figure 4. ORTEP diagram of 1 where two molecules of H2O forms bridge hydrogen bonds with lutidine's oxygen, driving their hydrogen atoms toward the oxygen atom. This Figure has been modified from Merino García et al.12 Please click here to view a larger version of this figure.

Figure 5. Pictures of suitable X-ray diffraction crystals of 1 in diethyl ether (top) and at open air (bottom). One of these crystals was confirmed in the X-ray diffractometer and showed a diffraction path of X-ray, which was traduced and refined by special computational programs in a molecular and crystalline structure24,25,26,27,28. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
The protocol presented here is a conventional method to link an oxygen atom to the nitrogen atom of the 3,5-lutidine as a functionalization method of substrates. This technique is also well established to yield X-ray suitable dehydrated crystals (Figure 5, pictures taken with a DSC-HX300 Cyber-shot Sony camera). As far as we are concerned, not many reports have described the production of such crystals16. Many compounds grow ideal crystals for X-ray analysis when they are chelated by various metals17,18,19,20. Once the crystalline powder is formed, it is important to extract it from their mother liquors using a Kitasato flask and a Buchner funnel. Using rubber hoses, the Kitasato flask is connected to a vacuum line and on top of it the Buchner funnel is placed with a filter paper. Once the vacuum has been turned on, the filter paper is moistened with a small amount of solvent from which the product crystallized. This prevents the crystalline powder from trickling into the Buchner funnel by the vacuum effect. After securing the filter paper, the solution containing the crystalline powder is shaken to ensure that all the crystalline powder is filtered, and none remains in the bottom of the flask. The solution is quickly poured over the Buchner funnel. The crystalline powder obtained is left for about 10 min on the filter paper, and then the vacuum is turned off and the crystalline powder is detached from the paper and stored in an opaque glass vial, labeled with its code and kept at 4 °C until further analysis. The filtrated liquid is poured into a glass Petri dish, leaving it to slowly evaporation at 4 °C to improve the formation of adequate crystals for X-ray analysis.
It is important to notice that this protocol uses solvents and materials that are easily obtainable and generally are found at any research laboratory. The pH adjustment by the addition of Na2CO3 and the consistent magnetic stirring are critical to the yield of the final product. However, it is important to pay extra careful attention in all process steps, especially in the extraction stage where no trace of starting material must be present to afford the formation of crystalline powder and subsequently crystals. Thus, this extraction/purification stage can be monitored by either NMR or IR spectroscopy to ensure the quality of the product.
To ensure the reproducibility of this protocol, NMR is an excellent tool. Even fine details are visible in the spectrum. All signals are shown as insets in Figure 1. These insets depict clearly a split, namely multiplicity, of all signals. For instance, the protons b (Jdipb ~ 0.75 Hz) show four peaks at the crest of the signal, with a separation among them (Δpeak-peak) queasy constant of ~ 0.0075 ppm. The 0.0075 ppm may be transformed to energy using the following equation21
 Equation 1
Equation 1

The transformation is recommended because the signals unfolding come from the dipolar spatial interaction among the three hydrogens nuclei of the methyl group, and even though they are further than the 4 single bonds with protons c and a, they are able to perceive their dipolar magnetic momentum interactions22. Additionally, the free sigma bonding rotation in the methyl group allows the super hyperfine proton-proton interaction to be visible in the multiplicity of the signal. The septuplets of protons a and c at 6.9 and 7.9 ppm, respectively are derived from the same dipolar nature phenomenon. In these cases, protons a and c can differentiate the protons in the methyl group for the same rotation dynamic. Last, as expected, the calculated Jdip for a, b, and c have barely the same value, ~ 0.75 Hz. These quantities of the interactions confirm the hydrogen nuclei spatial arrangement throughout the magnetic anisotropy.
On the other hand, the C2v symmetry of 1 makes equivalent carbons23. The 13C spectrum, Figure 2, shows the typical signal for methyl groups attached to aromatic rings, carbons d at 18 ppm. Moreover, a signal at 129 ppm is visible at this region due to the less electronegative element influenced carbon a. At high frequencies the signal for the more exposed carbons nuclei to the magnetic field are presented at 137 ppm22.
The presented methodology is very useful for the synthesis of pyridine N-oxides, providing good yields, in a reasonable time with soft reaction conditions and cheap and easy accessible reagents, that do not require additional catalysts. These conditions can be used for the scientific and educational community to obtain a broad range of pyridines N-oxides as precursors for other molecules of interest. The suitable methodology gives the opportunity to acquire basic experimental and conceptual tools in educational laboratories for students, proving a successful synthesis of compounds and the happiness to see the formation of crystals. However, it is important to emphasize that, like any chemical reaction, it is necessary to take all precautions since generally the reagents used are dangerous.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
All authors declare no conflict of interest.
Acknowledgments
The present work has been supported by Vicerrectoría de Investigación y Estudios de Posgrado from BUAP, Divulgation of Science, and Projects No. REOY-NAT14, 15, 16-G. HEAS-NAT17. RMG thanks CONACyT (Mexico) for scholarship 417887.
Materials
| Name | Company | Catalog Number | Comments |
| 3,5-lutidine | Sigma-Aldrich | L4206-500ML | |
| Glacial acetic acid | Fermont | 3015 | |
| Hidrogen peroxide (35%) | Sigma-Aldrich | 349887-500ML | |
| Na2CO3 anhydrous | Productos Químicos Monterrey | 1792 | |
| Na2SO4 anhydrous | Alfa reactivos | 25051-C | |
| CHCl3 | Fermont | 6205 | |
| Ethyl eter | Mercury Chemist | QME0309 | |
| Distilled water | Comercializadora Química Poblana | not-existent |
References
- Ochiai, E. Recent Japanese work on the chemistry of pyridine 1-oxide and related compounds. J. Org. Chem. 18 (5), 534-551 (1953).
- Solomons, T. W. G. Organic Chemistry 2nd Edition. , John Wiley & Sons. 1110 (1976).
- Albini, A., Pietra, S. Heterocyclic N-Oxides. , CRC Press. ISBN: 0849345529 328 (1991).
- Koukal, P., Ulc, J., Necas, D., Kotora, Heterocyclic N.-Oxides. Topics in Heterocyclic Chemistry. 53, 29-58 (2017).
- Wen-Man, Z., Jian-Jun, D., Xu, J., Jun, X., Huan-Jian, X. Visible-Light-Induced C2 alkylation of pyridine N.-oxides. J. Org. Chem. 82 (4), 2059-2066 (2017).
- Merino García, M. R., Ríos-Merino, F. J., Bernès, S., Reyes-Ortega, Y. Crystal structure of 3,5-dimethylpyridine N-oxide dihydrate. Acta Cryst. 72 (12), 1687-1690 (2016).
- Sarma, R., Karmakar, A., Baruah, J. B. N-Oxides in Metal-Containing Multicomponent Molecular Complexes. Inorg. Chem. 47 (3), 763-765 (2008).
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. ARKIVOC. 2001, 242-268 (2001).
- Chucholowski, A. W., Uhlendorf, S. Base catalyzed rearrangement of 5-cyanomethyl-2-isoxazolines; novel pathway for the formation of 2-aminopyridine N-oxides. Tetrahedron Lett. 31 (14), 1949-1952 (1990).
- Thellend, A., Battioni, P., Sanderson, W., Mansuy, D. Oxidation of N-Heterocycles by H2O2 Catalyzed by a Mn-Porphyrin: An Easy Access to N-Oxides Under Mild Conditions. Synthesis. 1997 (12), 1387-1388 (1997).
- Copéret, C., Adolfson, H., Tinh-Alfredo, V. K. h, Yudin, A. K., Sharpless, K. B. A simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 63 (5), 1740-1741 (1998).
- Ferrer, M., Sánchez-Baeza, F., Messeguer, A. On the preparation of amine N-oxides by using dioxiranes. Tetrahedron. 53 (46), 15877-15888 (1997).
- Adam, W., Briviba, K., Duschek, F., Golsch, D., Kiefer, W., Sies, H. Formation of singlet oxygen in the deoxygenation of heteroarene N-oxides by dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1995 (18), 1831-1832 (1995).
- Murray, R. W., Singh, M. A Facile One-Step Synthesis of C-Arylnitrones Using Dimethyldioxirane. J.Org.Chem. 55 (9), 2954-2957 (1990).
- Kim, S. W., Um, T., Shin, S. Brønsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides. Chem. Commun. 53 (18), 2733-2736 (2017).
- Campeau, L., Rousseaux, R., Fagnou, K. A solution to the 2-pyridyl organometallic cross-coupling problem: regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 127 (51), 18020-18021 (2005).
- Gang, L., et al. Metal-free methylation of a pyridine N-oxide C-H bond by using peroxides. Org. Biomol. Chem. 13 (46), 11184-11188 (2015).
- May, D., Nyman,, Hampden-Smith, M. J., Duesler, E. N. Synthesis, characterization, and reactivity of group 12 metal thiocarboxylates M(SOCR)2Lut2[M) Cd, Zn; R ) CH3, C(CH3)3; Lut ) 3,5-Dimethylpyridine (Lutidine)]. Inorg. Chem. 36 (10), 2218-2224 (1997).
- Cho, S. H., Hwang, S. J., Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 130 (29), 9254-9256 (2008).
- Ide, Y., et al. Spin-crossover between high-spin (S = 5/2) and low-spin (S = 1/2) states in six-coordinate iron(III) porphyrin complexes having two pyridine-N. oxide derivatives. Dalton Trans. 46 (1), 242-249 (2017).
- Drago, R. S. Physical Methods in Chemistry. , Saunders College Publishing USA. 750 (1977).
- Cervantes-Mejía, V., et al. Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies. American Journal of Analytical Chemistry. 5 (16), 1090-1101 (2014).
- Huheey, J. E., Keiter, E. A., Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th Edition. , Oxford University Press. Mexico. ISBN: 9706131620 1023 (1997).
- Rigaku, CrysAlisPRO. , (2013).
- Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. A short history of SHELX. Acta Cryst. 64 (1), 112-122 (2008).
- Macrae, C. F., et al. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 41 (2), 466-470 (2008).
- ChemBioDraw Ultra 13. , PerkinElmer. (2013).
