

Summary
Ici, nous rapportons la synthèse et cristallisation de 3,5-lutidine N -oxyde déshydratent par un protocole simple qui diffère de la synthèse classique de la pyridine N-oxyde. Ce protocole utilise des matériaux différent et comporte moins de temps pour donner une nouvelle structure supramoléculaire solvatés, qui cristallise par évaporation lente réaction.
Abstract
La synthèse de N -oxyde de 3,5-lutidine déshydrater, 1, ont été accomplis dans la voie de synthèse d’acide 2-amino-pyridine-3, 5-dicarboxylique. Ochiai a tout d’abord utilisé la méthodologie pour les pyridines substituées non en 1957 dans un processus de 12 h, mais aucun cristaux adapté aux rayons x ont été obtenus. Le cycle substitué utilisé dans la méthodologie présentée ici clairement influencé l’addition de molécules d’eau dans l’unité asymétrique, ce qui lui confère une force nucléophile différente dans 1. Les rayons x cristal approprié composé 1 a été possible grâce à la stabilisation de la charge négative de l’oxygène par la présence de deux molécules d’eau où les atomes d’hydrogène donnent charge positive dans l’anneau ; ces molécules d’eau servent bien à construire une interaction supramoléculaire. Les molécules hydratés peuvent être possibles pour le système alcalin qui intervient en ajustant le pH à 10. Ce qui est important, la double méthyle substitué anneau et un temps de réaction de 5 h, en fait une méthode plus souple et avec des applications chimiques plus larges pour les insertions future bague.
Introduction
De nos jours, les scientifiques du monde entier ont investi des ressources dans le développement de nouvelles voies de synthèse pour la fonctionnalisation des groupes aromatiques, qui sont connus pour front faible réactivité à l’addition des réactions1,2, 3. Pyridine, où un atome d’azote remplace un atome de carbone, présente une réactivité chimique semblable à analogique bagues composées uniquement d' atomes de carbone3et elle subit généralement un mécanisme de substitution plutôt que l’addition. N-oxydes sont distinguent par la présence d’un donneur de liaison entre l’azote et l’oxygène formé par la superposition de la paire d’électrons non liése sur l’azote avec une orbitale vide sur l’atome d’oxygen3. En particulier, la pyridine N-oxydes sont des bases de Lewis, parce que leur portion de N-O peut agir comme un donneur d’électrons, et ils peuvent combiner avec les acides de Lewis formant les paires d’acide-base de Lewis correspondantes. Cette propriété a une conséquence chimique essentielle, car il peut augmenter le caractère nucléophile des acides de Lewis vers électrophiles potentiels et ainsi leur permettre de réagir dans des conditions où normalement la réaction n’aurait pas lieu. L’utilisation plus fréquente de ces composés est probablement dans les réactions d’oxydation différents où ils agissent comme anti-oxydants4. Pyridine N-oxydes et plusieurs de leurs dérivés fonctionnalisés anneau sont des molécules récurrentes d’agents biologiquement actifs et pharmacologiques5, et établie une claire répartition par différents outils spectroscopiques pour certains d'entre eux6,7. Dans la recherche sur la fixation des différents groupes à l’anneau de la pyridine, les scientifiques ont testé diverses méthodes pour créer une méthode simple et conventionnelle, puisque isoxazolines nécessite une quantité catalytique de base comme la DBU en ébullition xylène pour former 6- par ce qui suit-2-aminopyridine N-oxydes8,9. Une variété de dérivés de la pyridine ont été converties en leurs correspondants N-oxydes en présence d’une quantité catalytique de manganèse tetrakis(2,6-diclorophenyl) porphyrine acétate d’ammonium dans le CH2Cl2/CH3 CN8,10. Autres pyridines sont oxydés à leurs oxydes à l’aide de H2O2 en présence d’une quantité catalytique de methyltrioxorhenium8,11, ou par l’addition du diméthyldioxirane excès en CH2Cl2 à 0 ° C, ce qui conduit à la correspondante N-oxydes8,12,13,14. Bis (triméthylsilyl) peroxyde en présence de trioxorhenium dans la CH2Cl2 a été utilisé pour la synthèse de la pyridine N-oxyde8,11. La synthèse d’aminopyridine N-oxydes d’acylation impliquant à l’aide de l’acide de Caro (peroxomonosulfuric acid) a également été rapporté8. Néanmoins, la méthodologie présentés ici, et qui utilise une partie de la méthodologie par Ochiai1, fournit de très bons résultats avec l’utilisation de réactifs moins chers et accessibles, de H2O2 et de l’acide acétique glacial. Cette pratique est plus appropriée pour une utilisation dans des préparations de grande échelle qui agissent sur les amines tertiaires, il produit des bons rendements dans une réaction qui ne nécessite que 30 % de peroxyde d’hydrogène et acide acétique glacial à une température comprise entre 70-80 ° C et il utilise un procédé de purification qui est disponible dans la plupart des laboratoires de synthèse comme la distillation, sans l’utilisation de catalyseur ou de réactifs plus cher1. La littérature rapporte qu’autres méthodologies impliquent souvent des délais de 10 à 24 h et des températures supérieures à 100 ° C 4,8, et le rendement des cristaux bien formés pour les analyses de rayons x est rarement rapporté.
Réactive, divers dérivés de N -oxyde sont utilisés pour activer correctement la bague lutidine, d’une manière ou l’autre nucléophile ou électrophile. Le facteur nucléophile ou électrophile est affecté par les substituants. Avec l’anneau de pyridine étant les groupes électroattracteurs, le principal facteur est la caractéristique nucléophile1. La libre N-oxydes sont rarement isolés sous forme de cristaux approprié pour analyse aux rayons x en raison de la charge délocalisée de l’anneau aromatique. Toutefois, le facteur de solvatation est critique pour stabiliser la densité négative de l' oxygène15.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. réaction
- Mettre dans une hotte de laboratoire un flacon ouvert rond 100 mL avec 0,5 mol (29,8 mL) d’acide acétique glacial et 0,051 mol (5,82 mL) de 3, 5-diméthylpyridine et 5 mL de H2O2 (35 %). Garder la réaction du mélange en agitant magnétique, à une température interne de 80 ° C pendant 5 h.
- Après le temps de réaction, refroidir le ballon à 24 ° C avec de la glace (do pas exposer l’acide acétique de gaz sur la glace) et branchez-le à une unité de distillation sous vide élevé pendant 90 à 120 minutes pour enlever l’excès d’acide acétique.
ATTENTION : N’utilisez pas de matériel chaud. Attendez que la verrerie atteint une température facile à gérer. Ceci évitera aussi des vapeurs entrant dans le haut de l’appareil de distillation. - Ajouter distillée (10 mL) d’eau deux fois pour assurer l’élimination de toute trace d’acide acétique et de concentrer le mélange beaucoup que possible.
2. extraction et ajustement de basicité
- Dissoudre dans de l’eau bi-distillée produit visqueux et transparent isolé et utiliser un potentiomètre pour ajuster le pH à 10 avec pure solid Na2CO3.
- Placez délicatement la solution dans une ampoule à décanter 250 mL et décompressez-le 5 fois avec 250 mL de CHCl3 afin d’améliorer le rendement. Récupérer la couche organique et séchez-le sur solide Na2SO4 pendant 30 min maximum, qui contiendra le produit. Si nécessaire, re-extraire la phase aqueuse avec la quantité désirée de CHCl3.
ATTENTION : CHCl3 peut provoquer somnolence et vertiges ; manipuler avec soin et à l’intérieur d’une hotte aspirante. - Éliminer le solvant sous pression réduite avec une unité de distillation sous vide élevé, jusqu'à la formation d’une très hygroscopique poudre cristalline claire beige (70 %).
3. processus de cristallisation
- Dissoudre 4,3 g de la poudre cristalline dans 50 mL d’éther diéthylique de froid haute effectué chromatographie en phase liquide (HPLC) grade. La solution pour supprimer toute trace de matière solide ou même poussière filtre à vide. Versez le filtrat dans un verre de Pétri, laissant à ralentir s’évaporent à 4 ° C dans un réfrigérateur de laboratoire.
- S’assurer que, après deux jours, on obtient des cristaux incolores clair. Mesurez ensuite le point de fusion, qui devrait être de l’ordre de 310 et 311 K.
4. analyse des 3,5-Lutidine N -oxyde Dehydrate
- Enlever les cristaux qui sont forment, de forme prismatique et incolore, par décantation des murs de la fiole pour analyse aux rayons x. Si pas immédiatement utilisé, gardez les cristaux dans l’éther diéthylique pour éviter hydratation de cristal.
- Dissoudre 0,010 g de N -oxyde de 3,5-lutidine déshydrater à 0,4 mL de CDCl3 H NMR1 et C13 analyse de prouver l’efficacité de la procédure.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Le protocole est essentiellement une extension de Ochiai technique1. Cependant, une température plus basse et moins de temps sont appliquées. Cette méthode simple permet d’obtenir un ligand polyvalent, qui est un substitués pyridine N-oxyde dérivé. Pour confirmer la formation du 1, 13C analyse NMR 1H et sont préférables pour tester l’efficacité de la procédure.
Le déplacement chimique montre la formation de 1. Le signal à 2,28 ppm (parties par million) correspond à six hydrogènes équivalentes des deux groupes méthyles en positions 3 et 5, qui perçoivent le champ magnétique en moins proportion que le magnétique permanent. Il existe deux ensembles de septuplets : l’un appartient à la proton en position c à 7,9, qui double la taille de l’autre signal à 6,9 qui appartient à la proton dans la position a. Figure 1 montre les déplacements chimiques provoquées par la présence de l’oxygène atome bonde d à l’atome d’azote du noyau pyridine. L’atome d’oxygène est électro-affinitaires et les atomes d’hydrogène plus étroites à l’atome d’oxygène (c et a) Voir la cylindrée à une fréquence plus élevée que celle pour les hydrogènes de méthyle (b).
Le même processus est tracé pour le spectre RMN de 13C, Figure 2, où les signaux pour les carbones plus étroites à l’atome d’oxygène (c et a) montrent séparation de fréquence entre leurs signaux de Δc = 1 300 Hz et Δun = 200 Hz. Une fois de plus, les carbones de méthyle ne montrent aucun changement. Le spectre infrarouge permet de voir le succès de la méthode aussi bien.
Le diagramme ORTEP, Figure 4, montre la présence de deux molécules d’eau entourant la molécule asymétrique. Ces molécules sont censés stabiliser la liaison N-O. Dans des cas similaires, il a été décrit pour la pyridine N-oxyde et oxydes aromatiques liés. Il y a une importante stabilisation de type π O→N dos-don, consignées dans un ordre de liaison calculée supérieur à 1 et un certain nombre de doublets d’électrons sur l’atome d’oxygène inférieur à 36.
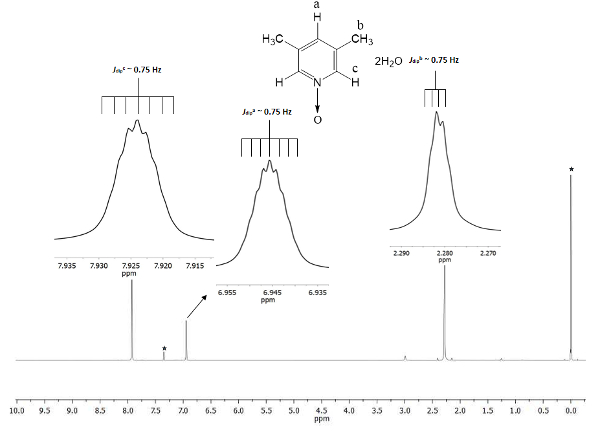
Figure 1 . TMS référencé CDCl3 spectre de 1H RMN 500 MHz de 1. Les intégrations et les déplacements chimiques des trois signaux d’accord avec trois différents types d’atomes d’hydrogène présents dans la lutine N-oxyde. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
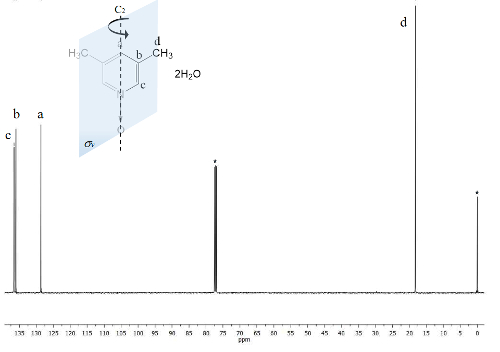
Figure 2 . TMS référencé CDCl3 100 MHz NMR 13C du spectre de 1. Trois signaux sont observés pour les cinq carbones aromatiques et une pour deux groupes méthyles. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
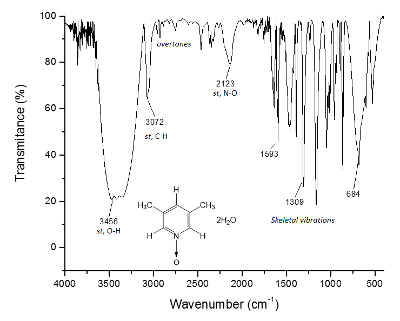
Figure 3 . Spectre IR du 1. Le O-H d’obligations, surtout de 3 300 cm-1, sont responsables de la formation des structures supramoléculaires et la formation de cristaux. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 4 . Diagramme ORTEP où deux molécules de formes de H2O pont des liaisons hydrogène avec l’oxygène de lutidine, conduisant leurs atomes d’hydrogène vers l’atome d’oxygène de 1. Ce chiffre a été modifié par Merino García et al. 12 S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 5 . Photos de cristaux de diffraction des rayons x adapté de 1 dans l’éther diéthylique (en haut) et au plein air (en bas). L’un de ces cristaux a été confirmé dans le diffractomètre de rayons x et ont montré un chemin d’accès de diffraction des rayons x, qui a été calomniée et affinée par des programmes informatiques spéciaux dans une structure moléculaire et cristalline24,25, 2627,de,28. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Le protocole présenté ici est une méthode conventionnelle pour lier un atome d’oxygène à l’atome d’azote de la 3, 5-lutidine comme une méthode de fonctionnalisation de substrats. Cette technique est également bien établie pour donner des cristaux déshydratés adapté de rayons x (Figure 5, photos prises avec un appareil photo Sony Cybershot DSC-HX300). Car ce qui nous concernés, pas beaucoup de rapports ont décrit la production de ces cristaux16. De nombreux composés croissance de cristaux idéal pour analyse aux rayons x lorsqu’ils sont chélatés par divers métaux17,18,19,20. Une fois la poudre cristalline est formé, il est important pour l’extraire de leurs liqueurs mères à l’aide d’une fiole de Kitasato et d’un entonnoir de Buchner. À l’aide de tuyaux en caoutchouc, la fiole de Kitasato est connectée à une ligne vide et couronner l’entonnoir Buchner est placé avec un filtre en papier. Une fois que le vide a été activé, le papier filtre est imprégné d’une petite quantité de solvant d'où le produit cristallisé. Cela empêche la poudre cristalline de goutte à goutte dans l’entonnoir de Buchner par l’effet de vide. Après s’être assuré le papier filtre, la solution contenant la poudre cristalline est secouée pour faire en sorte que toute la poudre cristalline est filtré, et n’en reste aucun dans le fond du flacon. La solution est rapidement versée sur l’entonnoir Buchner. La poudre cristalline obtenue est laissée pendant environ 10 min sur le papier filtre et puis le vide est éteint et la poudre cristalline est détaché du papier et stockée dans un flacon en verre opaque, étiqueté avec son code et conservé à 4 ° C jusqu'à une analyse plus approfondie. Le liquide filtré est versé dans un verre de Pétri, laissant l’évaporation lentement à 4 ° C afin d’améliorer la formation de cristaux adéquat pour l’analyse aux rayons x.
Il est important de noter que ce protocole utilise des solvants et des matériaux qui sont faciles à obtenir et sont généralement trouvés à n’importe quel laboratoire de recherches. L’ajustement du pH par l’ajout de Na2CO3 et de l’agitateur magnétique cohérente est essentielle pour le rendement du produit final. Cependant, il est important de faire attention très prudente dans toutes les étapes du processus, notamment dans la phase d’extraction où aucune trace de la matière première ne doit être présent pour permettre la formation de cristaux poudre cristalline et par la suite. Ainsi, cette étape d’extraction/purification peut être surveillée par spectroscopie NMR ou IR pour assurer la qualité du produit.
Afin d’assurer la reproductibilité du présent protocole, la RMN est un excellent outil. Même les petits détails sont visibles dans le spectre. Tous les signaux sont affichés sous forme d’encarts dans la Figure 1. Ces encarts représentent clairement une scission, à savoir la multiplicité, de tous les signaux. Par exemple, le b de protons (trempettedeJb ~ 0,75 Hz) montrent quatre pointes à la crête du signal, avec une séparation entre eux (Δcrête-crête) nausée constante de ~ 0,0075 ppm. Le 0,0075 ppm peut être transformé en énergie en utilisant le suivant équation21
 Équation 1
Équation 1

La transformation est recommandée parce que les signaux qui se déroulent proviennent de l’interaction dipolaire spatiale entre les noyaux de trois atomes d’hydrogène du groupe méthyle, et même s’ils sont plus loin que les 4 liaisons simples avec des protons c et a, ils sont capables de percevoir leur moment magnétique dipolaire interactions22. En outre, le sigma libre rotation du groupe méthyle de liaison permet l’interaction de proton-proton de super hyperfine soit visible dans la multiplicité du signal. Les septuplets de protons a et c à 6,9 et 7,9 ppm, respectivement sont issues de ce même phénomène de nature dipolaire. Dans ces cas, les protons un et c peut différencier les protons dans le groupe de méthyle pour la même rotation dynamique. Enfin, comme prévu, calculé Jdip pour a, b, et c ont à peine la même valeur, ~ 0,75 Hz. Ces quantités des interactions confirment l’arrangement spatial de noyaux d’hydrogène tout au long de l’anisotropie magnétique.
En revanche, la symétrie C2v 1 rend carbones équivalent23. Le spectre de 13C, Figure 2, montre le signal typique pour les groupes méthyle attaché à des noyaux aromatiques, carbones d 18 ppm. En outre, un signal ppm 129 est visible à cette région en raison de l’élément influencé carbone moins électronégatif une. À haute fréquence du signal pour les noyaux d’atomes de carbone plus exposés au champ magnétique sont présentés à 137 ppm22.
La méthodologie présentée est très utile pour la synthèse de la pyridine N-oxyde, offrant de bons rendements, dans un délai raisonnable avec conditions réactionnelles souples et réactifs accessibles bon marchés et faciles, ne nécessitant pas de catalyseurs supplémentaires. Ces conditions permet d’obtenir un large éventail de pyridines N-oxydes comme précurseurs pour d’autres molécules d’intérêt pour la communauté scientifique et éducative. La méthodologie appropriée donne la possibilité d’acquérir les outils expérimentaux et conceptuels de base dans les laboratoires d’enseignement pour les étudiants, prouvant une synthèse réussie des composés et le bonheur de voir la formation de cristaux. Toutefois, il est important de souligner que, comme toute réaction chimique, il est nécessaire de prendre toutes les précautions car les réactifs utilisés sont généralement dangereux.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Tous les auteurs ne déclarent aucun conflit d’intérêts.
Acknowledgments
Le présent travail a été soutenu par Vicerrectoría de Investigación y Estudios de Posgrado de BUAP, Divulgation de la Science et projets no REOY-NAT14, 15, 16.-G HEAS-NAT17. RMG Merci CONACyT (Mexique) pour la bourse 417887.
Materials
| Name | Company | Catalog Number | Comments |
| 3,5-lutidine | Sigma-Aldrich | L4206-500ML | |
| Glacial acetic acid | Fermont | 3015 | |
| Hidrogen peroxide (35%) | Sigma-Aldrich | 349887-500ML | |
| Na2CO3 anhydrous | Productos Químicos Monterrey | 1792 | |
| Na2SO4 anhydrous | Alfa reactivos | 25051-C | |
| CHCl3 | Fermont | 6205 | |
| Ethyl eter | Mercury Chemist | QME0309 | |
| Distilled water | Comercializadora Química Poblana | not-existent |
References
- Ochiai, E. Recent Japanese work on the chemistry of pyridine 1-oxide and related compounds. J. Org. Chem. 18 (5), 534-551 (1953).
- Solomons, T. W. G. Organic Chemistry 2nd Edition. , John Wiley & Sons. 1110 (1976).
- Albini, A., Pietra, S. Heterocyclic N-Oxides. , CRC Press. ISBN: 0849345529 328 (1991).
- Koukal, P., Ulc, J., Necas, D., Kotora, Heterocyclic N.-Oxides. Topics in Heterocyclic Chemistry. 53, 29-58 (2017).
- Wen-Man, Z., Jian-Jun, D., Xu, J., Jun, X., Huan-Jian, X. Visible-Light-Induced C2 alkylation of pyridine N.-oxides. J. Org. Chem. 82 (4), 2059-2066 (2017).
- Merino García, M. R., Ríos-Merino, F. J., Bernès, S., Reyes-Ortega, Y. Crystal structure of 3,5-dimethylpyridine N-oxide dihydrate. Acta Cryst. 72 (12), 1687-1690 (2016).
- Sarma, R., Karmakar, A., Baruah, J. B. N-Oxides in Metal-Containing Multicomponent Molecular Complexes. Inorg. Chem. 47 (3), 763-765 (2008).
- Youssif, S. Recent trends in the chemistry of pyridine N-oxides. ARKIVOC. 2001, 242-268 (2001).
- Chucholowski, A. W., Uhlendorf, S. Base catalyzed rearrangement of 5-cyanomethyl-2-isoxazolines; novel pathway for the formation of 2-aminopyridine N-oxides. Tetrahedron Lett. 31 (14), 1949-1952 (1990).
- Thellend, A., Battioni, P., Sanderson, W., Mansuy, D. Oxidation of N-Heterocycles by H2O2 Catalyzed by a Mn-Porphyrin: An Easy Access to N-Oxides Under Mild Conditions. Synthesis. 1997 (12), 1387-1388 (1997).
- Copéret, C., Adolfson, H., Tinh-Alfredo, V. K. h, Yudin, A. K., Sharpless, K. B. A simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 63 (5), 1740-1741 (1998).
- Ferrer, M., Sánchez-Baeza, F., Messeguer, A. On the preparation of amine N-oxides by using dioxiranes. Tetrahedron. 53 (46), 15877-15888 (1997).
- Adam, W., Briviba, K., Duschek, F., Golsch, D., Kiefer, W., Sies, H. Formation of singlet oxygen in the deoxygenation of heteroarene N-oxides by dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1995 (18), 1831-1832 (1995).
- Murray, R. W., Singh, M. A Facile One-Step Synthesis of C-Arylnitrones Using Dimethyldioxirane. J.Org.Chem. 55 (9), 2954-2957 (1990).
- Kim, S. W., Um, T., Shin, S. Brønsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides. Chem. Commun. 53 (18), 2733-2736 (2017).
- Campeau, L., Rousseaux, R., Fagnou, K. A solution to the 2-pyridyl organometallic cross-coupling problem: regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 127 (51), 18020-18021 (2005).
- Gang, L., et al. Metal-free methylation of a pyridine N-oxide C-H bond by using peroxides. Org. Biomol. Chem. 13 (46), 11184-11188 (2015).
- May, D., Nyman,, Hampden-Smith, M. J., Duesler, E. N. Synthesis, characterization, and reactivity of group 12 metal thiocarboxylates M(SOCR)2Lut2[M) Cd, Zn; R ) CH3, C(CH3)3; Lut ) 3,5-Dimethylpyridine (Lutidine)]. Inorg. Chem. 36 (10), 2218-2224 (1997).
- Cho, S. H., Hwang, S. J., Chang, S. Palladium-Catalyzed C-H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc. 130 (29), 9254-9256 (2008).
- Ide, Y., et al. Spin-crossover between high-spin (S = 5/2) and low-spin (S = 1/2) states in six-coordinate iron(III) porphyrin complexes having two pyridine-N. oxide derivatives. Dalton Trans. 46 (1), 242-249 (2017).
- Drago, R. S. Physical Methods in Chemistry. , Saunders College Publishing USA. 750 (1977).
- Cervantes-Mejía, V., et al. Branched Polyamines Functionalized with Proposed Reaction Pathways Based on 1H-NMR, Atomic Absorption and IR Spectroscopies. American Journal of Analytical Chemistry. 5 (16), 1090-1101 (2014).
- Huheey, J. E., Keiter, E. A., Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th Edition. , Oxford University Press. Mexico. ISBN: 9706131620 1023 (1997).
- Rigaku, CrysAlisPRO. , (2013).
- Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71 (1), 3-8 (2015).
- Sheldrick, G. M. A short history of SHELX. Acta Cryst. 64 (1), 112-122 (2008).
- Macrae, C. F., et al. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 41 (2), 466-470 (2008).
- ChemBioDraw Ultra 13. , PerkinElmer. (2013).
