

Summary
利用预装 Cas9 ribonucleoprotein 复合体 (RNP) 是精确、高效的基因组编辑的有力方法。在这里, 我们强调它的效用横跨广泛的细胞和有机体, 包括主要的人类细胞和经典和新兴的模型有机体。
Abstract
站点特定的真核基因组编辑与 CRISPR (聚集定期 interspaced 短回文重复)-Cas (CRISPR 相关) 系统已迅速成为一个普遍的研究人员寻求广泛的各种生物学问题。用户最常使用从链球菌化脓派生的 Cas9 蛋白, 并具有易于重新编程的指南 RNA (gRNA)。这些成分被引入细胞, 并通过与双链 DNA (dsDNA) 基因组的互补区域的基础配对, 该酶将两股形成双链断裂 (争端处理)。随后的修复将导致随机插入或删除事件 (indels), 或将实验者提供的 DNA 纳入断点地点。
使用纯化的单导 RNA 和 Cas9 蛋白, 预装形成 RNP 并直接传递给细胞, 是实现高效基因编辑的有效途径。RNP 编辑特别提高了基因插入的速度, 这一结果往往是难以实现的。与通过质粒进行传递相比, 细胞内 Cas9 RNP 的持续时间越短, 就会导致离靶事件减少。
尽管它的优势, 许多 CRISPR 基因编辑的休闲用户不太熟悉这种技术。为了降低进入门槛, 我们概述了在一系列背景下实施 RNP 战略的详细协议, 突出了其独特的优点和不同的应用。我们涵盖了两种类型的主要人类细胞, T 细胞和造血干细胞/祖细胞 (HSPCs) 的编辑。我们还展示了 Cas9 RNP 编辑如何使整个有机体的简单的基因操作, 包括经典模型蛔虫秀丽线虫和最近引入的模型甲壳类, Parhyale hawaiensis。
Introduction
fThe CRISPR-Cas9 系统允许科学家改变任何基因组的目标区域1。这种快速而廉价的技术革命性地进行了基础研究, 并承诺对个性化疾病治疗、精准农业和超过2的发展产生深远的影响。CRISPR 编辑是一种民主化的工具, 在一个新的实验室中实施该系统并不需要在基因组工程学方面的专门知识, 仅仅是基本的分子生物学技能。研究人员现在可以用一些替代基因操作的方法来研究以前顽固的有机体3,4。仅在过去的五年里, CRISPR 基因组编辑已经被用来设计超过200种不同的脊椎动物、无脊椎动物、植物和微生物物种。
根据 CRISPR 原核防御通路, 特定于地点的基因组编辑所需的核心元素是 Cas9 蛋白, 通常来自化脓和密码子-通过增加的核定位信号 (NLS) 进行优化, 其专门RNA 指南5,6。虽然这里没有讨论, 其他 Cas9 orthologues 或 CRISPR 内切酶也可能被使用。自然发生的 gRNA 由两个分开转录的片断、CRISPR RNA (crRNA) 和反激活 crRNA (tracrRNA)7组成。这些 rna 可以融合成一个单一的转录, 称为单导 RNA (sgRNA)8。大多数基因组编辑器都选择了流线型的 sgRNA9, 尽管双导则也经常使用10,11。实验者选择20核苷酸 (nt) 基因组 DNA 目标, 确保它位于 Cas9 识别所需的短授权签名旁边, 称为 protospacer 相邻的母题 (PAM), 并设计一个包含互补序列的 gRNA12.
一旦细胞内, RNP 复合体定位其基因组目标, gRNA 基对与互补 dna 链, 然后酶的两个 dna 链, 以产生双链断裂2。细胞修复机械通过至少两条路线之一修复了争端解决机构: 通过容易出错的非同源的端接 (NHEJ) 通路或同源定向修复 (HDR), 它无缝地将含有 "双臂" 的 DNA 包含在断裂的两边。前者的修复途径通常导致 indel 形成和随后的基因中断, 而后者则允许实验者插入或改变 DNA 序列1。
编辑效率和准确性取决于 Cas9 和 gRNA 进入单元格的方法。这些组件可以以核酸的形式传递给培养的细胞、胚胎或有机体, 或者作为预装 RNP 复杂的13、14、15。常见的核酸的传递方法包括病毒转导, 转染, 或电穿孔的 mRNA 或质粒 DNA。Cas9 蛋白和导 RNA 在细胞内产生, 它们联想形成一个复合体。
RNP 的直接传递需要分离纯化 Cas9 蛋白和引导 RNA。这可以在内部做, 或者蛋白质和 sgRNA 可以从几个商业供应商之一购买。一旦获得, Cas9 和 gRNA 混合形成酶能力的 RNP 复杂, 并引入细胞直接注射到受精卵/胚胎, 脂质为基础的转染16, 或电穿孔。RNP 编辑的第一个报告涉及注入C. 线虫性腺17。微注射仍然是将 RNP 引入胚胎和整个生物体的首选方法, 尽管在鼠标18、19和鼠20胚胎中已经显示有效的电穿孔。我们描述直接将 RNP 注入线虫性腺和P. hawaiensis胚胎的协议, 并推荐一种特殊类型的电穿孔, 用于在编辑主要人体细胞时提供 RNP。这种方法, nucleofection, 涉及优化的电穿孔程序和细胞类型特定的解决方案, 并允许 RNP 进入细胞质和核的21。
基因组编辑与 RNP 提供了几个明显的优势。由于蛋白质和 RNA 成分是预先组装, 质量可以确保在交付之前, RNP 编辑避免了许多陷阱与核酸的交付。也就是说, 没有 Cas9-encoding DNA 整合到宿主基因组的风险, mRNA 永远不会被暴露为退化, 它绕过了体内gRNA 或蛋白质表达、折叠和关联22、23的问题。此外, 使用 RNP 导致毒性降低, 离目标事件远少于以质粒为基础的表达式, 这是由于 RNP 在单元格中的半衰期更短的半生命周期24,25,26,27。
最后, RNP 编辑可明显导致在各种人类细胞系的高编辑率, 主要细胞, 如成纤维细胞, 胚胎干细胞 (ESCs), 诱导多能干细胞 (iSPCs), HSPCs 和 T 细胞16,24, 25,26,27,28,29;在无脊椎动物, 包括线虫, P. hawaiensis, 果蝇3,17,30;如斑马鱼、老鼠和老鼠的脊椎动物,31,32;植物种类包括拟南芥、烟草、生菜、大米、葡萄、苹果、玉米和小麦33、34、35、36;在衣藻、青霉和念珠菌种类37,38,39。当使用 RNP 与质粒传递相比, indel 形成的频率会更高, 而 HDR 介导的 DNA 插入可以更容易实现25,27,29。
此处描述的协议使用 Cas9 RNP, 是一种有效的、易于适应的技术, 可直接应用于各种生物系统40、41, 特别是在其他难于工作的单元格中。与和在有机体, 不用完善的系统为精确基因操作。我们首先描述如何设计、获取和组装 Cas9 RNP, 然后再覆盖其在不同模型细胞类型和有机体中的使用。造血干/祖细胞 (HSPCs) 和 T 细胞的编辑使用相同的方法, nucleofection, 因此, 它们被覆盖在该协议的步骤2和3。编辑C的过程。线虫在步骤4和5以及P中进行描述。hawaiensis编辑包括在步骤6和7中。最后, 由于任何有机体的基因编辑实验的成功都可以通过基因型测序来评估, 子步骤描述了《议定书》中所描述的所有细胞和有机体可能的分析方法, 步骤8中概述了这一点。
Protocol
1. RNP 总成
-
提前做好实验设计, 提前获取所有的 RNA、DNA 和蛋白质成分。作为第一个通行证, 尝试在表1中列出的积极控制之一, 并使用材料表中描述的商业试剂, 以确保可靠的实验设计和材料的完整性。有关规划新的基因组编辑实验的其他提示, 请参阅本主题的论文12,42,43。
注: 一旦按照后续步骤进行组装, 预先准备好的 RNPs 可以存储在-80 摄氏度。- 选择目标的基因后, 使用一个免费的在线工具设计最佳 gRNA44,45,46,47,48。一定要瞄准一个外显子, 如果希望产生一个淘汰赛。
注意: 这些工具将有助于识别具有相邻化脓PAM 序列、高质量评分和低目标得分的目标站点。 - 通过已发布的方法8纯化化脓Cas9 蛋白, 或从商业供应商购买它。
- 准备一个典型的 Cas9 缓冲, 为 RNA 稀释, RNP 准备和蛋白质储藏, 其中包含20毫米 HEPES pH 值 7.5, 150 毫米氯化钾, 10% 甘油和1毫米 TCEP。总是使用核酸酶的水在缓冲, 将用于并用重悬或稀释 RNA, 以防止退化。
- 通过使用已发布方法的体外转录生成导 RNA (tracrRNA 和 crRNA 或 sgRNA), 或从核酸合成公司 (17,21,49, ) 中购买它。50,51。
- 如果插入基因, 合成或购买捐赠者 DNA 模板。
- 储存蛋白质和 RNA 整除数在-80 摄氏度和解冻后立即在冰上使用。
注: 每次冻融略有降低效率。Cas9 纯化52和体外转录 sgRNAs53的详细的开放访问协议在别处可用。
- 选择目标的基因后, 使用一个免费的在线工具设计最佳 gRNA44,45,46,47,48。一定要瞄准一个外显子, 如果希望产生一个淘汰赛。
- 如果使用C. 线虫, 请跳到步骤1.5。对于hawaiensis协议, 请跳到步骤1.6。如果使用 sgRNA, 请跳到步骤1.4。继续步骤 1.3, 为主单元格编辑装配 gRNA。
-
通过混合摩尔数量的 tracrRNA 和 crRNA 来组装 gRNA。制作100µL 80 µM gRNA 股票, 约50个基因组编辑实验。
- 孵化的 gRNA 在37°c 30 分钟, 然后让它慢慢冷却到室温。
-
RNP 准备 HSPC 和 T 细胞编辑: 通过混合 1-2x 摩尔量 gRNA 到 pmol 蛋白质的总容积 10 Cas9, 组装一个 RNP 复合体. 非常缓慢, 添加集中µL Cas9 (预稀释在 gRNA 缓冲) 约三十年代, 用吸管快速绕圈, 使最终的 Cas9 浓度达到20µM。
- 准备电穿孔小试管。
注意: 此协议特定于材料表中提到的商业系统, 但 RNP 编辑也可以与其他电穿孔设备一起完成。 - 添加5µL (100 pmols, T 细胞) 或10µL (200 pmol, HSPCs) RNP 到每个试管。
- 如果插入新的 DNA 而不是做一个挖空, 添加1µL 100 µM (100 pmol) 单链寡核苷酸捐献者 DNA (ssODN)25,54,55到小试管或水井的板块。
- 对于主单元格编辑协议中的下一个指令, 请跳到步骤 2 .
- 准备电穿孔小试管。
-
RNP 准备C. 线虫编辑: 通过添加以下试剂来组装 RNP 复合体, 以创建最终体积为20µL (最后浓度在括号中指出): Cas9 (2 µM), HEPES pH 7.5 (10 µM), 氯化钾 (115 µM), crRNA (12 µM)、tracrRNA (40 µM) 和修复模板 (如果需要) (0.5 µM ssDNA 或最多 350 ng/µL dsDNA)。
注: Cas9-mediated 的模板修复的效率与 dsDNA 修复结构的浓度成正比;因此, 修复模板的浓度越高, 模板修复的效率就越低。然而, 已证明注射含有大于350µL 的 dsDNA 的混合物可以降低注射蠕虫的生存能力。因此, 最好使用最多, 但不超过350µL 的 dsDNA 在混合, 以最大限度地提高维修效率, 同时尽量减少其杀伤力。- 根据步骤5.4 中描述的共同 CRISPR/联合转换筛选方法, 在需要时添加多个 crRNAs 以同时针对多个定位点。添加多个 crRNA 时, 将每个顺序添加到主组合中。
注: 每个 crRNA 的数量不需要相同, 甚至加倍 crRNAs 的总浓度, 而不改变 Cas9 的浓度, 似乎不会干扰在特定的部位突变的频率。示例在et中详细描述。56。 - 混合吹打和旋转的 RNP 溶液在 1.6万 x g 5 秒, 以确保解决方案收集在底部的管。
- 孵化溶液在37°c 为15米。
- 离心样品在 1.6万 x g 1 分钟, 以颗粒任何可能堵塞薄钻孔注射针的微粒。在后续步骤中使用上清。
- 跳到步骤 4, 以执行C. 线虫协议的其余部分。
- 根据步骤5.4 中描述的共同 CRISPR/联合转换筛选方法, 在需要时添加多个 crRNAs 以同时针对多个定位点。添加多个 crRNA 时, 将每个顺序添加到主组合中。
-
RNP 准备P. hawaiensis编辑: 使用核酸酶水和苯酚红 (用于可视化注射) 稀释6.25 µM 和0.15% 苯酚红色的最后浓度, 制备单用途 Cas9 整除数。
- 通过将2-5x 摩尔过量的 gRNA 混合到 Cas9 蛋白质中, 组装 RNP 复合体, 总容积为6µl. 将 pmol 的 12 Cas9 添加到 gRNA, 使最终 Cas9 浓度达到2µM, gRNA 浓度为 4-8 µM, 苯酚红浓度为0.05%。
- 在室温下孵育混合物10分钟, 使 RNP 复杂。
- 跳到步骤 6, 以执行hawaiensis编辑协议中的下一个指令.
2. 细胞培养和制备
注意: 在生物安全柜中执行2.1.1 到3.3.3 的步骤。
-
从供应商处购买冻存的人工外周血 CD34+ HSPCs。
- 解冻 ~ 1 x106 HSPCs 在37°c 水浴为3分钟并且转移他们到15毫升圆锥管。添加10毫升的无血清膨胀培养基从商业来源和旋转的混合物在 100 x g 为10分钟. 移除上清和并用重悬在2毫升补充随机有限元的细胞。在 RNP 电穿孔前, 将细胞在6井板中, 并在37摄氏度孵化箱中培养成24-48 小时。
- 用 hemocytometer 计数单元格, 并将所需的 HSPCs 总数 (每试管 1.5亿 HSPCs 转) 转换为离心管。
- 旋转管在 100 x g 10 分钟, 以颗粒细胞。
-
从供应商处购买人类主 CD4+ T 单元格, 或者通过密度梯度离心29将它们从人的全血中分离出来。
- 在 T 细胞激活之前, 前涂层48井培养板材与αCD3 (UCHT1) 和αCD28 (CD28.2)。用500µL 10 µg/毫升αCD3 和10µg/毫升αCD28 在 PBS 上涂上板材, 至少2小时37摄氏度。
注意: 对于某些位点, NHEJ 可以在没有预刺激的情况下实现, 但包括这一步将最大化其效率。 - 培养 T 细胞为48小时在37°c 在αCD3 或αCD28 抗体束缚的板在 RPMI 完全媒介 [RPMI-1640 补充与5毫米 HEPES, 2 毫米的商业替代 l-谷氨酰胺, 50 µg/毫升青霉素/链霉素, 50 µM 2 巯基乙醇, 5 毫米的非必需氨基酸, 丙酮酸钠的5毫米, 和 10% (卷/卷) 的血清]。在 200万 t 细胞的密度上培养 t 细胞在500µL 的媒介每井的48井板材。
- 用 hemocytometer 计数 t 细胞并将电穿孔实验所需的 t 细胞总数转移 (每试管100,000-1、000,000 T 细胞转) 到离心管。
- 旋转管在 90 x g 8 分钟, 以颗粒细胞。如果细胞密度梯度-在2天内分离, 旋转他们在 200 x g 为8分钟。
- 在 T 细胞激活之前, 前涂层48井培养板材与αCD3 (UCHT1) 和αCD28 (CD28.2)。用500µL 10 µg/毫升αCD3 和10µg/毫升αCD28 在 PBS 上涂上板材, 至少2小时37摄氏度。
-
对于两种细胞类型, 用吸管/真空吸入上清液, 除去任何气泡。
- 每试管用20µL 的电穿孔缓冲器轻轻并用重悬细胞。
- 将20µL 的细胞 (1.5亿 HSPCs 或 100,000-1, 000,000 T 细胞) 添加到每个试管, 它已经包含了 RNP 的10µL, 并通过吹打上下混合而不产生气泡。
3. RNP 电穿孔
- Electroporate 把小试管放在 nucleofector 后。对于 HSPCs, 使用脉冲编码 ER100。对于 T 细胞, 使用脉冲编码 EH-115。
-
仅 HSPCs:在电穿孔后立即向每个试管添加100µL 的补充随机有限元培养基 (加热到37°c), 并让细胞恢复 10-15分钟。
- 转移细胞培养他们在一个96井的圆底板和增加另外100µL 补充的随机有限元培养基为 24 h。
- 将它们改成新鲜的补充随机有限元培养基, 再孵化 24-72 小时。
- 取出细胞进行基因分型48-96 小时后电穿孔。旋转细胞在 300 x g 5 分钟, 并删除上清之前开始的 DNA 提取 (步骤 8.2)。
-
T 细胞只: 添加80µL 的 RPMI 完全培养基预热到37°c 从水库到每试管或好, 使用多通道吸管 (如果需要)。
- 孵化他们在37°c 15 分钟。
- 将适当的培养基、抗体、细胞因子、等添加到目标板上, 并在37摄氏度孵化器中预热。
- 使用多通道吸管 (如有必要) 将107µL 的转细胞从油井转移到圆底96井板。
- 有关评估编辑结果的信息, 请跳到步骤8。
4.线虫制剂
-
注射前1天: 为微注射准备琼脂糖垫。
- 在水中加入琼脂糖, 使溶液在热板或微波炉中煮沸, 以 3% (w/v) 琼脂糖溶液。
- 在桌子上排列24毫米 x 50 毫米 x 1.5 毫米的玻璃幻灯片, 并使用玻璃巴斯德吸管将一个小 (~ 15 µL) 滴琼脂糖溶液放到滑梯上。通过将另一盖玻片放在上面, 快速地拼合琼脂糖滴。允许琼脂糖固化, 然后删除其中一个盖玻片。
- 把琼脂糖涂层的盖玻片脸放在桌上过夜晾干。24小时后, 将琼脂糖垫存放在干净、干燥的容器中。
注意: 这些可以无限期地使用。
- 拉注射针: 使用硅硅酸盐玻璃毛细血管与花丝 (外径1.0 毫米和内径0.58 毫米), 拉针基于梅洛和火57和其他资源58。针头可以立即使用, 也可以储存在一个干净的, 干燥的容器中, 由粘土支撑支撑。
- 为维护蠕虫, 准备一个线虫生长培养基 (NGM) 琼脂倒入培养板和发现与 OP50 细菌 (对于标准的C. 线虫维护和食谱的增长媒体, 请参见 Stiernagle59)。
- 对微注射的蠕虫进行12-24 小时的显微注射, 选择 L4-staged 雌雄同体到一个新的 NG 琼脂板上, OP50 细菌并在20摄氏度过夜。对于每个 Cas9 的目标/注射组合, 选择30蠕虫的盘子。
-
微注射日:使用步骤1.5 中准备的 RNP 溶液上清液加载拉入的微注射针.
- 将上清液吸管从步骤1.5.4 进入被拔出的毛细管吸管中, 将毛细管吸管中的溶液回填到所制备的微注射针头中 (通常加载少于0.1 µL)。
- 将载入的针装入附着在机器人上的微注射装置上。将注射器具压力设置为250帕, 平衡压力为25帕。
-
折断已加载的针尖, 产生锋利的针缘。放置一个15毫米 x 15 毫米 x 1.5 毫米正方形盖玻片在24毫米 x 50 毫米 x 1.5 毫米盖玻片的顶部。
- 覆盖正方形盖玻片的一边缘与卤化碳油700。
- 把针放在油中, 在15毫米盖玻片的边缘。
- 用手指导显微镜的舞台和盖玻片, 刷上和沿针的边缘, 同时压抑注射踏板/按钮。把针尖向后折断, 增加液体流出针的流量。通过使注射混合沿针边流动, 形成1气泡/s, 达到最佳流速。
- 证实 L4 蠕虫在注射前注射12-24 小时是发育阶段的年轻成年人。把幼虫带到一个缺乏 OP50 细菌的五琼脂盘子里, 让它们在5分钟内爬行。这可以减少转移到注射垫的细菌数量, 减少针头堵塞。
- 将琼脂糖注射垫/盖玻片放在解剖范围内。使用蠕虫采摘, 放置一个小轨道的卤化碳油沿一个边缘的垫。
-
利用在油中涂抹的蠕虫, 将几条蠕虫从五琼脂板上提起, 进入油的轨道。一个细的头发附着在吸管上, 如睫毛或猫晶须, 平行放置蠕虫, 轻轻地将蠕虫推入琼脂糖垫。在使用微注射的过程中, 一次只能装入一个蠕虫。
注: 干琼脂糖将从蠕虫的水分, 使他们坚持到垫。因此, 你必须迅速工作, 因为蠕虫可以脱水。- 一旦在位置和附加到垫, 覆盖蠕虫与另几滴卤化碳油 (~ 20 µL) 从顶端的蠕虫采摘。
5.线虫性腺注射 RNPs 和注射后护理
注: 微注射协议由梅洛和火57改编, 并在其他地方详细描述60,61。
-
将盖玻片与安装的蠕虫放到注射显微镜上。在低放大率 (5X 目标, 10X 眼), 定位蠕虫垂直于注射针。
- 切换到高放大倍数 (40X 目标, 10X 眼), 重新定位靠近性腺臂的针, 对应于中部至晚期粗线期的细胞核附近的区域。
- 使用机器人, 移动针对蠕虫, 轻微地压抑角质层。然后, 用一只手, 点击显微镜阶段的一侧, 通过角质层震动针。压低注射踏板/按钮, 慢慢填充性腺手臂与注射混合, 并删除针。
- 与另一只性腺手臂重复这一步。
-
一旦蠕虫被注射, 取出盖玻片/琼脂糖垫, 并将其置于解剖显微镜下。
- 使用被拉扯的毛细管吸管, 通过吹打一个 M9 缓冲在他们附近替换油从蠕虫。执行这种治疗, 以释放蠕虫从琼脂。
- 10分钟后, 当蠕虫在缓冲区中颠簸时, 用所拉毛细管吸管将它们移到 OP50 细菌的 NG 板上。把盘子放在20摄氏度, 2-3 小时, 直到蠕虫恢复并四处走动。
- 一旦恢复, 单独转移蠕虫到 NG 琼脂板块与 OP50 和转移板块到25°c 孵化器。
-
允许 P0注入的蠕虫生长并放置后代3天。筛选 F1子代。
- 如果使用 co CRISPR 或共转换62、63、64、65, 则根据它们是否具有参考基因的突变型, 选择候选蠕虫进行筛选。将这些标记的蠕虫单独转移到新的 OP50, 并允许它们在20摄氏度放置 F2子代。
注: 用于联合 CRISPR 筛选或选择的表型应为 Cas9 编辑的成功提供早期估计。 - 如果 CRISPR 表型不存在, microinject 一个积极的控制质粒, 以协助提高注射效率。
注: 例如, 包括注射混合中的质粒, 编码 mCherry 标记的 MYO-2 将有助于评估注射效率。成功注射 pCFJ90 的蠕虫将会有一些具有荧光 pharynxes 的后代。
- 如果使用 co CRISPR 或共转换62、63、64、65, 则根据它们是否具有参考基因的突变型, 选择候选蠕虫进行筛选。将这些标记的蠕虫单独转移到新的 OP50, 并允许它们在20摄氏度放置 F2子代。
- 检查 F1蠕虫是否存在所需的编辑。选择 F1母亲到一个96井板的个人井, 溶解她, 并检查她的 DNA, 无论是插入特异 PCR 放大, DNA 序列分析, 或测量师核酸酶化验 (CEL-1) 66.
注意: 这些测试可以在使用 CRISPR/co 转换或其他筛选或选择机制65,66,67,68时执行。 - 有关评估编辑结果的信息, 请跳到步骤8。
6. hawaiensis准备
- 1天前的微注射, 丰富的早期胚胎通过建立一个 ' 一对坦克 ' 前一天晚上;新分离的雌性将含有新鲜受精的胚胎。请参见雷姆et 。69以了解详细信息。
- 在注射的当天, 用0.02% 丁香油在海水中收集单细胞的Parhyale胚胎 (0-4 小时后受精), 然后用火焰拉动的方法轻轻地刮掉她腹窝囊中的胚胎。圆形玻璃吸管和一双钝 #3 钳。
7. hawaiensis胚胎显微注射 RNPs 和注射后护理
- 回填一被拉扯的毛细管与大约1µL RNP 注射液混合物上面描述。
-
使用压缩氮气 microinject 每个胚胎, 如雷姆et . 中所述。69。
- 使用 microinjector 和机器人在解剖显微镜下注入Parhyale胚胎。使用 microloader 吸管尖, 将注射剂的1.5 µL 插入毛细管的背部 (4 inches-1.0 毫米和花丝, 用微拉动装置拉动)。
- 在注射装置上设置针头, 在解剖范围内用一对钳折断针 (非常小的数量) 的针尖。通过注入卤化碳油700和测量气泡直径来校准所交付的体积。
- 用剃刀刀片从固化剂中切下一个 "槽"。用过滤消毒的海水将其填入一半, 并将Parhyale胚胎排在槽中以稳定。
- 注射时, 用注射剂对胚胎进行注射, 用一对镊子稳定每个胚胎。注射后, 用玻璃转移吸管将胚胎转移到一个新鲜的60毫米培养皿中, 用过滤灭菌的海水在中途填充。
-
如果第一个分裂已经发生形成一个2细胞胚胎 (4-6 小时后受精), 通过注射两个卵裂球产生完全突变的动物。为确保2细胞阶段的完全分裂, 与 FITC 或 TRITC 葡聚糖共注入卵裂球, 观察在注射后荧光解剖范围内的信号仅限于单个卵裂球。
- 或者, 通过在2细胞阶段只注射两个卵裂球中的一个来生成 ' 半变种 ' 动物 (大致上根据 P 轴上的组织和位置来划分左右)。
- 在8细胞胚胎 (7.5-9 小时后受精) 中注入一个细胞, 以限制编辑到单一的胚芽层。请参见 Gerberding et 。70用于早期卵裂球血统的地图。
-
在60毫米培养皿中孵化胚胎 (不超过25每道菜), 用过滤消毒的海水半填充, "预氧化" 使用水族馆涌泉或剧烈摇晃。
- 将胚胎的盘子放在一个松散密封的 plasticware 内, 用湿纸巾衬里, 以保持湿度, 并将它们放在一个26摄氏度的恒温箱中, 12 小时的光暗循环。
- 每隔几天把幸存的胚胎转移到清洁海水的盘子里。
注意: 胚胎可以在室温下培养, 尽管它们的发育速度会慢得多。
-
通过原位杂交或抗体染色, 解剖和修复不同阶段的胚胎, 以进行表达分析 (见布朗et 。71用于暂存指南, 以及用于解剖和固定的其他引用72、原位杂交73和抗体染色74)。
- 通过将一根弯曲的钨丝长度大约0.5 到胰岛素针的末端进行解剖针。在电流下将氢氧化钠中的针削尖。使用1毫升注射器作为解剖针的手柄。
- 在一半的3井玻璃盘子中, 用一个新的解决方案9部件 PEM 缓冲器 (0.1 米的管道 pH 值 6.95, 2 毫米的 EGTA, 1 毫米的 MgSO4), 1-部分 10x PBS, 1 部分32% 粉煤灰。将3-5 个胚胎放入盘子中, 然后将一个小孔戳进每个胚胎, 用一根锋利的钨针戳一下, 稍微迟钝一点, 以稳定, 允许蛋黄流出, 固定器跑进去。
- 使用一对削尖的钨针, 轻轻地将周围的两个膜包围在Parhyale胚上。解剖它们在固定, 使胚胎更健壮, 但迅速工作, 以保持膜不变的胚胎, 这使得膜去除更加困难。允许胚胎修复15-20 分钟的抗体染色或40-50 分钟的原位杂交。
- 图像活孵化和分析他们的形态学和行为表型或修复和染色, 以更详细的分析。在2-3 月内提高幼仔的性成熟度, 建立淘汰赛和转基因线 (见 Kontarakis 和 Pavlopoulos75幼体护理和其他有用的细节)。
8. 评估编辑结果
- 如果适用, 在编辑过的细胞或生物体中寻找视觉或功能表型。
注意: 此过程将因应用程序而异, 在上面的相关协议步骤结束时将介绍一些示例。在纠正 HSPCs 镰状细胞突变后, 用 HPLC 法分析不同孵育的血红蛋白产生情况 (图 1A)。IL-2 受体基因在 T 细胞中的挖空可以通过表面染色和流式细胞术 (图 1B) 来确定。要评估线虫和hawaiensis表型, 请观察在光或荧光显微镜下的动物形态学和行为 (图 1C和1D)。 - 为了确定生成的基因组编辑的效率和类型, 溶解编辑过的细胞池并使用商业提取套件21提取其基因组 DNA。
-
为快速估计 indel 形成, PCR-放大至少200基对在切口站点附近并且执行 T7 endonuclease1 (T7E1)76或测量师 (CEL-1 核酸酶) 化验 77.
- 如果 Cas9-cut 站点上的 indel 组或成功的 HDR 将创建或删除已知的限制站点, 请考虑使用限制酶消化来估计编辑效率6。限制片段长度多态性 (限制性) 法可以作为一种方便的方法来检查效率, 如果它碰巧是可用的。
- 为准确量化编辑效率和主要编辑结果的确定, 发送 PCR 扩增子的标准的桑格测序与正反引物。
注意: 如果分析单个克隆或有机体, 则对桑格结果的分析很简单, 如图 2a所示。如果分析单元格池, 则使用联机工具78分析图谱, 如图 2B所示。 - 要进行完整的量化和编辑结果序列, 请执行深度排序27、54, 如图 2C中所示。
- 为了评估一组特定的非目标变化, PCR-放大预测的非目标站点, 并将它们发送给。要启用对染色体易位的检测, 请执行引导序列79或高通量、全基因组移位顺序 (HTGTS)80。有关克隆填充中的非目标编辑的完整图片, 请执行全基因组排序 (WGS)81,82,83。
注意: 有多种方法来量化对和非目标基因组的编辑, 在各种评论文章中进一步解释了84,85,86。
Representative Results
这些实验表明, 预组装 Cas9 RNP 可以用来操纵基因组的原细胞和整个有机体。研究人员纯化或购买 Cas9 蛋白和 sgRNA, 将这两种成分结合在一起预先形成复合物, 并将 RNP 引入其细胞或生物体中。在允许足够的时间进行编辑和下一代后代出生 (如果适用), 检查表型和/或收集细胞的基因分型。表型可以通过功能化验, 表达分析, 可视化 (眼睛或显微镜), 或其他方法, 根据实验。
例如, 经过编辑以纠正导致镰状细胞疾病的β-球蛋白突变的 HSPCs 可以分化为红细胞, 用于生产健康或镰状血红蛋白27,87 (图1). T 细胞编辑, 以敲出高亲和 IL-2 受体基因, CD25 (IL2RA), 可以分析的表面染色和流式细胞术88, 并功能分析, 以检测信号响应 IL-2 刺激 (图 1B).t 细胞也可以重新编程在许多临床重要的方式, 需要评估不同的表型, 包括艾滋病毒感染的功效89和体内抗肿瘤疗效的车 T 细胞11。
使用共同 CRISPR/联合转换筛选方法, C. 线虫蠕虫在两个位点62同时编辑。使用 ssODN 修复模板的dpy-10引用基因的 HDR 会导致一个容易得分的显性dpy-10增益函数突变。异型 F1dpy-10 (gof)动物是滚筒 (卷) 和合合dpy-10 (gof)动物是矮胖的 (dpy)。表型的存在表明, Cas9 编辑发生在这些动物, 并提高了识别一个编辑事件的可能性, 在第二个轨迹在轮或 Dpy F1动物。成功的编辑实验应导致33-50% 注入的 P0蠕虫产生20或更多 F1子代, 它们是 Dpy90。然后, 可以选择非 "动物" 返回dpy-10以 wildtype 并选择用于纯合编辑的兴趣。作为一个经验法则, crRNA 靶向 CRISPR 参考基因的浓度应该是 crRNA 靶向感兴趣基因的一半。如果不恢复兴趣基因的编辑, 可以调整两个 CRISPR rna 的比值, 以增加恢复所需突变的可能性。例如, 增加兴趣基因的 crRNA 的数量相对于参考基因 crRNA 将增加蠕虫的数量在蠕虫的人口的兴趣基因编辑的百分比也拥有编辑在参考基因位置。Co 转换频率各不相同, 但速率通常为 20-60%, 通常在 F1生成中产生纯合编辑 (图 1C)。
P. hawaiensis已编辑以敲除腹部 b基因 (和 b) 的幼仔显示清晰的形态学异常3 (图 1D)。这种基因是需要正确的腹部模式, 它的中断导致胸部类型的跳跃和步行腿取代游泳和锚腿, 通常存在的腹部。
在基因型层面确定基因组编辑结果需要测序或检测序列变化的体外检测。在这里, 我们显示了典型的序列数据从我们的模型细胞类型和有机体, 强调不同的方法来编辑量化。请注意, 图形标签是一般化的, 因为此处显示的所有方法都可以应用于任何生物系统。
基于排序的方法因技术复杂性和结果深度而异。对于克隆编辑的种群或容易分离的个体有机体, 编辑的个体可以在基因组 DNA 提取之后进行排序。标准的 Cas9-cut 排序结果将在给定的个体中显示该站点中的序列更改, 假设 frameshifts 会中断其功能 (图 2a)。用于排序的在线工具是另一个基于排序的方法, 可应用于混合种群, 而不是单个突变体78。序列的分析与在线工具, 可以近似整体编辑效率和主要顺序结果。代表数据显示在图 2B中。
这里描述的最彻底的测序方法是深度测序 (有时称为高通量或下一代测序)。该方法提供了混合种群中单个基因组的 DNA 序列。这些数据可以用多种方式加以说明。在这里, 我们根据编辑结果 (图 2C) 将单个排序读取从编辑的单元格中分类。大多数细胞通过 NHEJ 通路进行编辑, 这通常会导致基因中断。在其他方面, 通过 HDR27, 目标基因已被替换为备用版本。
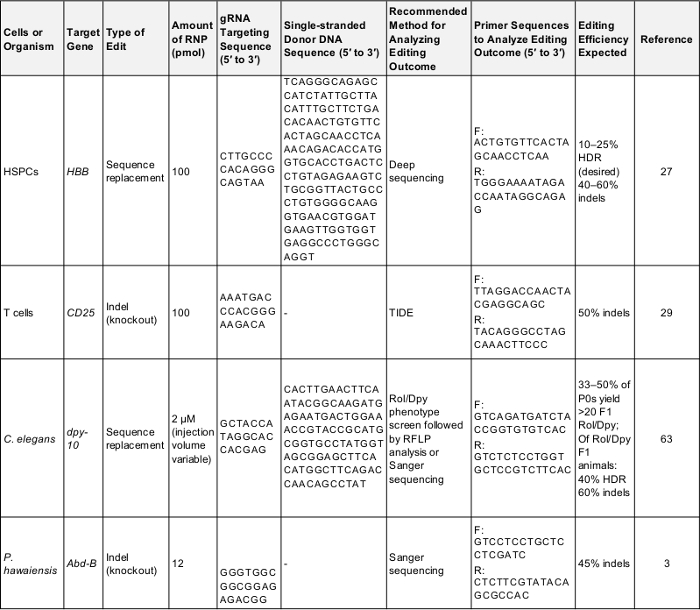
表 1: 对初步基因组编辑实验的积极控制.本表显示了在本议定书所述的每个细胞和有机体中执行第一次基因组编辑实验所需的关键信息。以下这些参数很可能产生一个成功的结果, 可以用来测试协议或作为比较基准, 一旦实验者的目标是自己感兴趣的基因。F: 向前, R: 反向, HDR: 同源定向修复。请单击此处下载此表.

图 1: 具有代表性的表型结果来自 Cas9 RNP 编辑的主要人类细胞和有机体。(A)这是一种高效液相色谱示踪, 表明在基因组编辑成功后, 分化为晚期孵育的 HSPCs 将产生比镰状血红蛋白更多的功能性血红蛋白。突变红细胞产生镰状血红蛋白 (哈佛商学院), 而成功编辑的细胞将产生健康的血红蛋白 (HbA 和 HbA2) 以及胎儿血红蛋白 (HbF)。吸光度以任意单位 (au) 为图表。本小组首次在德威特电子杂志上发表.t 。27. 它在美国科学促进协会的允许下重印。(B)在左侧, 对于每个条件, 此面板显示流式细胞仪数据, 表明在CD25基因被 RNP 击倒后, 表面染色的 T 细胞不会表达 CD25。CD25 丰度绘制在 x 轴上, 单元格大小在 y 轴上。在右侧, 对于每个条件, 此面板显示 Phospho-Stat5 (pStat5) 量化后, 诱导与 IL-2。当 IL-2 受体缺失时, 信号减少 (CD25 )。在 x 轴上绘制 pStat5 丰度, 并垂直比较三个不同级别的 IL-2 输入所产生的数据。(C)此面板显示秀丽线虫co CRISPR/共转换屏幕, 其目标是dpy-10作为协转换标记。两个引导 rna 目标两个位点, dpy-10和您最喜爱的基因 (yfg), 在同一 P0注射的动物。在dpy-10上的 HDR 结果为 dpy 表型。选择 Dpy1动物会增加在第二个轨迹上识别编辑的几率。(D)此面板显示 wildtype Parhyale hawaiensis幼仔有正常的腹部与游泳和锚腿。B 型敲出的幼仔 (F0个体) 开发腹部向胸部转变。因此, 游泳和锚腿已经消失, 并取代了跳跃和步行腿与正常的胸部。请单击此处查看此图的较大版本.

图 2: 编辑结果分析方法的典型结果。(A)此面板显示了来自单个 F1 hawaiensis有机体的桑格测序结果的示例, 包括 wildtype 序列和三种不同的 indels, 通过移开阅读框架来破坏基因功能。(B)这些潮汐结果显示在序列化 T 单元格池中的 Cas9-target 站点上发生的插入和删除事件的范围。x 轴指示核苷酸中给定插入或删除的长度。(C)这些深度测序结果显示没有 nucleofection 或 gRNA 的基因组编辑, 并且成功编辑了完整的 Cas9 RNP, 按 HSPCs 的 DNA 修复结果分组.请单击此处查看此图的更大版本.
Discussion
在细胞系或感兴趣的生物体中建立一个健壮的基因组编辑协议, 需要对这一节中讨论的几个关键参数进行优化和经验测试。对这里提出的一般方法有一些不同的尝试是非常鼓励的。该协议的主要局限性是, 将这些方法应用于其他细胞或生物体可能会导致不同的结果, 这取决于所研究的物种, 而导致高效基因敲除的实验设计可能不会促进 DNA 的插入。因此, 我们建议从此处介绍的方法开始, 并按照下面的说明进行故障排除。
基因组编辑试剂质量疑难解答:
生成或购买高质量的试剂是任何基因组编辑协议的关键步骤。Cas9 蛋白可以在实验室纯化或商业化购买。许多协议注意到 Cas9 在 RNP 食谱中的最终浓度, 但最佳的基因编辑活动将取决于任何个体 Cas9 蛋白制剂的特定活动, 这取决于来源的不同。一旦在这里提出的协议是工作, 考虑优化 RNP 使用的数量滴定 Cas9 水平, 以建立一个最佳浓度: 一个提供高度特异的目标 DNA 裂解, 而不必要的离靶分裂造成的过度 Cas940。
指南 RNA 纯度和同质性也可以是基因组编辑成功的决定因素22。购买的 sgRNAs 或单独的 crRNA 和 tracrRNA 组件通常是高质量的试剂, 并且有多种化学修饰可用于消除 RNA 降解的问题或将附加功能灌输给 RNP91。虽然化学修饰的 gRNAs 可能不是标准基因组编辑实验所必需的, 但有些组观察到了更高的编辑效率与这种试剂, 所以他们可能值得尝试后, 掌握的过程和/或当 gRNA 降解似乎是一个问题22,91。体外转录和随后的凝胶纯化是一种廉价的替代品, 它可能足以用于常规基因组编辑实验17,21,49,50。此外, 通常适用于产生同质 gRNA 种群在体内的几种方法, 包括核酶和基于 tRNA 的单个参考线的切除, 可扩展到体外RNA 制备, 以生成清洁剂。产品92。
指南 RNA 和捐赠者 DNA 设计提示:
引导 RNA 的选择是实现高效的目标编辑的关键因素, 同时最大限度地减少了离靶解的几率。为了帮助引导选择, 一些研究使用高通量屏幕加上下一代排序, 以编译成功的参考线47,79,93,94的序列特征, 95,96。这些功能已用于开发预测算法和在线工具, 以帮助引导选择44,45,46,47,48。这种算法是基于基于 DNA 的制导 RNA 表达系统的屏幕上进行的。指南是使用一个三级启动子表示的, 因此它们的表达式很容易与第三级转录相关的限制, 例如遇到尿97、98的曲目时过早终止, 99。然而, 使用与体外的 RNPs 合成的指南 rna 绕过这些问题, 并简化了指导设计的约束。从这些算法中产生的一个共同特征, 并已被证实在许多研究与高效的基因组编辑, 是存在嘌呤, 特别是鸟嘌呤, 在3′结束的指南的目标特定序列。该指南的特点是非常成功的有机体之间, 从哺乳动物到线虫, 果蝇和斑马鱼65,100,101。此外, 对于C. 线虫, 在指南目标区域的3′端设计具有 GG 二核苷酸的参考线是预测高效导 rna65的有效策略。理想情况下, 并行测试多个参考线, 以确定给定应用程序中哪个最成功。
当试图向基因组引入 dna 序列时, 捐赠者或模板 dna 的设计也至关重要。单链寡核苷酸捐赠者 (ssODNs) 比其他典型的修复模板, 线性双绞线和质粒 DNA54,55,102更可靠地插入。在某些位点上, HDR 效率可以通过与非目标或移位的 DNA 链互补的 ssODNs 来改进, 并且拥有长度不对称的同源臂27,55。由于修复模板正在入到剪切站点并包含目标序列 , 因此必须采取步骤防止 Cas9 在基因组插入之前或之后裂解捐赠者的 DNA 。这是通过对 PAM 序列或种子区域进行无声的突变来实现的, 它避免了 Cas9 的识别, 同时保留了插入基因21、103的功能。即使单核苷酸改变 PAM 可能会取消绑定104, 尝试改变至少四核苷酸是安全的。
意义和未来应用:
基因组编辑与 CRISPR-Cas9 已经成为一种强有力的方法, 使任何有机体的巧妙的基因操作。使用 Cas9 RNP 进行编辑时, 首先需要做一些努力, 但在实验室中建立试剂和协议后, 就可以直接使用了。用预组装的 RNP (而不是质粒 DNA) 编辑细胞可以提高总体编辑效率, 包括通过 HDR 难以实现的基因插入, 较少的目标效果24,25,26,27,29. 此外, 实验者避免了基因表达、RNA 降解、蛋白质折叠等问题, 以及在细胞22、23中分别合成的 gRNA 和 Cas9 分子之间的关联。RNP 编辑还绕过了在临床上使用病毒传递方法14时可能出现的插入突变和持续表达的安全问题。由于这些优势, 许多科学家进行了临床前, 概念验证的实验, 有利于 RNP 编辑的人类治疗应用。体内和前体基于 RNP 的基因组编辑方法正在开发中以治疗甚至治愈各种条件, 从诸如杜氏肌营养不良症105和镰状细胞疾病27的遗传疾病到HIV29和癌症11。有趣的是, Cas9 RNP 越来越被用作农业工程的交付方法, 因为它使 "无 DNA" 编辑植物33,34,36。
Disclosures
作者亚历山大马森和雅各布 e 玉米是聚光灯疗法的共同创始人。雅各布玉米是特派团治疗的顾问, 他的实验室得到了阿斯利康和辉瑞赞助的研究支持。亚历山大马森是朱诺疗法和契约疗法的顾问, 他的实验室获得了来自朱诺疗法、Epinomics 和赛诺菲的赞助研究支持。他的实验室也申请了与 Cas9 RNP 技术有关的专利。
Acknowledgments
我们感谢许多以前的实验室成员和湾区基因组编辑社区为这些方法的发展做出的贡献。我们感谢罗斯. 威尔逊批判地阅读了这篇手稿。
亚历山大马森的研究得到了来自杰克 Aronov 和全国多发性硬化社会补助金 (CA 1074-21) 的礼物的支持。亚历山大马森为来自斯勒威威威基金的医学科学家颁发了职业奖, 是一位 Biohub 调查员。雅各布玉米的研究得到了李嘉诚基金会、遗产医学研究医学院和加利福尼亚再生医学研究所的支持。Behnom Farboud 和芭芭拉 j. 迈耶的研究部分是由研究院补助金 R01 GM030702 给芭芭拉 j. 迈耶, 他是霍华德休斯医学研究所的调查员。艾琳. 贾维斯和 Nipam h. 帕特尔的研究部分由 nsf 资助 IOS-1257379 和艾琳. 贾维斯承认来自 nsf GRFP 和 Philomathia 研究生奖学金的支持。
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
