

Summary
एक जुदा Cas9 ribonucleoprotein परिसर (RNP) का उपयोग सटीक, कुशल जीनोम संपादन के लिए एक शक्तिशाली तरीका है । यहां, हम प्राथमिक मानव कोशिकाओं और दोनों क्लासिक और उभरते मॉडल जीवों सहित कोशिकाओं और जीवों की एक विस्तृत रेंज भर में इसकी उपयोगिता पर प्रकाश डाला ।
Abstract
साइट-CRISPR के साथ विशिष्ट eukaryotic जीनोम संपादन (नियमित रूप से अंतराल लघु palindromic दोहराता संकुल)-कैस (CRISPR-संबद्ध) सिस्टम जल्दी से जैविक प्रश्नों की एक विस्तृत विविधता का पीछा शोधकर्ताओं के बीच एक आम हो गया है । उपयोगकर्ता सबसे अधिक बार आसानी से reCas9ed गाइड आरएनए (gRNA) के साथ एक जटिल में स्ट्रेप्टोकोकस pyogenes से व्युत्पंन प्रोटीन को रोजगार । इन घटकों को कोशिकाओं में पेश कर रहे हैं, और डबल फंसे डीएनए (dsDNA) जीनोम के एक पूरक क्षेत्र के साथ बाँधना आधार के माध्यम से, एंजाइम दोनों किस्में एक डबल-किनारा तोड़ (DSB) उत्पन्न करने के लिए सट. बाद में सुधार या तो यादृच्छिक सम्मिलन या हटाने की घटनाओं (indels) या प्रयोगकर्ता के शामिल करने की ओर जाता है-तोड़ने की साइट पर डीएनए प्रदान की ।
एक शुद्ध एकल गाइड आरएनए और Cas9 प्रोटीन, एक RNP फार्म और कोशिकाओं को सीधे दिया करने के लिए इकट्ठे के उपयोग, अत्यधिक कुशल जीन संपादन को प्राप्त करने के लिए एक शक्तिशाली दृष्टिकोण है । RNP संपादन विशेष रूप से जीन प्रविष्टि, एक परिणाम है कि अक्सर प्राप्त करने के लिए चुनौतीपूर्ण है की दर को बढ़ाता है । एक प्लाज्मिड के माध्यम से वितरण की तुलना में, सेल के भीतर Cas9 RNP के छोटे हठ कम बंद लक्ष्य घटनाओं की ओर जाता है ।
अपने फायदे के बावजूद, CRISPR जीन संपादन के कई आकस्मिक उपयोगकर्ताओं को इस तकनीक से कम परिचित हैं । प्रवेश के लिए बाधा कम करने के लिए, हम संदर्भों की एक श्रेणी में RNP रणनीति को लागू करने के लिए विस्तृत प्रोटोकॉल रूपरेखा, अपने अलग लाभ और विविध अनुप्रयोगों पर प्रकाश डाला । हम दो प्रकार के प्राथमिक मानव कोशिकाओं, टी कोशिकाओं और टेम स्टेम/जनक कोशिकाओं (HSPCs) में संपादन को कवर । हम यह भी बताते है कि कैसे Cas9 RNP संपादन पूरे जीवों की सतही आनुवंशिक हेरफेर सक्षम बनाता है, क्लासिक मॉडल roundworm Caenorhabditis एलिगेंस और अधिक हाल ही में पेश मॉडल जलीय, Parhyale hawaiensisसहित ।
Introduction
fThe CRISPR-Cas9 प्रणाली वैज्ञानिकों के किसी भी जीनोम1के लक्षित क्षेत्रों को बदलने के लिए अनुमति देता है । इस त्वरित और सस्ती प्रौद्योगिकी बुनियादी अनुसंधान में क्रांति ला दिया है और व्यक्तिगत रोग चिकित्सा, परिशुद्धता कृषि के विकास पर गहरा प्रभाव बनाने का वादा किया है, और2से परे । CRISPR संपादन एक democratizing उपकरण है और एक नई प्रयोगशाला में प्रणाली को लागू करने के जीनोम इंजीनियरिंग में कोई विशेष विशेषज्ञता की आवश्यकता है, सिर्फ बुनियादी आणविक जीवविज्ञान कौशल । शोधकर्ताओं अब आनुवंशिक हेरफेर के लिए कुछ वैकल्पिक साधन के साथ पहले से असभ्य जीवों का अध्ययन कर सकते हैं3,4. पिछले पांच वर्षों में अकेले, CRISPR जीनोम संपादन २०० विभिंन रीढ़, invertebrate, संयंत्र, और माइक्रोबियल प्रजातियों पर इंजीनियर के लिए इस्तेमाल किया गया है ।
CRISPR prokaryotic रक्षा मार्ग से अनुकूलित, कोर साइट विशेष जीनोम संपादन के लिए आवश्यक तत्वों Cas9 प्रोटीन हैं, आमतौर पर एस. pyogenes और codon से-एक जोड़ा परमाणु स्थानीयकरण संकेत (एनएलएस) के साथ अनुकूलित, और उसके विशेष आरएनए गाइड5,6. हालांकि यहां पर चर्चा नहीं की गई, अन्य Cas9 orthologues या CRISPR endonucleases का भी इस्तेमाल हो सकता है । स्वाभाविक रूप से होने वाली gRNA दो अलग से लिखित टुकड़ों से बना है, CRISPR आरएनए (crRNA) और ट्रांस सक्रिय crRNA (tracrRNA)7। इन RNAs एक प्रतिलिपि, एकल गाइड आरएनए (sgRNA)8के रूप में जाना जाता है में इनकार किया जा सकता है । सबसे जीनोम संपादकों सुव्यवस्थित sgRNA9चुनते हैं, हालांकि दोहरे गाइड भी नियमित रूप से इस्तेमाल किया जाता है10,11। experimenters एक 20-न्यूक्लियोटाइड (nt) जीनोमिक डीएनए लक्ष्य का चयन, यह सुनिश्चित करना है कि यह Cas9 मांयता के लिए आवश्यक एक छोटी लाइसेंसिंग हस्ताक्षर के बगल में है, एक protospacer आसंन आकृति (पाम) कहा जाता है, और एक gRNA है कि पूरक अनुक्रम शामिल है डिजाइन12 .
एक बार कक्ष के अंदर, RNP परिसर अपने जीनोमिक लक्ष्य, पूरक डीएनए किनारा के साथ gRNA आधार जोड़े को रेखांकित करता है, और फिर एंजाइम दोनों डीएनए किस्में एक डबल-किनारा तोड़2उत्पंन करने के लिए सट । सेल मरंमत मशीनरी को ठीक से कम दो मार्गों में से एक द्वारा DSB: त्रुटि प्रवण गैर मुताबिक़ अंत में शामिल होने (NHEJ) मार्ग या समरूपता-निर्देशित मरंमत (HDR) है, जो मूल समरूपता के ' हथियार ' युक्त डीएनए को तोड़ने के दोनों ओर के द्वारा । पूर्व मरंमत मार्ग आम तौर पर indel गठन और फलस्वरूप जीन व्यवधान की ओर जाता है, जबकि बाद experimenters डालने या डीएनए दृश्यों को बदलने के लिए अनुमति देता है1।
संपादन दक्षता और सटीकता मतलब है जिसके द्वारा Cas9 और gRNA कक्ष में प्रवेश पर निर्भर करते हैं । इन घटकों न्यूक्लिक एसिड के रूप में या एक जुदा RNP जटिल13,14,15के रूप में प्रसंस्कृत कोशिकाओं, भ्रूण, या जीवों को दिया जा सकता है । कॉमन न्यूक्लिक एसिड आधारित डिलिवरी तरीकों में शामिल है वायरल transduction, अभिकर्मक, या electroporation mRNA या प्लाज्मिड डीएनए । Cas9 प्रोटीन और गाइड आरएनए तो सेल के भीतर उत्पादन कर रहे हैं और वे एक जटिल बनाने के लिए सहयोगी.
RNP के प्रत्यक्ष वितरण Cas9 प्रोटीन और गाइड आरएनए के अलग शुद्धि की आवश्यकता है । यह घर में किया जा सकता है, या प्रोटीन और sgRNA कई वाणिज्यिक विक्रेताओं में से एक से खरीदा जा सकता है । एक बार प्राप्त कर लिया, Cas9 और gRNA enzymatically सक्षम RNP परिसर के रूप में मिश्रित और निषेचित अंडे में प्रत्यक्ष इंजेक्शन द्वारा कोशिकाओं को शुरू की है/भ्रूण, लिपिड आधारित अभिकर्मक16, या electroporation । RNP संपादन की पहली रिपोर्ट सी. एलिगेंस gonads17में इंजेक्शन शामिल है । Microinjection अभी भी भ्रूण और पूरे जीवों में RNP शुरू करने का पसंदीदा साधन है, हालांकि प्रभावी electroporation माउस18,19 और चूहे20 भ्रूण में प्रदर्शन किया गया है । हम सीधे सी. एलिगेंस gonads और पी. hawaiensis भ्रूण में RNP इंजेक्शन के लिए प्रोटोकॉल का वर्णन है और electroporation के एक विशेष प्रकार की सिफारिश करने के लिए जब प्राथमिक मानव कोशिकाओं संपादन RNP उद्धार । इस विधि, nucleofection, अनुकूलित electroporation कार्यक्रमों और सेल प्रकार विशेष समाधान शामिल है और RNP दोनों कोशिका द्रव्य और नाभिक 21 दर्ज करने के लिए अनुमति देता है ।
RNP के साथ जीनोम संपादन कई विशिष्ट लाभ प्रदान करता है । क्योंकि प्रोटीन और आरएनए घटक पूर्व इकट्ठे होते हैं, और गुणवत्ता के वितरण से पहले सुनिश्चित किया जा सकता है, RNP संपादन न्यूक्लिक एसिड आधारित वितरण के साथ जुड़े कई नुकसान से बचा जाता है । अर्थात्, वहां Cas9 का कोई खतरा नहीं है मेजबान जीनोम में डीएनए एकीकरण एंकोडिंग, mRNA क्षरण के लिए कभी नहीं उजागर है, और यह vivo gRNA या प्रोटीन अभिव्यक्ति, तह में के साथ समस्याओं को दरकिनार, और22एसोसिएशन,23। इसके अलावा, RNP का उपयोग करने के लिए कम विषाक्तता की ओर जाता है और दूर से कम लक्ष्य की घटनाओं से प्लाज्मिड आधारित अभिव्यक्ति, RNP के छोटे आधे सेल के अंदर जीवन का परिणाम24,25,26,27।
अंत में, RNP संपादन प्रदर्शन मानव कोशिका लाइनों की एक किस्म में उच्च संपादन दरों की ओर जाता है, fibroblasts के रूप में प्राथमिक कोशिकाओं, भ्रूण स्टेम सेल (ESCs), प्रेरित pluripotent स्टेम सेल (iSPCs), HSPCs, और टी कोशिकाओं16,24, 25,26,27,28,29; C. एलिगेंस, P. hawaiensisसहित अकशेरूकीय में, और फल मक्खियों3,17,30; zebrafish, चूहों, और चूहों की तरह हड्डीवाला प्रजातियों में31,३२; Arabidopsis, तंबाकू, सलाद पत्ता, चावल, दाखलता, सेब, मक्का, और गेहूं३३,३४,३५,३६सहित संयंत्र प्रजातियों में; और Chlamydomonas, Penicillium, और Candida प्रजातियों में३७,३८,३९। indel गठन की आवृत्ति अधिक हो सकता है जब प्लाज्मिड वितरण की तुलना में RNP का उपयोग कर, और HDR-मध्यस्थता डीएनए प्रविष्टि25,27,29प्राप्त करने के लिए आसान हो सकता है ।
प्रोटोकॉल यहां वर्णित Cas9 RNP का उपयोग करता है और एक प्रभावी, आसानी से अनुकूलनीय तकनीक है कि जैविक प्रणालियों की एक विस्तृत विविधता४०,४१, विशेष रूप से कोशिकाओं है कि अंयथा काम करने के लिए मुश्किल है में लागू करने के लिए सरल है साथ और जीवों में अच्छी तरह से बिना सटीक आनुवंशिक हेरफेर के लिए सिस्टम की स्थापना की । हम कैसे डिजाइन करने के लिए, प्राप्त करते हैं, और विभिंन मॉडल सेल प्रकार और जीवों के पार इसके उपयोग को कवर करने से पहले Cas9 RNP इकट्ठा वर्णन द्वारा शुरू करते हैं । टेम स्टेम/जनक कोशिकाओं (HSPCs) और टी कोशिकाओं को एक ही विधि का उपयोग कर संपादित कर रहे हैं, nucleofection, ताकि वे एक साथ चरण 2 और 3 में इस प्रोटोकॉल को कवर कर रहे हैं । Cके लिए संपादन कार्यविधियां । एलिगेंस चरण 4 और 5, और Pमें वर्णित हैं । hawaiensis संपादन चरण 6 और 7 में शामिल किया गया है । अंत में, के बाद से किसी भी जीव में एक जीन संपादन प्रयोग की सफलता जीनोटाइप अनुक्रमण द्वारा मूल्यांकन किया जा सकता है, सभी कोशिकाओं और प्रोटोकॉल में वर्णित जीवों के लिए संभव विश्लेषण विधियों का वर्णन उप चरण 8 में उल्लिखित हैं ।
Protocol
1. RNP विधानसभा
-
समय से आगे सभी आरएनए, डीएनए, और प्रोटीन घटक प्राप्त करने, अग्रिम में अच्छी तरह से प्रयोग डिजाइन । पहले पास के रूप में, एक सकारात्मक नियंत्रण तालिका 1 में सूचीबद्ध है और एक विश्वसनीय प्रायोगिक डिजाइन और सामग्री की अखंडता को सुनिश्चित करने के लिए सामग्री की तालिका में वर्णित वाणिज्यिक रिएजेंट का उपयोग की कोशिश करें । एक नया जीनोम संपादन प्रयोग की योजना बना पर अतिरिक्त सुझावों के लिए, इस विषय12,४२,४३पर कागजात देखें ।
नोट: एक बार बाद के चरणों में वर्णित के रूप में इकट्ठे, अग्रिम में तैयार RNPs-८० डिग्री सेल्सियस पर संग्रहीत किया जा सकता है ।- चुनने के बाद जो जीन को लक्षित करने के लिए, एक मुफ्त ऑनलाइन उपकरण का उपयोग एक इष्टतम gRNA४४,४५,४६,४७,४८डिजाइन । हो एक एक्सॉन लक्ष्य यकीन है कि अगर एक नॉकआउट उत्पंन उंमीद है ।
नोट: इन उपकरणों के लिए एक आसंन एस pyogenes पाम अनुक्रम, उच्च गुणवत्ता स्कोर, और कम ऑफ लक्ष्य स्कोर के साथ एक लक्ष्य साइट की पहचान करने में मदद मिलेगी । - प्रकाशित तरीकों8के माध्यम से एस pyogenes Cas9 प्रोटीन शुद्ध, या यह एक वाणिज्यिक विक्रेता से खरीद ।
- आरएनए कमजोर पड़ने के लिए एक ठेठ Cas9 बफर तैयार, RNP तैयारी, और प्रोटीन भंडारण, जिसमें HEPES पीएच ७.५, KCl के १५० मिमी, 10% ग्लिसरॉल, और TCEP के 1 मिमी के 20 मिमी शामिल हैं । हमेशा nuclease-मुक्त पानी का उपयोग करें buffers कि reसस्पेंड करने के लिए या पतन को रोकने के लिए आरएनए पतला करने के लिए इस्तेमाल किया जाएगा ।
- गाइड आरएनए (tracrRNA और crRNA या sgRNA) के माध्यम से एक इन विट्रो प्रतिलेखन में प्रकाशित तरीकों का उपयोग कर उत्पादन, या यह एक न्यूक्लिक एसिड संश्लेषण कंपनी से खरीद17,21,४९, ५० , ५१.
- यदि एक जीन डालने, संश्लेषित या एक दाता डीएनए टेंपलेट खरीद ।
- का उपयोग करने से पहले तुरंत बर्फ पर-८० डिग्री सेल्सियस और गल पर प्रोटीन और आरएनए aliquots की दुकान ।
नोट: प्रत्येक फ्रीज-गल थोड़ा दक्षता कम करती है । विस्तृत, खुला Cas9 शुद्धि के लिए उपयोग प्रोटोकॉल५२ और इन विट्रो sgRNAs५३ की प्रतिलिपि कहीं और उपलब्ध हैं ।
- चुनने के बाद जो जीन को लक्षित करने के लिए, एक मुफ्त ऑनलाइन उपकरण का उपयोग एक इष्टतम gRNA४४,४५,४६,४७,४८डिजाइन । हो एक एक्सॉन लक्ष्य यकीन है कि अगर एक नॉकआउट उत्पंन उंमीद है ।
- यदि C. एलिगेंसके साथ कार्य कर रहा है, तो चरण १.५ पर जाएं । P. hawaiensis प्रोटोकॉल के लिए, चरण १.६ पर जाएं । sgRNA का उपयोग करते हैं, तो चरण १.४ पर जाएं । १.३ चरण के लिए प्राथमिक सेल संपादन के लिए एक gRNA इकट्ठा करने के लिए आगे बढ़ें ।
-
tracrRNA और crRNA की equimolar मात्रा को मिलाकर एक gRNA को इकट्ठा करें । ८० µ एम gRNA शेयर के १०० µ एल बनाओ, के बारे में ५० जीनोम संपादन प्रयोगों के लिए ।
- 30 मिनट के लिए ३७ ° c पर gRNA मशीन और फिर यह धीरे से कमरे के तापमान को शांत करने के लिए अनुमति देते हैं ।
-
HSPC और टी सेल संपादन के लिए RNP तैयारी: gRNA के २०० pmol के लिए एक 1-2x दाढ़ राशि के मिश्रण से एक RNP परिसर इकट्ठा Cas9 प्रोटीन की कुल मात्रा में 10 µ एल बहुत धीरे से, Cas9 के लिए ध्यान केंद्रित gRNA जोड़ने (पूर्व Cas9 बफर में पतला) के बारे में 30 एस के लिए , पिपेट के साथ त्वरित हलकों बनाने, 20 µ एम के लिए अंतिम Cas9 एकाग्रता लाने ।
- electroporation cuvettes तैयार करें ।
नोट: इस प्रोटोकॉल के लिए विशिष्ट है वाणिज्यिक प्रणाली में संदर्भित करने के लिए सामग्री की तालिका, लेकिन RNP संपादन भी अंय electroporation उपकरणों के साथ प्राप्त किया जा सकता है । - इसमें 5 µ l (१०० pmols, T cells) या 10 µ l (२०० pmol, HSPCs) RNP के प्रत्येक cuvette को जोड़ें ।
- अगर नया डीएनए डालने के बजाय एक नॉकआउट, १०० µ एम (१०० pmol) एकल असहाय oligonucleotide दाता डीएनए (ssODN)25,५४,५५ की cuvettes या प्लेट के कुओं के 1 µ एल जोड़ें ।
- प्राथमिक कक्ष संपादन प्रोटोकॉल में अगले निर्देशों के लिए चरण 2 पर जाएं .
- electroporation cuvettes तैयार करें ।
-
सी. एलिगेंस संपादन के लिए RNP तैयारी: 20 µ एल के एक अंतिम मात्रा बनाने के क्रम में निम्नलिखित एजेंट जोड़कर RNP परिसर को इकट्ठा (अंतिम सांद्रता कोष्ठक में नोट किया जाता है): Cas9 (२ µ मी), HEPES पीएच ७.५ (१० µ मी), KCl (११५ µ मीटर), crRNA (१२ µ मीटर) , tracrRNA (४० µ m), और मरंमत टेम्पलेट्स अगर जरूरत (०.५ µ m ssDNA या अप करने के लिए ३५० एनजी/µ l dsDNA).
नोट: एक Cas9 की दक्षता-मध्यस्थता DSB-टेम्पलेट की मरम्मत dsDNA मरम्मत के निर्माण की एकाग्रता के लिए आनुपातिक है; इस प्रकार, मरंमत के उच्च एकाग्रता टेंपलेट, और अधिक कुशल टेंपलेट मरंमत । हालांकि, dsDNA के ३५० एनजी/µ एल से अधिक से अधिक युक्त घोला जा सकता है इंजेक्शन कीड़े की व्यवहार्यता को कम करने के लिए दिखाया गया है । इस प्रकार, यह करने के लिए उपयोग करने के लिए सबसे अच्छा है, लेकिन कोई अधिक से अधिक ३५० एनजी/µ एल मिश्रण में dsDNA की मरंमत क्षमता को अधिकतम करने के लिए जबकि अपनी घातकता को कम करने ।- चरण ५.४ में वर्णित सह-CRISPR/सह-रूपांतरण जांच दृष्टिकोण के लिए आवश्यकतानुसार एकाधिक crRNAs को एक साथ टार्गेट करने के लिए एकाधिक loci जोड़ें । एक से अधिक crRNA जोड़ते समय, प्रत्येक क्रमिक रूप से मास्टर मिश्रण को जोड़ें ।
नोट: प्रत्येक crRNA की मात्रा एक ही होने की जरूरत नहीं है, और यहां तक कि Cas9 की एकाग्रता को बदलने के बिना मास्टर मिश्रण में crRNAs की कुल एकाग्रता दोहरीकरण एक विशिष्ट लोकस में mutagenesis की आवृत्ति के साथ हस्तक्षेप करने के लिए प्रकट नहीं होता है. उदाहरण Paix एट अल में विस्तार से वर्णित हैं । ५६. - pipetting द्वारा मिक्स और 5 एस के लिए १६,००० x g पर RNP समाधान स्पिन सुनिश्चित करें कि समाधान ट्यूब के तल पर एकत्र की है ।
- 15 एम के लिए ३७ ° c पर समाधान मशीन ।
- 1 मिनट के लिए १६,००० x g पर नमूना केंद्रापसारक किसी भी कण कि पतली ऊब microinjection सुई रोकना सकता है गोली । क्रमिक चरणों में supernatant का उपयोग करें ।
- C. एलिगेंस प्रोटोकॉल के शेष के लिए चरण 4 पर जाएं ।
- चरण ५.४ में वर्णित सह-CRISPR/सह-रूपांतरण जांच दृष्टिकोण के लिए आवश्यकतानुसार एकाधिक crRNAs को एक साथ टार्गेट करने के लिए एकाधिक loci जोड़ें । एक से अधिक crRNA जोड़ते समय, प्रत्येक क्रमिक रूप से मास्टर मिश्रण को जोड़ें ।
-
पी hawaiensis संपादन के लिए RNP तैयारी: कमजोर और ०.१५% nuclease लाल के ६.२५ phenol मीटर की एक अंतिम एकाग्रता के लिए (इंजेक्शन visualizing के लिए) के साथ उन्हें µ द्वारा एकल उपयोग Cas9 aliquots तैयार करें ।
- एक 2-5x दाढ़ gRNA के एक कुल मात्रा में Cas9 प्रोटीन के लिए अतिरिक्त 6 µ एल के मिश्रण से RNP परिसर को इकट्ठा pmol के 12 Cas9 जोड़ें gRNA, 2 Cas9 एम के लिए अंतिम µ एकाग्रता लाने, gRNA एकाग्रता को 4-8 µ m, और phenol लाल एकाग्रता ०.०५% करने के लिए ।
- RNP जटिल करने के लिए 10 मिनट के लिए कमरे के तापमान पर मिश्रण की मशीन ।
- P. hawaiensis संपादन प्रोटोकॉल में अगले निर्देशों के लिए चरण 6 पर जाएं।
2. सेल संस्कृति और तैयारी
नोट: एक जैविक सुरक्षा कैबिनेट में 3.3.3 करने के लिए 2.1.1 कदम प्रदर्शन ।
-
एक वेंडर से cryopreserved ह्यूमन मैटीरियल पेरिफेरल ब्लड CD34+ HSPCs खरीदे ।
- गल ~ 1 x106 HSPCs में एक ३७ ° c पानी स्नान 3 मिनट के लिए और एक 15 मिलीलीटर शंकु ट्यूब के लिए उन्हें हस्तांतरण । एक वाणिज्यिक स्रोत से एक सीरम मुक्त विस्तार मध्यम के 10 मिलीलीटर जोड़ें और 10 मिनट के लिए १०० x g पर मिश्रण स्पिन । supernatant को निकालें और अनुपूरक SFEM के 2 मिलीलीटर में कोशिकाओं को फिर से सस्पेंड करें । प्लेट 6 में कोशिकाओं-अच्छी तरह से प्लेटें और उन्हें एक ३७ डिग्री सेल्सियस में संस्कृति 24-48 एच के लिए पहले RNP electroporation.
- एक hemocytometer के साथ कोशिकाओं की गणना और HSPCs की जरूरत की कुल संख्या (150000-200000 HSPCs प्रति cuvette) एक केंद्रापसारक ट्यूब करने के लिए स्थानांतरण ।
- 10 मिनट के लिए १०० x g पर ट्यूब स्पिन कोशिकाओं को गोली ।
-
खरीद मानव प्राथमिक CD4+ टी कोशिकाओं एक विक्रेता से या उंहें मानव पूरे रक्त से घनत्व ढाल केंद्रापसारक29द्वारा अलग ।
- टी सेल सक्रियकरण करने से पहले, पूर्व कोट ४८-αCD3 (UCHT1) और αCD28 (सीडी 28.2) के साथ अच्छी तरह से संस्कृति प्लेटें । कोट ५०० µ एल के साथ प्लेटें 10 µ g/एमएल αCD3 और 10 µ g/एमएल αCD28 के लिए पंजाब के ३७ डिग्री सेल्सियस से कम 2 ज के लिए ।
नोट: कुछ loci के लिए, NHEJ पूर्व उत्तेजना के बिना प्राप्त किया जा सकता है, लेकिन इस कदम सहित अपनी दक्षता अधिकतम । - संस्कृति αCD3 पर ३७ डिग्री सेल्सियस पर ४८ ज के लिए टी कोशिकाओं/αCD28 एंटीबॉडी-बाउंड प्लेट्स एक RPMI पूर्ण माध्यम में [RPMI-१६४० HEPES के 5 मिमी के साथ पूरक, 2 मिमी के लिए वाणिज्यिक वैकल्पिक के एल-Glutamine, ५० µ g/एमएल के streptomycin, ५० µ एम के 2-mercaptoethanol, 5 मिमी के गैर आवश्यक अमीनो एसिड, सोडियम पाइरूवेट के 5 मिमी, और 10% (vol/FBS]. संस्कृति एक ४८ अच्छी तरह से थाली के प्रति अच्छी तरह से मीडिया के ५०० µ एल में २,०००,००० टी कोशिकाओं के घनत्व पर टी कोशिकाओं ।
- एक hemocytometer का उपयोग कर टी कोशिकाओं गिनती और electroporation प्रयोग के लिए आवश्यक टी कोशिकाओं की कुल संख्या (100000-1000000 टी कोशिकाओं को electroporated किया जा करने के लिए प्रति cuvette) एक केंद्रापसारक ट्यूब करने के लिए स्थानांतरण ।
- 8 मिनट के लिए ९० x g पर ट्यूब स्पिन कोशिकाओं को गोली । यदि 2 दिनों के भीतर कोशिकाओं को घनत्व ढाल-अलग कर दिया गया है, उंहें २०० x g पर 8 मिनट के लिए स्पिन ।
- टी सेल सक्रियकरण करने से पहले, पूर्व कोट ४८-αCD3 (UCHT1) और αCD28 (सीडी 28.2) के साथ अच्छी तरह से संस्कृति प्लेटें । कोट ५०० µ एल के साथ प्लेटें 10 µ g/एमएल αCD3 और 10 µ g/एमएल αCD28 के लिए पंजाब के ३७ डिग्री सेल्सियस से कम 2 ज के लिए ।
-
दोनों कक्ष प्रकार के लिए, किसी भी बुलबुले को हटाने, एक पिपेट/निर्वात के साथ supernatant महाप्राण ।
- धीरे cuvette प्रति electroporation बफर के 20 µ एल के साथ कोशिकाओं reसस्पेंड ।
- कोशिकाओं के 20 µ एल जोड़ें (150000-200000 HSPCs या 100000-1000000 टी कोशिकाओं) प्रत्येक cuvette है, जो पहले से ही µ के 10 RNP एल शामिल हैं, और अच्छी तरह से ऊपर और नीचे बुलबुले बनाने के बिना pipetting द्वारा मिश्रण ।
3. RNP Electroporation
- Electroporate को एक nucleofector में रखने के बाद cuvettes । HSPCs के लिए, पल्स कोड ER100 का उपयोग करें । T कक्षों के लिए, का उपयोग करें पल्स कोड एह-११५ ।
-
HSPCs: एक पूरक SFEM मध्यम के १०० µ एल जोड़ें (३७ डिग्री सेल्सियस के लिए गर्म) प्रत्येक cuvette के लिए तुरंत electroporation के बाद और कोशिकाओं 10-15 मिनट के लिए ठीक कर दें.
- उंहें एक ९६-अच्छी तरह से गोल-नीचे प्लेट में संस्कृति के लिए कोशिकाओं को हस्तांतरण और पूरक SFEM मध्यम के 24 ज के लिए एक अतिरिक्त १०० µ एल जोड़ें ।
- उंहें एक ताजा पूरक SFEM मध्यम करने के लिए बदलें और उंहें एक अतिरिक्त 24-72 एच के लिए मशीन ।
- genotyping के लिए उन कक्षों को निकालें 48-96 h post-electroporation. 5 मिनट के लिए ३०० x g पर कोशिकाओं स्पिन और डीएनए निष्कर्षण (८.२ कदम) शुरू करने से पहले supernatant हटा दें ।
-
केवल टी कोशिकाओं: RPMI पूरा संस्कृति मीडिया पूर्व के ८० µ एल जोड़ें जलाशय से प्रत्येक cuvette या अच्छी तरह के लिए ३७ डिग्री सेल्सियस से गरम, एक मल्टी चैनल पिपेट (यदि आवश्यक हो) का उपयोग कर ।
- उंहें 15 मिनट के लिए ३७ ° c पर मशीन ।
- उपयुक्त मीडिया, एंटीबॉडी, साइटोकिंस, आदि गंतव्य प्लेट (ओं) को जोड़ें और पूर्व उन्हें एक ३७ डिग्री सेल्सियस मशीन में गर्म ।
- स्थानांतरण १०७ कुओं से electroporated कोशिकाओं के µ एल एक गोल नीचे ९६-अच्छी तरह से एक बहु चैनल पिपेट का उपयोग कर प्लेट (यदि आवश्यक हो) ।
- संपादन परिणामों का आकलन करने के बारे में जानकारी के लिए, चरण 8 पर जाएं ।
४. ग. एलिगेंस तयारी
-
1 microinjection से पहले दिन: microinjection के लिए agarose पैड तैयार करें ।
- पानी में agarose जोड़ने और एक गर्म थाली पर या एक माइक्रोवेव में एक फोड़ा करने के लिए समाधान लाने के द्वारा एक 3% (डब्ल्यू/
- 24 मिमी x ५० मिमी x १.५ मिमी एक मेज पर कवर ग्लास स्लाइड की व्यवस्था और एक गिलास पाश्चर पिपेट का उपयोग करने के लिए स्लाइड पर agarose समाधान की एक छोटी (~ 15 µ एल) ड्रॉप जगह है । जल्दी से शीर्ष पर एक और coverslip रखकर agarose ड्रॉप समतल । agarose को जमना और फिर coverslips में से किसी एक को निकालने की अनुमति दें ।
- शुष्क करने के लिए रातोंरात एक तालिका के ऊपर agarose लेपित coverslip चेहरा छोड़ दें । 24 घंटे के बाद, एक साफ, सूखी कंटेनर में agarose पैड की दुकान ।
नोट: ये अनिश्चित काल के लिए इस्तेमाल किया जा सकता है ।
- microinjection सुई खींचो: रेशा के साथ borosilicate ग्लास केशिकाओं का उपयोग (बाहरी व्यास १.० मिमी और भीतरी व्यास ०.५८ मिमी), मेलो और आग के आधार पर सुई खींच५७ और अन्य संसाधनों५८. सुई तुरंत इस्तेमाल किया जा सकता है या एक साफ, सूखी कंटेनर में संग्रहित किया जा सकता है, मिट्टी से ब्रेस का समर्थन करता है ।
- कीड़े के रखरखाव के लिए, एक निमेटोड विकास मीडिया (NGM) पेट्री प्लेटों में डाला आगर तैयार है और OP50 बैक्टीरिया के साथ देखा (मानक सी. एलिगेंस रखरखाव और विकास मीडिया के लिए व्यंजनों पर प्रोटोकॉल के लिए, देखें Stiernagle५९) ।
- microinjection के लिए कीड़े स्टेज: 12-24 एच microinjection से पहले, उठाओ L4-OP50 बैक्टीरिया के साथ एक नया एनजी-आगर प्लेट के लिए hermaphrodites मंचन और 20 डिग्री सेल्सियस पर रात भर उन्हें गर्मी । प्रत्येक Cas9 लक्ष्य के लिए/इंजेक्शन मिश्रण, थाली के लिए ~ 30 कीड़े उठाओ ।
-
microinjection का दिन: RNP समाधान supernatant चरण १.५ में तैयार के साथ खींचा microinjection सुई लोड ।
- पिपेट एक खींच केशिका पिपेट में कदम 1.5.4 से supernatant और backfill तैयार पिपेट सुई में केशिका microinjection से समाधान (आम तौर पर ०.१ µ एल से कम लोड हो रहा है) ।
- एक micromanipulator से जुड़ी microinjection तंत्र पर लोड सुई माउंट । २५० केपीए और 25 केपीए के लिए संतुलन दबाव इंजेक्शन तंत्र दबाव सेट करें ।
-
एक तेज सुई बढ़त उत्पन्न करने के लिए वापस भरी हुई सुई टिप तोड़. एक 24 मिमी x ५० मिमी x १.५ मिमी coverslip के शीर्ष पर एक 15 मिमी x 15 मिमी x १.५ मिमी वर्ग coverslip प्लेस ।
- halocarbon तेल ७०० के साथ वर्ग coverslip के एक किनारे ओवरले ।
- तेल में सुई की स्थिति, 15 मिमी वर्ग coverslip के किनारे पर ।
- एक हाथ का उपयोग करने के लिए खुर्दबीन स्टेज और coverslip गाइड, स्लाइड ब्रश और सुई के किनारे के साथ जबकि निराशाजनक इंजेक्शन पेडल/ सुई टिप वापस तोड़, तरल की सुई से बाहर के प्रवाह में वृद्धि. सुई के किनारे के साथ इंजेक्शन मिश्रण प्रवाह बनाने के द्वारा एक इष्टतम प्रवाह दर पाना, ~ 1 बुलबुला बनाने/
- पुष्टि करें कि L4 कीड़े उठाया 12-24 microinjection करने से पहले एच इंजेक्शन के दिन पर विकास के युवा वयस्कों का मंचन कर रहे हैं. एक एनजी-आगर प्लेट कि OP50 बैक्टीरिया का अभाव है और उंहें 5 मिनट के लिए चारों ओर क्रॉल करने की अनुमति के लिए युवा वयस्क कीड़े उठाओ । यह इंजेक्शन पैड को हस्तांतरित बैक्टीरिया की मात्रा कम कर देता है, सुई को कम करता है ।
- एक विच्छेदन क्षेत्र पर एक agarose इंजेक्शन पैड/coverslip रखें । एक कीड़ा लेने का उपयोग करना, पैड के एक किनारे के साथ halocarbon तेल का एक छोटा सा ट्रैक करना ।
-
कीड़ा उठाओ तेल में लेपित का उपयोग करना, एनजी-आगर प्लेट से कई कीड़े उठा और तेल के ट्रैक में । एक ठीक एक पिपेट से जुड़े बालों के साथ, ऐसे एक बरौनी या बिल्ली मूंछ के रूप में, समानांतर में कीड़े की स्थिति, धीरे agarose पैड में कीड़े धक्का । microinjection प्रक्रिया के साथ सहज जब तक, केवल माउंट और एक समय में एक कीड़ा इंजेक्षन.
नोट: सूखी agarose कीड़े से नमी बाती, उंहें पैड का पालन करने के लिए कारण होगा । नतीजतन, एक जल्दी से काम करना चाहिए के रूप में कीड़े desiccate कर सकते हैं ।- एक बार स्थिति में और पैड से जुड़ी, कीड़ा लेने की नोक से halocarbon तेल (~ 20 µ एल) की एक और कुछ बूंदों के साथ कीड़े ओवरले ।
5. ग. एलिगेंस Gonad Microinjection के साथ RNPs और पोस्ट-इंजेक्शन की देखभाल
नोट: microinjection प्रोटोकॉल मेलो और आग५७से अनुकूलित और विस्तार से६०,६१कहीं और वर्णित है ।
-
इंजेक्शन माइक्रोस्कोप पर घुड़सवार कीड़ों के साथ coverslip रखें । एक कम आवर्धन (5x उद्देश्य, 10x आंख) के तहत, इंजेक्शन सुई को सीधा कीड़े की स्थिति ।
- स्विच करने के लिए एक उच्च आवर्धन (40X उद्देश्य, 10x नेत्र), gonad हाथ से सटे सुई की स्थिति नाभिक के पास इस क्षेत्र के लिए इसी के मध्य में देर से pachytene ।
- micromanipulator का प्रयोग, कीड़ा के खिलाफ सुई चाल, छल्ली थोड़ा निराशाजनक । फिर, एक हाथ से, छल्ली के माध्यम से सुई झटका करने के लिए माइक्रोस्कोप चरण की ओर नल. दबाना इंजेक्शन पेडल/बटन और धीरे से इंजेक्शन मिश्रण के साथ gonad हाथ भरने और सुई हटा दें ।
- अन्य gonad भुजा के साथ इस चरण को दोहराएँ.
-
एक बार कीड़े इंजेक्ट कर रहे हैं, coverslip/agarose पैड को हटाने और यह एक विदारक खुर्दबीन के नीचे जगह है ।
- एक खींचा केशिका पिपेट का प्रयोग, उन पर एक M9 बफर pipetting द्वारा कीड़े से तेल विस्थापित । इस उपचार के लिए आगर से कीड़े जारी करने के लिए प्रदर्शन ।
- 10 मिनट के बाद, जब कीड़े बफर में चारों ओर पिटाई कर रहे हैं, उंहें खींच लिया केशिका पिपेट का उपयोग कर OP50 बैक्टीरिया के साथ एक एनजी-आगर प्लेट में ले जाएं । 2-3 एच के लिए 20 ° c पर थाली प्लेस जब तक कीड़े बरामद कर लिया है और चारों ओर बढ़ रहे हैं ।
- एक बार बरामद, व्यक्तिगत रूप से एनजी के लिए कीड़े हस्तांतरण-आगर OP50 के साथ प्लेटें और एक 25 डिग्री सेल्सियस मशीन के लिए प्लेटों हस्तांतरण ।
-
पी0की अनुमति दें कीड़े बढ़ने के लिए और 3 दिनों के लिए संतति करना. च1 वंश स्क्रीन ।
- यदि सह-CRISPR या सह-रूपांतरण६२,६३,६४,६५का उपयोग कर रहे हैं, तो क्या वे संदर्भ जीन के उत्परिवर्ती phenotype है के आधार पर स्क्रीनिंग के लिए उंमीदवार कीड़े का चयन करें । व्यक्तिगत रूप से OP50 के साथ नए एनजी-आगर प्लेटों के लिए इन चिह्नित कीड़े हस्तांतरण और उन्हें 20 डिग्री सेल्सियस पर च2 संतान रखना करने के लिए अनुमति देते हैं ।
नोट: phenotype एक सह CRISPR स्क्रीनिंग या चयन के लिए इस्तेमाल किया Cas9 संपादन की सफलता के लिए एक प्रारंभिक अनुमान प्रदान करना चाहिए । - यदि कं CRISPR phenotype मौजूद न हो, तो microinjection दक्षता में सुधार लाने में सहायता करने के लिए एक सकारात्मक नियंत्रण प्लाज्मिड microinject ।
नोट: उदाहरण के लिए, इंजेक्शन मिश्रण में एक प्लाज्मिड है कि सांकेतिक शब्दों में बदलना mCherry-टैग MYO-2 इंजेक्शन दक्षता का आकलन करने में मदद मिलेगी शामिल है । कीड़े सफलतापूर्वक pCFJ90 के साथ इंजेक्शन फ्लोरोसेंट pharynxes के साथ कुछ वंश होगा ।
- यदि सह-CRISPR या सह-रूपांतरण६२,६३,६४,६५का उपयोग कर रहे हैं, तो क्या वे संदर्भ जीन के उत्परिवर्ती phenotype है के आधार पर स्क्रीनिंग के लिए उंमीदवार कीड़े का चयन करें । व्यक्तिगत रूप से OP50 के साथ नए एनजी-आगर प्लेटों के लिए इन चिह्नित कीड़े हस्तांतरण और उन्हें 20 डिग्री सेल्सियस पर च2 संतान रखना करने के लिए अनुमति देते हैं ।
- वांछित संपादन की उपस्थिति के लिए एफ1 कीड़े की जांच करें । एक ९६-अच्छी तरह से प्लेट के एक व्यक्ति के लिए एफ1 मां उठाओ, उसे लाइसे, और या तो डालने के विशिष्ट पीसीआर प्रवर्धन, डीएनए अनुक्रम विश्लेषण, या सर्वेयर nuclease परख (CEL-1)६६द्वारा उसके डीएनए की जांच ।
नोट: इन परख प्रदर्शन किया जा सकता है जब एक सह का उपयोग कर-CRISPR/सह रूपांतरण या अंय स्क्रीनिंग या चयन शासन६५,६६,६७,६८। - संपादन परिणामों का आकलन करने के बारे में जानकारी के लिए, चरण 8 पर जाएं ।
६. पी. hawaiensis तयारी
- 1 दिन पहले microinjection के लिए, ' एक जोड़ी ' टैंक की स्थापना के द्वारा जल्दी भ्रूण के लिए समृद्ध रात; नव अलग महिलाओं हौसले से निषेचित भ्रूण शामिल होंगे । देखें Rehm एट अल । विवरण के लिए ६९ ।
- microinjection के दिन पर, एकल सेल Parhyale भ्रूण (0-4 एच के बाद निषेचन) ०.०२% समुद्री जल में लौंग के तेल के साथ anesthetizing gravid महिलाओं द्वारा इकट्ठा करने और धीरे उसे ventral चिंता थैली के बाहर एक लौ का उपयोग कर भ्रूण scraping-खींच लिया और गोल ग् पिपेट और #3 संदंश की नीरस जोड़ी ।
7. P. hawaiensis भ्रूण Microinjection के साथ RNPs और पोस्ट-इंजेक्शन की देखभाल
- Backfill एक खींचा केशिका ट्यूब RNP इंजेक्शन मिश्रण के लगभग 1 µ एल के साथ ऊपर वर्णित है ।
-
Rehm एट अल के रूप में वर्णित प्रत्येक भ्रूण को microinject करने के लिए संपीड़ित नाइट्रोजन का उपयोग करें । ६९.
- एक microinjector और एक micromanipulator का उपयोग कर एक विदारक माइक्रोस्कोप के तहत Parhyale भ्रूण सुई । एक खींचा केशिका ट्यूब के पीछे में इंजेक्शन मिश्रण के १.५ µ एल लोड (4 इंच-१.० मिमी रेशा के साथ, एक micropipette खींच उपकरण का उपयोग कर खींच लिया) एक microloader पिपेट टिप का उपयोग कर.
- इंजेक्शन उपकरण पर सुई सेट और सुई की नोक तोड़ (एक बहुत छोटी राशि) विदारक गुंजाइश के तहत संदंश की एक जोड़ी का उपयोग कर । halocarbon तेल ७०० में इंजेक्शन और बुलबुला के व्यास को मापने के द्वारा वितरित की मात्रा जांचना ।
- काट एक ' गर्त ' इलाज एजेंट के बाहर एक उस्तरा ब्लेड का उपयोग कर । यह आधे रास्ते फ़िल्टर के साथ भरें-समुद्र के पानी निष्फल, और रेखा के गर्त में Parhyale भ्रूण को स्थिर करने के लिए ।
- इंजेक्शन के दौरान संदंश की एक जोड़ी के साथ प्रत्येक भ्रूण स्थिर, microinjection सेटअप का उपयोग कर भ्रूण इंजेक्षन । इंजेक्शन के बाद, एक गिलास स्थानांतरण पिपेट का उपयोग करने के लिए एक ताजा ६० मिमी संस्कृति आधी फिल्टर के साथ आधे रास्ते भर-निष्फल समुद्र के पानी को भ्रूण हस्तांतरण ।
-
यदि पहले विभाजन पहले से ही एक 2-सेल भ्रूण (4-6 एच के बाद निषेचन), दोनों blastomeres इंजेक्शन द्वारा पूरी तरह से उत्परिवर्ती पशुओं उत्पंन करने के लिए हुआ है । 2-सेल चरण की कुल दरार सुनिश्चित करने के लिए, सह blastomeres FITC या TRITC dextran के साथ सुई और निरीक्षण है कि संकेत इंजेक्शन के बाद एक फ्लोरोसेंट विदारक गुंजाइश के तहत एक एकल blastomere करने के लिए प्रतिबंधित है ।
- वैकल्पिक रूप से, 2-सेल स्टेज पर दो blastomeres में से सिर्फ एक इंजेक्शन द्वारा ' हाफ ' उत्परिवर्ती जानवरों उत्पन्न (मोटे तौर पर एक-पी धुरी के साथ ऊतक और स्थिति के आधार पर बाएँ-दाएँ विभाजित).
- एक 8 सेल भ्रूण में एक सेल सुई (7.5-9 एच के बाद निषेचन) एक एकल रोगाणु परत करने के लिए संपादन को प्रतिबंधित करने के लिए । देखें Gerberding एट अल । जल्दी blastomere वंश के एक नक्शे के लिए ७०
-
६० mm संस्कृति व्यंजन में भ्रूण की मशीन (कोई 25 से अधिक पकवान प्रति), आधे रास्ते फ़िल्टर के साथ भर-निष्फल समुद्री जल, ' पूर्व oxygenated ' एक मछलीघर bubbler का उपयोग कर या जोरदार झटकों से ।
- एक ढीले-सील गीले कागज तौलिए के साथ लाइन में खड़ा करने के लिए नमी बनाए रखने और उंहें एक 26 डिग्री सेल्सियस के साथ एक 12 एच प्रकाश अंधेरे चक्र में जगह plasticware में भ्रूण के व्यंजन रखें ।
- हर कुछ दिनों में समुद्री जल व्यंजन स्वच्छ करने के लिए जीवित भ्रूण स्थानांतरण ।
नोट: भ्रूण कमरे के तापमान पर प्रसंस्कृत किया जा सकता है, हालांकि वे बहुत अधिक धीरे विकसित होगा ।
-
काटना और एक अभिव्यक्ति विश्लेषण के लिए विभिंन चरणों में सीटू संकरण या एंटीबॉडी धुंधला में द्वारा भ्रूण को ठीक (ब्राउन एट अल देखें । ७१ स्टेजिंग मार्गदर्शिका के लिए, और विच्छेदन और निर्धारण के लिए अतिरिक्त संदर्भ७२, में सीटू संकरण७३, और एंटीबॉडी धुंधलान७४) ।
- लगभग ०.५ में लंबाई में एक इंसुलिन सुई के अंत में टंगस्टन तार का एक तुला टुकड़ा थ्रेडिंग द्वारा विच्छेदन सुई बनाओ । एक मौजूदा के तहत सोडियम हीड्राकसीड में सुई पैनापन । विच्छेदन सुई के हैंडल के रूप में एक 1 मिलीलीटर सिरिंज का उपयोग करें ।
- एक 3-अच्छी तरह से कांच पकवान आधे रास्ते के एक नए सिरे से 9 भागों PEM बफर (पाइप के ०.१ मीटर पीएच ६.९५, EGTA के 2 मिमी, MgSO4के 1 मिमी), 1 भाग 10x पंजाबियों, और 1 भाग का हल के साथ एक अच्छी तरह से भरें ३२% पीएफए । पकवान में 3-5 भ्रूण प्लेस और प्रत्येक भ्रूण में एक छोटे से छेद प्रहार, प्रहार करने के लिए एक तेज टंगस्टन सुई का उपयोग करने और एक थोड़ा सुस्त एक स्थिर करने के लिए, जर्दी बाहर प्रवाह और निर्धारण में चलाने के लिए अनुमति देता है
- तेज टंगस्टन सुई की एक जोड़ी का उपयोग करना, धीरे दूर दो Parhyale भ्रूण आसपास की झिल्ली को चिढ़ाने । भ्रूण को और अधिक मजबूत बनाने के लिए उन्हें काटना निर्धारण में अधिक मेहनत करनी पड़ती है लेकिन भ्रूण को निश्चित बनने से झिल्ली को रखने में जल्दी काम आता है, जो झिल्ली हटाने को अधिक कठिन बना देता है. भ्रूण की अनुमति के लिए 15-20 min. एंटीबॉडी धुंधला या सीटू संकरण में के लिए 40-50 मिनट के लिए कुल के लिए ठीक ।
- छवि लाइव hatchlings और रूपात्मक और व्यवहार phenotypes या फिक्स के लिए उन्हें विश्लेषण और अधिक विस्तृत विश्लेषण के लिए उन्हें दाग. 2-3 महीने में यौन परिपक्वता को hatchlings उठाएं नॉकआउट और ट्रांसजेनिक लाइनों की स्थापना (hatchling देखभाल और अंय उपयोगी जानकारी के लिए Kontarakis और Pavlopoulos७५ देखें) ।
8. संपादन परिणामों का आकलन
- यदि लागू हो, तो संपादित कक्षों या जीवों में विज़ुअल या कार्यात्मक phenotype देखें.
नोट: यह प्रक्रिया विस्तृत रूप से अनुप्रयोग द्वारा भिन्न हो जाएगी, और कुछ उदाहरण ऊपर उनके प्रासंगिक प्रोटोकॉल चरणों के अंत में वर्णित हैं । HSPCs में सिकल सेल उत्परिवर्तन को सही करने के बाद, HPLC (चित्रा 1) का उपयोग करके विभेदित erythroblasts द्वारा हीमोग्लोबिन उत्पादन का विश्लेषण करें । टी कोशिकाओं में आईएल-2 रिसेप्टर जीन की एक नॉकआउट सतह धुंधला और फ्लो cytometry (चित्रा 1बी) द्वारा पुष्टि की जा सकती है । C. एलिगेंस और P. hawaiensis phenotypes का आकलन करने के लिए, एक प्रकाश या फ्लोरोसेंट माइक्रोस्कोपी (आंकड़े 1C और 1 डी) के तहत पशु आकृति विज्ञान और व्यवहार का निरीक्षण करें । - जीनोमिक संपादन उत्पन्न की दक्षता और प्रकार का निर्धारण करने के लिए, संपादित कोशिकाओं के पूल लाइसे और एक वाणिज्यिक निष्कर्षण किट21का उपयोग कर अपने जीनोमिक डीएनए निकालने ।
-
indel गठन का एक त्वरित आकलन के लिए, पीसीआर-कम २०० कट साइट के आसपास आधार जोड़े को बढ़ाना और एक T7 endonuclease1 (T7E1)७६ या सर्वेयर (CEL-1 nuclease) परख७७प्रदर्शन ।
- यदि Cas9-कट साइट या सफल HDR पर एक indel गठन बनाने के लिए या एक ज्ञात प्रतिबंध साइट को दूर करेगा, एक प्रतिबंध एंजाइम पाचन का उपयोग करने के लिए संपादन क्षमता6का अनुमान लगाने पर विचार करें । प्रतिबंध टुकड़ा लंबाई बहुरूपता (RFLP) परख एक सुविधाजनक तरीका है अगर यह उपलब्ध होना होता है की जांच करने के लिए हो सकता है ।
- संपादन दक्षता और प्रमुख संपादन परिणामों के निर्धारण का एक सटीक ठहराव के लिए, दोनों आगे और रिवर्स प्राइमरों के साथ एक मानक सैंज अनुक्रमण के लिए पीसीआर amplicon भेजें ।
नोट: यदि एक एकल क्लोन या जीव का विश्लेषण, सैंज परिणामों के विश्लेषण सरल है, के रूप में चित्रा 2एमें प्रदर्शन किया । यदि कक्षों का एक पूल विश्लेषण कर रहा है, तो ऑनलाइन उपकरण७८के साथ chromatograms का विश्लेषण करें, जैसा कि चित्र 2Bमें दिखाया गया है । - एक पूर्ण ठहराव और परिणामों के संपादन के दृश्यों के लिए, गहरी अनुक्रमण27,५४, के रूप में चित्रा 2सीमें चित्रित प्रदर्शन ।
- बंद लक्ष्य परिवर्तन की एक विशेष सेट का आकलन करने के लिए, पीसीआर-अनुमानित बंद लक्ष्य साइटों को बढ़ाना और उन्हें NGS के लिए भेजते हैं । गुणसूत्र अनुवादन का पता लगाने को सक्षम करने के लिए, मार्गदर्शिका-seq७९ या उच्च-प्रवाह, जीनोम-वाइड translocation sequencing (HTGTS)८०निष्पादित करें । एक क्लोनल जनसंख्या में बंद लक्ष्य संपादन की एक पूरी तस्वीर के लिए, पूरे जीनोम अनुक्रमण (WGS)८१,८२,८३प्रदर्शन करते हैं ।
नोट: वहां पर और ऑफ लक्ष्य जीनोम संपादन को बढ़ाता है, विभिंन समीक्षा लेख८४,८५,८६में आगे समझाया के लिए तरीकों की एक किस्म है ।
Representative Results
इन प्रयोगों से पता चलता है कि कैसे पूर्व इकट्ठे Cas9 RNP को प्राथमिक कोशिकाओं और पूरे जीवों के जीनोम में हेरफेर किया जा सकता है. शोधकर्ताओं शुद्ध या Cas9 प्रोटीन और sgRNA खरीद, दो घटकों के संयोजन के लिए पूर्व फार्म का परिसर, और उनकी कोशिकाओं या ब्याज की जीव में RNP परिचय । संपादन के लिए पर्याप्त समय की अनुमति के बाद होने के लिए और अगली पीढ़ी के वंश के लिए पैदा हो (यदि लागू हो), phenotypes के लिए जांच करें और/या genotyping के लिए कोशिकाओं को इकट्ठा । Phenotypes कार्यात्मक परख, अभिव्यक्ति परख, दृश्य के द्वारा देखा जा सकता है (आंख या माइक्रोस्कोप के साथ), या अंय तरीकों, प्रयोग पर निर्भर करता है ।
उदाहरण के लिए, HSPCs कि सिकल सेल रोग का कारण बनता है β-ग्लोबिन उत्परिवर्तन सही करने के लिए संपादित किया गया है एरिथ्रोसाइट्स में विभेदित किया जा सकता है और स्वस्थ या सिकल हीमोग्लोबिन के उत्पादन के लिए परख27,८७ (चित्रा 1 अ). टी कोशिकाओं को बाहर उच्च संबंध IL-2 रिसेप्टर जीन दस्तक संपादित, CD25 (IL2RA), सतह धुंधला और प्रवाह cytometry८८द्वारा विश्लेषण किया जा सकता है, और कार्यात्मक IL-2 उत्तेजना के लिए एक संकेतात्मक प्रतिक्रिया का पता लगाने के लिए विश्लेषण (चित्रा 1बी ). टी कोशिकाओं को भी कई नैदानिक महत्वपूर्ण तरीके है कि विभिंन phenotypes के आकलन की आवश्यकता है, एचआईवी संक्रमण की प्रभावकारिता८९ और कार टी कोशिकाओं11के vivo में अर्बुदरोधी प्रभावकारिता में शामिल किया जा सकता है ।
एक सह-CRISPR/सह-रूपांतरण जांच दृष्टिकोण का उपयोग करके, C. एलिगेंस वर्म एक साथ दो loci६२में संपादित किए जाते हैं । dpy पर HDR -10 संदर्भ जीन एक ssODN मरंमत का उपयोग कर एक आसानी से-प्रमुख dpy-10 लाभ-समारोह उत्परिवर्तन बनाए में टेंपलेट परिणाम । Heterozygous च1dpy-10 (गोफ) जानवर हैं रोलर (Rol) और homozygous dpy-10 (गोफ) पशुओं को डंपिंग (dpy) कर रहे हैं । phenotype की उपस्थिति इंगित करता है कि Cas9 संपादन इन जानवरों में हुई और Rol या Dpy च1 जानवरों में दूसरी लोकस में एक संपादन घटना की पहचान की बाधाओं को बेहतर बनाता है । एक सफल संपादन प्रयोग इंजेक्शन पी के 33-50% में परिणाम चाहिए0 कीड़े 20 या अधिक F1 वंश कि Rol या Dpy९०है उपज । यह तो गैर Rol पशुओं का चयन करने के लिए wildtype के लिए dpy-10 वापस करने के लिए और ब्याज की homozygous संपादित करने के लिए चयन संभव है. अंगूठे का एक नियम के रूप में, सह लक्ष्यीकरण crRNA की एकाग्रता-CRISPR संदर्भ जीन आधा होना चाहिए कि crRNA के हित के जीन लक्ष्यीकरण । यदि ब्याज की जीन में एक संपादित करें बरामद नहीं है, दो CRISPR RNAs के अनुपात में वांछित उत्परिवर्तन उबरने की संभावना को बढ़ाने के लिए समायोजित किया जा सकता है । उदाहरण के लिए, संदर्भ जीन crRNA के सापेक्ष ब्याज के जीन के लिए crRNA की मात्रा बढ़ाने से कीड़े की आबादी के भीतर ब्याज के जीन में संपादन रखने वाले कीड़े का प्रतिशत बढ़ेगा जो कि संदर्भ जीन लोकस पर भी संपादनों के अधिकारी हैं । सह-रूपांतरण आवृत्तियों बदलती हैं, लेकिन दरों आमतौर पर 20-60%, अक्सर एफ1 जनरेशन (चित्रा 1सी) में homozygous संपादन उपज रहे हैं ।
पी. hawaiensis hatchlings कि उदर-बी जीन (एबीडी-बी) का प्रदर्शन स्पष्ट रूपात्मक विषमताओं3 (चित्रा 1डी) बाहर दस्तक करने के लिए संपादित किया गया है. यह जीन सही पेट के नमूनों के लिए आवश्यक है, और इसके विघटन का परिणाम वक्ष-प्रकार में कूदते और पैदल चलने वाले पैरों की जगह तैराकी और लंगर वाले पैरों का होता है जो आमतौर पर पेट पर मौजूद होते हैं.
genotypic स्तर पर जीनोम संपादन परिणामों का निर्धारण या तो अनुक्रमण या इन विट्रो परख है कि अनुक्रम परिवर्तन का पता लगाता है की आवश्यकता है । यहाँ, हम हमारे मॉडल सेल प्रकार और जीवों से प्रतिनिधि अनुक्रमण डेटा दिखाएँ, संपादन ठहराव के लिए विभिन्न दृष्टिकोण पर प्रकाश डाला. ध्यान दें कि आंकड़ा लेबल सामान्यीकृत है क्योंकि सभी तरीकों यहां दिखाया गया है किसी भी जैविक प्रणाली के लिए लागू किया जा सकता है ।
sequencing-आधारित दृष्टिकोण तकनीकी जटिलता और परिणामों की गहराई में बदलती हैं । क्लोनेड संपादित आबादी या आसानी से-अविभाज्य व्यक्ति जीवों के लिए, संपादित व्यक्तियों जीनोमिक डीएनए निष्कर्षण के बाद अनुक्रम किया जा सकता है । मानक सैंज अनुक्रमण परिणाम किसी दिए गए व्यक्ति में Cas9-कट साइट पर अनुक्रम परिवर्तन प्रकट होगा, काल्पनिक frameshifts है कि इसके समारोह (चित्रा 2ए) को बाधित करेगा के साथ । अनुक्रमण के लिए इस्तेमाल किया ऑनलाइन उपकरण एक और सैंज अनुक्रमण आधारित दृष्टिकोण है कि मिश्रित आबादी के बजाय व्यक्तिगत म्यूटेंट७८के लिए लागू किया जा सकता है । अनुक्रम एक ऑनलाइन उपकरण है कि लगभग समग्र संपादन दक्षता के रूप में अच्छी तरह से प्रमुख अनुक्रम परिणामों के साथ कर सकते है विश्लेषण कर रहे हैं । प्रतिनिधि डेटा चित्रा 2Bमें दिखाए जाते हैं ।
यहाँ वर्णित सबसे गहन अनुक्रमण विधि गहरी अनुक्रमण है (कभी-कभार उच्च प्रवाह या अगली पीढ़ी के अनुक्रमण के रूप में जाना जाता है) । यह विधि मिश्रित जनसंख्या वाले व्यक्ति जीनोम से डीएनए अनुक्रम प्रदान करती है । इस तरह के डेटा को कई तरह से सचित्र किया जा सकता है । यहाँ, हम वर्गीकृत संपादन परिणाम (चित्रा 2सी) के आधार पर संपादित कोशिकाओं से व्यक्तिगत अनुक्रमण पढ़ता है. अधिकांश कोशिकाओं NHEJ मार्ग है, जो आम तौर पर जीन व्यवधान में परिणाम के माध्यम से संपादित कर रहे हैं । दूसरों में, लक्ष्य जीन है HDR27के माध्यम से एक वैकल्पिक संस्करण के लिए बाहर बदली गई है ।
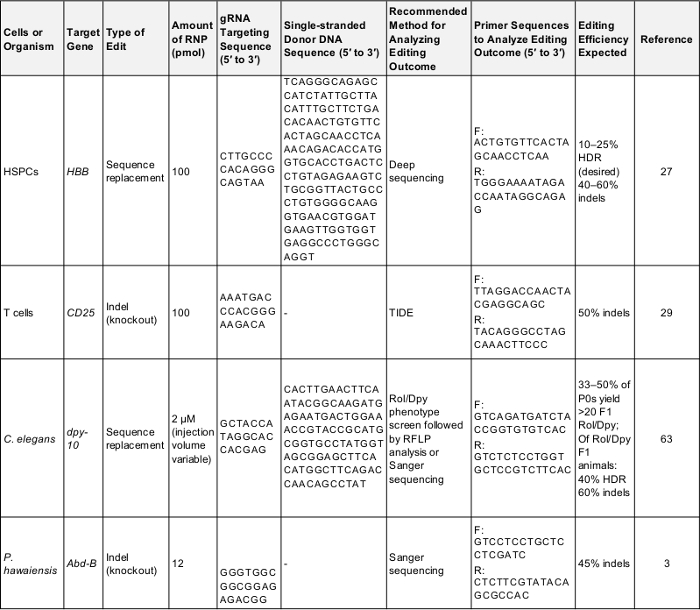
तालिका 1: प्रारंभिक जीनोम संपादन प्रयोगों के लिए सकारात्मक नियंत्रण. यह तालिका इस प्रोटोकॉल में वर्णित कक्षों और जीवों में से प्रत्येक में पहली बार जीनोम संपादन प्रयोग को करने के लिए आवश्यक महत्वपूर्ण जानकारी दिखाती है । इन पैरामीटर्स के बाद एक सफल परिणाम है जो प्रोटोकॉल या तुलना के लिए एक आधार रेखा के रूप में परीक्षण करने के लिए उपयोग किया जा सकता है एक बार प्रयोगकर्ता अपनी रुचि का एक जीन को लक्षित कर रहा है उपज होने की संभावना है । एफ: फॉरवर्ड, आर: रिवर्स, HDR: समरूपता-मरंमत का निर्देश दिया । इस तालिका को डाउनलोड करने के लिए कृपया यहां क्लिक करें.

चित्र 1 : प्रतिनिधि phenotypic परिणाम प्राथमिक मानव कोशिकाओं और जीवों के Cas9 RNP संपादन से । (क) यह एक HPLC ट्रेस दिखा रहा है कि सफल जीनोम संपादन के बाद, HSPCs कि देर चरण erythroblasts में अंतर कर रहे है सिकल हीमोग्लोबिन से अधिक कार्यात्मक हीमोग्लोबिन का उत्पादन होगा । उत्परिवर्ती एरिथ्रोसाइट्स सिकल हीमोग्लोबिन (एचबीएस) का उत्पादन, जबकि सफलतापूर्वक संपादित कोशिकाओं स्वस्थ हीमोग्लोबिन (बांधणी और HbA2) के रूप में के रूप में अच्छी तरह से भ्रूण हीमोग्लोबिन (HbF) का उत्पादन होगा. अवशोषक मनमाना इकाइयों (au) में ग्राफ है । यह पैनल सबसे पहले डेविट ईटी अल में प्रकाशित हुआ था । 27. यह विज्ञान की उंनति के लिए अमेरिकन एसोसिएशन से अनुमति के साथ पुनर्मुद्रित है । छोड़ दिया पर (ख) , प्रत्येक शर्त के लिए, इस पैनल प्रवाह cytometry डेटा दिखा रहा है कि सतह से सना हुआ टी कोशिकाओं CD25 व्यक्त नहीं है के बाद CD25 जीन है RNP के साथ बाहर खटखटाया गया है । CD25 बहुतायत x-अक्ष पर y-अक्ष पर कक्ष आकार के साथ प्लॉट किए गए हैं । सही पर, प्रत्येक शर्त के लिए, इस पैनल Phospho-Stat5 (pStat5) ठहराव आईएल-2 के साथ एक प्रेरण के बाद पता चलता है । संकेतन कम है जब IL-2 रिसेप्टर अनुपस्थित है (CD25 KO) । pStat5 बहुतायत x-अक्ष पर और तीन आईएल-2 इनपुट के विभिंन स्तरों से उत्पंन डेटा खड़ी तुलना कर रहे है पर साजिश रची है । (ग) यह पैनल सह-रूपांतरण मार्कर के रूप में एक Caenorhabditis एलिगेंस सह-CRISPR/सह-रूपांतरण स्क्रीन लक्ष्यीकरण dpy-10 दिखाता है. दो गाइड RNAs लक्ष्य दो loci, dpy-10 और अपने पसंदीदा जीन (yfg), एक ही पी में0इंजेक्शन जानवर । HDR at dpy-10 result a Rol or dpy phenotype. Rol-या Dpy-F1 पशुओं के चयन से दूसरे लोकस में संपादनों की पहचान करने की संभावना बढ़ जाती है । (घ) इस पैनल से पता चलता है कि wildtype Parhyale hawaiensis hatchlings में तैराकी और एंकर पैरों के साथ सामान्य पेट होता है । एबीडी-बी नॉक आउट hatchlings (एफ0 व्यक्ति) छाती की दिशा में परिवर्तित पेट विकसित करते हैं. इस प्रकार, तैराकी और लंगर पैर चला रहे है और कूद और एक सामांय छाती के साथ जुड़े पैर से बदल दिया । कृपया यहां क्लिक करें इस आंकड़े का एक बड़ा संस्करण को देखने के लिए ।

चित्र 2 : संपादन परिणाम विश्लेषण विधियों से विशिष्ट परिणाम । (क) यह पैनल व्यक्तिगत एफ१ पी. hawaiensis जीवों से wildtype अनुक्रम और तीन विभिन्न indels सहित, जो कि ओपन रीडिंग फ्रेम को शिफ्ट करके जीन फंक्शन को बाधित करता है, से सैंज अनुक्रमणक परिणामों के उदाहरण दिखाता है. (B) ये ज्वार परिणाम किसी Cas9-लक्ष्य साइट पर sequenced T कक्षों के किसी पूल में हुई सम्मिलन और हटाने की घटनाओं की श्रेणी दिखाते हैं । x-अक्ष न्यूक्लियोटाइड में दिए गए सम्मिलन या हटाने की लंबाई को इंगित करता है. (ग) इन गहरी अनुक्रमण परिणाम nucleofection या gRNA बिना कोई जीनोम संपादन, और बरकरार Cas9 RNP के साथ सफल संपादन, HSPCs में डीएनए की मरंमत के परिणाम द्वारा समूहीकृत दिखा । कृपया यहां क्लिक करें इस आंकड़े का एक बड़ा संस्करण देखने के लिए ।
Discussion
एक सेल लाइन या ब्याज की जीव में एक मजबूत जीनोम संपादन प्रोटोकॉल की स्थापना के अनुकूलन और कई प्रमुख मापदंडों के अनुभवजंय परीक्षण, इस खंड में चर्चा की आवश्यकता है । यहां प्रस्तुत सामांय दृष्टिकोण के कुछ रूपों की कोशिश कर रहा है अत्यधिक प्रोत्साहित किया । इस प्रोटोकॉल की कुंजी सीमा है कि अंय कोशिकाओं या जीवों के लिए इन तरीकों को लागू करने का अध्ययन प्रजातियों के आधार पर एक अलग परिणाम के लिए नेतृत्व कर सकते हैं, और एक प्रयोगात्मक डिजाइन कि एक उच्च दक्षता जीन नॉकआउट करने के लिए सुराग डीएनए प्रविष्टि को बढ़ावा नहीं कर सकते हैं । इस प्रकार, हम अनुशंसा करते है यहां प्रस्तुत तरीकों के साथ शुरू करने और नीचे वर्णित के रूप में समस्या निवारण ।
जीनोम संपादन एजेंट गुणवत्ता समस्या निवारण:
उत्पादन या क्रय उच्च गुणवत्ता वाले एजेंट किसी भी जीनोम संपादन प्रोटोकॉल में एक महत्वपूर्ण कदम है । Cas9 प्रोटीन लैब में शुद्ध किया जा सकता है या व्यावसायिक रूप से खरीदा है । कई प्रोटोकॉल RNP व्यंजनों में Cas9 के लिए एक अंतिम एकाग्रता ध्यान दें, लेकिन इष्टतम जीन संपादन गतिविधि किसी भी व्यक्ति Cas9 प्रोटीन की तैयारी है, जो स्रोत के आधार पर बदलता है की विशिष्ट गतिविधि पर निर्भर करेगा । एक बार प्रोटोकॉल यहां प्रस्तुत काम कर रहा है, titrating Cas9 स्तर द्वारा प्रयुक्त RNP की राशि के अनुकूलन पर विचार के लिए एक इष्टतम एकाग्रता की स्थापना: एक कि अनावश्यक बंद लक्ष्य दरार के बिना अत्यधिक विशिष्ट लक्ष्य डीएनए दरार प्रदान करता है के कारण अत्यधिक Cas9४०।
गाइड आरएनए शुद्धता और एकरूपता भी जीनोम संपादन सफलता22के निर्धारकों हो सकता है । खरीदा sgRNAs या अलग crRNA और tracrRNA घटकों को आम तौर पर उच्च गुणवत्ता के एजेंट हैं और रासायनिक संशोधनों की एक किस्म आरएनए गिरावट के साथ समस्याओं का मुकाबला करने के लिए या RNP९१के लिए अतिरिक्त सुविधाओं को रंगना करने के लिए उपलब्ध हैं । जबकि रासायनिक संशोधित gRNAs मानक जीनोम संपादन प्रयोगों के लिए आवश्यक नहीं हो सकता है, कुछ समूहों में ऐसे रिएजेंट के साथ बहुत अधिक संपादन क्षमता देखा है, तो वे प्रक्रिया माहिर के बाद की कोशिश कर रहा लायक हो सकता है और/या जब gRNA क्षरण एक समस्या22,९१प्रतीत होता है । इन विट्रो प्रतिलेखन और बाद में जेल शुद्धिकरण एक सस्ता विकल्प है, जो नियमित जीनोम संपादन प्रयोगों17,21,४९,५०के लिए पर्याप्त हो सकता है । इसके अलावा, कई दृष्टिकोण है कि सामांयतः vivo मेंसमरूप gRNA आबादी का उत्पादन करने के लिए लागू कर रहे हैं, सहित ribozyme-और tRNA-व्यक्तिगत गाइड के आधारित उत्पाद, इन विट्रो में करने के लिए बढ़ाया जा सकता है आरएनए तैयारी क्लीनर उत्पन्न करने के लिए उत्पाद९२।
गाइड आरएनए और दाता डीएनए डिजाइन युक्तियां:
गाइड आरएनए चयन बंद लक्ष्य दरार की संभावना को कम करते हुए अत्यधिक कुशल पर लक्ष्य संपादन को प्राप्त करने में एक महत्वपूर्ण कारक है । गाइड के चयन में सहायता करने के लिए, कई अध्ययनों से सफल गाइड के अनुक्रम सुविधाओं को संकलित करने के लिए उच्च प्रवाह अगली पीढ़ी के अनुक्रमण के साथ युग्मित स्क्रीन का उपयोग किया है४७,७९,९३,९४, ९५,९६. इन सुविधाओं के लिए पूर्वानुमानित एल्गोरिदम और ऑनलाइन उपकरण गाइड चयन में सहायता करने के लिए विकसित करने के लिए उपयोग किया गया है४४,४५,४६,४७,४८। इस तरह के एल्गोरिदम गाइड आरएनए अभिव्यक्ति के लिए डीएनए आधारित सिस्टम का उपयोग कर स्क्रीन पर जमीन कर रहे हैं. गाइड एक पोल iii प्रमोटर का उपयोग कर व्यक्त कर रहे हैं, और उनकी अभिव्यक्ति इसलिए ऐसे समयपूर्व समाप्ति के रूप में पॉल तृतीय प्रतिलेखन, के साथ जुड़े सीमाओं से ग्रस्त है जब uracil की पटरियों का सामना कर रहे हैं९७,९८, ९९. हालांकि, RNPs के उपयोग के साथ बनाया इन विट्रो-संश्लेषित गाइड RNAs उन चिंताओं को दरकिनार और गाइड डिजाइन पर बाधाओं को सरल । एक आम सुविधा है कि इन एल्गोरिदम से उभरा है और अत्यधिक प्रभावी जीनोम संपादन के साथ कई अध्ययनों में पुष्टि की गई है, एक प्यूरीन की उपस्थिति है, विशेष रूप से एक guanine, ' 3 गाइड लक्ष्य विशेष अनुक्रम के अंत में । इस गाइड सुविधा स्तनधारी से सी. एलिगेंस, फल मक्खियों, और zebrafish६५,१००,१०१से लेकर जीवों के बीच बहुत सफल रहा है । इसके अलावा, C. एलिगेंसके लिए, मार्गदर्शिका के लक्ष्यीकरण क्षेत्र के 3 ' अंत में एक जीजी dinucleotide के साथ मार्गदर्शिकाएं डिज़ाइन करना अत्यधिक प्रभावी गाइड RNAs६५की भविष्यवाणी के लिए एक प्रभावी रणनीति है । आदर्श रूप में, परीक्षण कई गाइड समानांतर में निर्धारित करने के लिए जो किसी दिए गए आवेदन के लिए सबसे सफल है ।
जब जीनोम में एक डीएनए अनुक्रम परिचय का प्रयास, दाता या टेंपलेट डीएनए के डिजाइन भी महत्वपूर्ण है । एकल असहाय oligonucleotide दाताओं (ssODNs) अंय ठेठ मरंमत टेंपलेट्स, रैखिक डबल फंसे और प्लाज्मिड डीएनए५४,५५,१०२से अधिक मज़बूती से डाला जाता है । कुछ loci पर, HDR दक्षता ssODNs कि गैर-लक्ष्य या विस्थापित डीएनए कतरा के पूरक हैं और समरूपता हथियार है कि लंबाई27,५५में असममित हैं अधिकारी के साथ सुधार किया जा सकता है. चूंकि मरंमत टेम्पलेट कट साइट पर डाला जा रहा है और लक्षित अनुक्रम भी शामिल है, कदम Cas9 से पहले या जीनोमिक प्रविष्टि के बाद दाता डीएनए सट से रोकने के लिए लिया जाना चाहिए । इस पाम अनुक्रम या बीज क्षेत्र में मूक उत्परिवर्तनों बनाने के द्वारा पूरा किया है, Cas9 द्वारा मांयता से परहेज करते हुए डाला जीन21,१०३के समारोह को बनाए रखने । हालांकि पाम में भी एकल न्यूक्लियोटाइड परिवर्तन के लिए बाध्यकारी१०४को समाप्त करने की संभावना है, को बदलने की कोशिश कम से चार न्यूक्लियोटाइड सुरक्षित हो ।
महत्व और भविष्य अनुप्रयोगों:
CRISPR के साथ जीनोम संपादन-Cas9 किसी भी जीव के सतही आनुवंशिक हेरफेर को सक्षम करने के लिए एक शक्तिशाली विधि के रूप में उभरा है । Cas9 RNP के साथ संपादन पहले थोड़ा और अधिक प्रयास लेता है, लेकिन एक बार रिएजेंट और प्रोटोकॉल एक प्रयोगशाला में स्थापित कर रहे है का उपयोग करने के लिए सीधा है । प्लाज्मिड डीएनए के बजाय पूर्व इकट्ठे RNP के साथ संपादन कोशिकाओं उच्च समग्र संपादन क्षमता की ओर जाता है, HDR के माध्यम से मुश्किल से प्राप्त जीन प्रविष्टि सहित, कम से दूर लक्ष्य प्रभाव के साथ24,25,26 , 27 , 29. इसके अलावा, प्रयोगकों जीन अभिव्यक्ति, आरएनए गिरावट, प्रोटीन तह, और gRNA और Cas9 सेल के भीतर अलग से संश्लेषित अणुओं के बीच संघ22,23के साथ समस्याओं से बचें । RNP संपादन भी संमिलनत्मक mutagenesis और निरंतर अभिव्यक्ति है कि जब वायरल वितरण तरीकों नैदानिक14इस्तेमाल कर रहे है पैदा हो सकता है के बारे में सुरक्षा चिंताओं को दरकिनार । इन फायदों की वजह से, कई वैज्ञानिकों का आयोजन पूर्व नैदानिक, सबूत की अवधारणा प्रयोगों मानव चिकित्सीय अनुप्रयोगों के लिए RNP संपादन एहसान । दोनों vivo में और पूर्व vivo RNP-आधारित जीनोम संपादन दृष्टिकोण विकास में है या यहां तक कि शर्तों की एक किस्म का इलाज, Duchenne पेशी dystrophy१०५ और सिकल सेल रोग की तरह आनुवंशिक रोगों से27 करने के लिए एचआईवी29 और कैंसर11। दिलचस्प है, Cas9 RNP तेजी से कृषि इंजीनियरिंग के लिए एक वितरण पद्धति के रूप में कार्यरत है, क्योंकि यह ' सक्षम बनाता है डीएनए मुक्त ' पौधों के संपादन३३,३४,३६।
Disclosures
लेखक अलेक्जेंडर Marson और याकूब ई. मकई स्पॉटलाइट चिकित्सकीय के सह-संस्थापक हैं । याकूब ई. मकई एक मिशन के चिकित्सीय सलाहकार है और उसकी प्रयोगशाला AstraZeneca और फाइजर से प्रायोजित अनुसंधान समर्थन प्राप्त हुआ है । अलेक्जेंडर Marson चिकित्सकीय और समझौता चिकित्सकीय जूनो के लिए एक सलाहकार है, और उसकी प्रयोगशाला जूनो चिकित्सकीय, Epinomics, और सनोफी से प्रायोजित अनुसंधान समर्थन प्राप्त हुआ है । उनकी प्रयोगशाला ने Cas9 RNP तकनीक से संबंधित पेटेंट के लिए भी आवेदन किया है ।
Acknowledgments
हम अपने प्रयोगशालाओं के कई पिछले सदस्यों और इन तरीकों के विकास के लिए उनके योगदान के लिए खाड़ी क्षेत्र जीनोम संपादन समुदाय का धंयवाद । हम गंभीर रूप से इस पांडुलिपि पढ़ने के लिए रॉस विल्सन धंयवाद ।
अलेक्जेंडर है Marson अनुसंधान जेक Aronov और एक राष्ट्रीय मल्टीपल स्केलेरोसिस सोसायटी अनुदान से एक उपहार द्वारा समर्थित है (सीए १०७४-a-21) । अलेक्जेंडर Marson बरोज वेलकम फंड से मेडिकल वैज्ञानिकों के लिए करियर अवार्ड रखती है और एक चान नॐ Biohub खोजी है । याकूब ई. मकई अनुसंधान ली Ka शिंग फाउंडेशन द्वारा समर्थित है, विरासत चिकित्सा अनुसंधान चिकित्सा संस्थान, और कैलिफोर्निया संस्थान के लिए अपक्षयी चिकित्सा । Behnom Farboud और बारबरा जे है मेयेर अनुसंधान NIGMS अनुदान R01 GM030702 बारबरा जे मेयेर, जो हावर्ड ह्यूजेस चिकित्सा संस्थान के एक अंवेषक है द्वारा भाग में वित्त पोषित है । आयलैंड जार्विस और Nipam एच पटेल अनुसंधान NSF अनुदान IOS-१२५७३७९ और आयलैंड जार्विस द्वारा भाग में वित्त पोषित है एक NSF GRFP और एक Philomathia स्नातक फैलोशिप से समर्थन स्वीकार करता है ।
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
