

Summary
Bruker en prefabrikkerte Cas9 er ribonucleoprotein kompleks (RNP) en kraftig metode for presis og effektiv genomet redigering. Her markere vi sin verktøyet over et bredt spekter av celler og organismer, inkludert primære menneskelige celler og både klassiske og nye modell organismer.
Abstract
Områdespesifikke eukaryote genomet redigering med CRISPR (gruppert regelmessig interspaced kort palindromic gjentar)-Cas (CRISPR-tilknyttet) systemer har raskt blitt en vanlig blant forskere forfølge en rekke biologiske spørsmål. Brukerne benytter oftest Cas9 protein avledet fra Streptococcus pyogenes i et kompleks med en enkelt omprogrammeres guide RNA (gRNA). Disse komponentene er introdusert i celler, og gjennom en base sammenkobling med en utfyllende område double-strandet DNA (dsDNA) genomet, enzymet kløyver både tråder for å generere en dobbel-strand pause (DSB). Etterfølgende reparasjon fører til tilfeldige innsetting eller sletting hendelser (indeler) eller innlemmelse av eksperimentator gitt DNA på stedet av pausen.
Bruk av en renset single-guide RNA og Cas9 protein, prefabrikkerte for å danne en RNP og levert direkte til celler, er en potent tilnærming for å oppnå svært effektiv genet redigering. RNP redigering øker spesielt frekvensen av genet innsetting, et utfall som er ofte vanskelig å oppnå. Sammenlignet med levering via en plasmider, fører kortere utholdenhet av Cas9 RNP i cellen til færre off-målet hendelsene.
Til tross for sine fordeler er mange tilfeldige brukere av CRISPR genet mindre kjent med denne teknikken. Lavere barrieren til oppføring, skissere vi detaljert protokoller for implementering av strategien er RNP i en rekke sammenhenger, fremhever sin distinkte fordeler og forskjellige programmer. Vi dekker redigering i to typer primære menneskelige celler, T celler og blodkreft stem/stamfar celler (HSPCs). Vi viser også hvordan Cas9 RNP redigering gir lettvinte genetisk manipulering av hele organismer, inkludert den klassiske modellen rundorm Caenorhabditis elegans og mer nylig introdusert modell krepsdyr, Parhyale hawaiensis.
Introduction
fThe CRISPR-Cas9 systemet tillater forskere å endre målrettede regioner av alle genomet1. Denne rask og billig teknologi har revolusjonert grunnforskning og lover å gjøre en betydelig innflytelse på utviklingen av personlig sykdom behandling, presisjon landbruk, og utover2. CRISPR redigering er et demokratiserende verktøy og implementere systemet i et nytt laboratorium krever ingen bestemt kompetanse i genomet engineering, bare grunnleggende molekylærbiologi ferdigheter. Forskere kan nå studere tidligere uløselige organismer noen alternative måter for genetisk manipulasjon3,4. I de siste fem årene alene, har CRISPR genomet redigering blitt brukt til ingeniør over 200 forskjellige virveldyr, virvelløse dyr, plante og mikrobielle arter.
Tilpasset fra CRISPR prokaryote forsvar veien, kjerneelementene kreves for områdespesifikke genomet redigering er Cas9 protein, vanligvis fra S. pyogenes og codon-optimalisert med et ekstra kjernefysiske lokalisering signal (NLS) og dens spesialiserte RNA guide5,6. Selv om ikke diskutert her, kan andre Cas9 orthologues eller CRISPR endonucleases også brukes. Den naturlig forekommende gRNA består av to separat transkribert stykker, CRISPR RNA (crRNA) og det trans-aktivert crRNA (tracrRNA)7. Disse RNAs kan være smeltet sammen til en enkelt transkripsjon, kjent som enkelt-guide RNA (sgRNA)8. De fleste genomet redaktører velge strømlinjeformet sgRNA9, men dual-guiden er også brukt regelmessig10,11. Forskere Velg et 20-nukleotid (nt) genomisk DNA mål, slik at det ligger en kort lisensiering signatur kreves for Cas9 anerkjennelse, kalt en protospacer tilstøtende motiv (PAM), og utforme en gRNA som inneholder komplementære rekkefølgen12 .
Når inne i cellen, RNP komplekse finner genomisk målet gRNA base parene med komplementære DNA strand og deretter enzymet kløyver bryter både DNA tilnærmingene til å generere en dobbel-strand2. Celle reparasjon maskiner løser DSB av en av minst to ruter: via feilutsatte ikke-homologe slutten-begynte (NHEJ) veien eller homologi-rettet reparasjon (HDR), som sømløst omfatter DNA som inneholder "armer" homologi på begge sider av pausen. Tidligere reparasjon veien vanligvis fører til indel dannelse og påfølgende gen avbrudd, mens sistnevnte tillater forskere å sette inn eller endre DNA-sekvenser1.
Redigering effektivitet og nøyaktighet, avhenger av midler som Cas9 og gRNA inn i cellen. Disse komponentene kan leveres til kulturperler celler, embryo eller organismer i form av nukleinsyrer eller som en prefabrikkerte RNP komplekse13,14,15. Vanlige nucleic acid-baserte leveringsmåter inkludere viral signaltransduksjon, hva, eller electroporation av mRNA eller plasmider DNA. Cas9 protein og guide RNA da produsert i cellen og de knytte danner et kompleks.
Direkte levering av RNP krever separate rensing av Cas9 protein og guide RNA. Dette kan gjøres internt, eller protein og sgRNA kan kjøpes fra en av flere kommersielle leverandører. Når kjøpte, er Cas9 og gRNA blandet for å danne enzymatisk kompetent RNP komplekset og introdusert til celler ved direkte injeksjon i befruktede egg/embryo, lipid-baserte transfection16eller electroporation. Den første rapporten RNP redigering involvert injeksjon i C. elegans gonader17. Microinjection er fortsatt foretrukket måte å introdusere RNP inn embryoer og hele organismer, men effektive electroporation har vist i mouse18,19 og rotten20 embryoer. Vi beskriver protokoller for direkte injeksjon RNP C. elegans gonader og P. hawaiensis embryoer og anbefaler en spesialisert type electroporation å levere RNP når du redigerer primære menneskelige celler. Denne metoden, nucleofection, optimalisert electroporation programmer og celle type-spesifikk løsninger og lar RNP både cytoplasma og kjernen21.
Genomet redigering med RNP tilbyr flere forskjellige fordeler. Fordi protein og RNA komponenter er ferdigmontert og kvalitet kan sikres før levering, unngår RNP redigering mange feller som er forbundet med nukleinsyre-baserte levering. Nemlig, det er ingen risiko for Cas9-koding DNA integrering i vert genomet mRNA er aldri utsatt til fornedrelse og det omgår problemer med i vivo gRNA eller protein uttrykk, bretting og foreningen22,23. Videre fører bruker RNP til lavere toksisitet og langt færre off-målet hendelser enn plasmider-baserte uttrykket, et resultat av den RNP kortere half-life inni cellen24,25,26,27.
Endelig fører RNP redigering påviselig til høye redigering priser i en rekke humane cellelinjer, primær celler som fibroblaster, embryonale stamceller (ESCs), indusert pluripotent stamceller (iSPCs), HSPCs, og T celler16,24, 25,26,27,28,29; i dyr inkludert C. elegans, P. hawaiensisog frukt fluer3,17,30; i virveldyr arter som sebrafisk, mus og rotter31,32; i plante arter inkludert Arabidopsis, tobakk, salat, ris, grapevine, apple, mais og hvete33,34,35,36; og i Chlamydomonas, penicillinog Candida arter37,38,39. Hyppigheten av indel formasjon kan være høyere ved RNP sammenlignet med plasmider levering, og HDR-mediert DNA innsetting kan være lettere å oppnå25,27,29.
Protokollen beskrevet her bruker Cas9 RNP og er en effektiv, lett tilpasses teknikk som er enkelt å bruke en rekke biologiske systemer40,41, spesielt i celler som er vanskelig å arbeide med og i organismer uten godt etablerte systemer for presis genetisk manipulasjon. Vi starter ved å beskrive hvordan design, hente og sette sammen Cas9 RNP før dekker bruk over annen modell celletyper og organismer. Blodkreft stem/stamfar celler (HSPCs) og T-celler redigeres ved hjelp av samme metode, nucleofection, så de er dekket sammen i trinn 2 og 3 i denne protokollen. Redigere prosedyrer for C. elegans er beskrevet i trinn 4 og 5, og P. hawaiensis redigering er dekket i trinn 6 og 7. Til slutt, siden suksessen til et gen-redigering eksperiment i noen organismer vurderes av genotype sekvensering, undertrinn beskriver mulige dataanalyse metoder for alle celler og organismer beskrevet i protokollen er beskrevet i trinn 8.
Protocol
1. RNP montering
-
Utforme eksperimentet i forveien anskaffe alle RNA og DNA protein komponentene forhånd. Som en første pass, prøve en positiv kontrollene oppført i tabell 1 og bruke kommersielle reagensene beskrevet i tabellen for materiale for å sikre et pålitelig eksperimentell design og integriteten av materialet. Flere tips om planlegging et nytt genomet-redigering eksperiment, kan du se papirer på dette emnet12,42,43.
Merk: Når samlet som beskrevet i de etterfølgende trinnene, RNPs forberedt på forhånd kan lagres på-80 ° C.- Etter å velge hvilket gen mål, kan du bruke en av gratis online verktøy til å utforme en optimal gRNA44,45,46,47,48. Husk å målrette en ekson hvis håper å generere en knockout.
Merk: Disse verktøyene vil hjelpe til å identifisere et målområde med en nærliggende S. pyogenes PAM sekvens, høy kvalitet score og lav off-målet score. - Rense S. pyogenes Cas9 protein gjennom publiserte metoder8, eller kjøpe det fra en kommersiell leverandør.
- Forberede en typisk Cas9 buffer RNA fortynning, RNP, og protein lagring, som inneholder 20 mM HEPES pH 7.5, 150 mM KCl, 10% glyserol og 1 mM TCEP. Bruk alltid nuclease-fritt vann i buffere som brukes til å resuspend eller fortynne RNA for å hindre nedbrytning.
- Produsere guiden RNA (tracrRNA og crRNA eller sgRNA) gjennom en i vitro transkripsjon bruke publiserte metoder, eller kjøpe det fra en nukleinsyre syntese selskapet17,21,49, 50 , 51.
- Hvis du setter inn et gen, syntetisere eller kjøpe en donor DNA mal.
- Lagre proteiner og RNA dele-80 ° C og tine på is umiddelbart før bruk.
Merk: Hver fryse-tine litt lavere effektivitet. Detaljert, åpen tilgang protokoller for Cas9 rensing52 og i vitro transkripsjon av sgRNAs53 er tilgjengelig andre steder.
- Etter å velge hvilket gen mål, kan du bruke en av gratis online verktøy til å utforme en optimal gRNA44,45,46,47,48. Husk å målrette en ekson hvis håper å generere en knockout.
- Hvis arbeider med C. elegans, hopper du til trinn 1.5. For P. hawaiensis protokollen, hopper du til trinn 1.6. Hvis bruker sgRNA, hopper du til trinn 1.4. Fortsett til trinn 1.3 å montere en gRNA for redigering primære celler.
-
Sett sammen en gRNA ved å blande ekvimolare mengder tracrRNA og crRNA. Gjøre 100 µL av 80 µM gRNA aksjer, for 50 genomet redigering eksperimenter.
- Inkuber gRNA på 37 ° C i 30 min og deretter la den sakte avkjøles til romtemperatur.
-
RNP prep for HSPC og T celle redigering: sette sammen en RNP kompleks ved å blande en 1-2 x molar mengde gRNA til 200 pmol Cas9 protein i et totalt volum på 10 µL. veldig sakte, legge konsentrert Cas9 til gRNA (pre utvannet i Cas9 buffer) for om 30 s , gjør rask sirkler med pipette, bringe den endelige Cas9 konsentrasjonen til 20 µM.
- Forberede electroporation cuvettes.
Merk: Denne protokollen gjelder kommersielle systemet i Tabellen for materiale, men RNP redigering kan også oppnås med andre electroporation enheter. - Legge til 5 µL (100 pmols, T celler) eller 10 µL (200 pmol, HSPCs) av RNP hver cuvette.
- Hvis å sette inn nye DNA i stedet for å lage en knockout, legge 1 µL 100 µM (100 pmol) single-strandet oligonucleotide donor DNA (ssODN)25,54,55 til cuvettes eller brønner av platen.
- Gå til trinn 2 for neste instruksjonene i primære celle redigering protokollen.
- Forberede electroporation cuvettes.
-
RNP prep for C. elegans redigering: montere RNP kompleks ved å legge til følgende reagensene for å skape et endelig antall 20 µL (de siste konsentrasjonene er angitt i parentes): Cas9 (2 µM), HEPES pH 7.5 (10 µM), KCl (115 µM), crRNA (12 µM) , tracrRNA (40 µM), og i reparasjon maler hvis nødvendig (0,5 µM ssDNA eller opptil 350 ng/µL dsDNA).
Merk: Effektiviteten av en Cas9-mediert DSB mal reparasjon er proporsjonal med konsentrasjonen av dsDNA reparasjon Konstruer; Således, jo høyere konsentrasjon av reparasjon malen, jo mer effektiv mal reparasjon. Men har en injeksjon av mikser som inneholder større enn 350 ng/µL av dsDNA blitt vist å redusere levedyktigheten til injisert ormer. Derfor er det best å bruke opptil, men ikke mer enn 350 ng/µL av dsDNA i miksen å maksimere reparasjon effektiviteten samtidig minimere sin dødelighet.- Legge til flere crRNAs å målrette flere loci samtidig, behov for co-CRISPR/co-conversion screening tilnærming beskrevet i trinn 5.4. Når du legger til flere crRNA, Legg hver sekvensielt til master mix.
Merk: Mengden av hver crRNA trenger ikke være det samme, og selv dobler den totale konsentrasjonen av crRNAs i master blanding uten å endre konsentrasjonen av Cas9 synes ikke å forstyrre frekvensen av mutagenese på et bestemte locus. Eksempler er beskrevet i detalj i Paix et al. 56. - Blande av pipettering og spinne den RNP løsningen 16.000 x g for 5 å sikre at løsningen er samlet i bunnen av røret.
- Ruge i løsning på 37 ° C i 15 m.
- Sentrifuge prøven 16.000 x g for 1 min til pellets eventuelle partikler som kan tette tynn-kjeder microinjection nålen. Bruk nedbryting i de etterfølgende trinnene.
- Gå til trinn 4 for resten av C. elegans protokollen.
- Legge til flere crRNAs å målrette flere loci samtidig, behov for co-CRISPR/co-conversion screening tilnærming beskrevet i trinn 5.4. Når du legger til flere crRNA, Legg hver sekvensielt til master mix.
-
RNP prep for P. hawaiensis redigering: forberede bruke én Cas9 dele fortynne dem med nuclease-fritt vann og fenol rød (for å visualisere injeksjoner) til en siste konsentrasjon av 6,25 µM Cas9 og 0,15% fenol rød.
- Montere RNP kompleks ved å blande en 2-5 x molar overskudd av gRNA til Cas9 proteinet i et totalt volum på 6 µl. legge til 12 pmol av Cas9 til gRNA, bringe den endelige Cas9 konsentrasjonen 2 µM, gRNA konsentrasjon til 4-8 µM og fenol red konsentrasjon til 0,05%.
- Inkuber blandingen ved romtemperatur for 10 min å kompleks i RNP.
- Gå til trinn 6 for neste instruksjonene i P. hawaiensis redigeringprotokollen.
2. celle kultur og forberedelse
Merk: Utføre trinnene 2.1.1 til 3.3.3 i en biologisk sikkerhet regjering.
-
Kjøp cryopreserved menneskelige mobilisert perifert blod CD34+ HSPCs fra en leverandør.
- Tine ~ 1 x106 HSPCs i en 37 ° C vann bad for 3 min og overføre dem til en 15 mL konisk rør. Legge til 10 mL av en serum-free ekspansjon medium fra en kommersiell kilde og spinne blandingen på 100 x g for 10 min. fjerne nedbryting og resuspend cellene i 2 mL supplert SFEM. Plate cellene i 6-vel plater og kultur dem i en 37 ° C inkubator 24-48 h før RNP electroporation.
- Telle celler med en hemocytometer og overføre totalt antall HSPCs nødvendig (150.000-200 000 HSPCs per cuvette skal electroporated) til en sentrifuge rør.
- Spinne røret 100 x g for 10 min å pellets cellene.
-
Kjøpe menneskelige primære CD4+ T celler fra en leverandør eller isolere dem fra menneskelig fullblod av tetthet gradert sentrifugering29.
- Før T celle aktivering, pre pels 48-brønnen plater med αCD3 (UCHT1) og αCD28 (CD28.2). Pels platene med 500 µL av 10 µg/mL αCD3 og 10 µg/mL αCD28 i PBS minst 2 h på 37 ° C.
Merk: For noen loci,-NHEJ, kan oppnås uten pre stimulering, men inkludert dette trinnet maksimerer effektiviteten. - Kultur T celler for 48 h på 37 ° C på αCD3/αCD28 antistoff-bundet plater i et RPMI komplett medium [RPMI-1640 supplert med 5 mM av HEPES, 2 mM av kommersielle alternativ til L-Glutamine, 50 µg/mL av penicillin/streptomycin, 50 µM av 2-mercaptoethanol, 5 mM ikke-essensielle aminosyrer, 5 mM av natrium pyruvate og 10% (vol/vol) FBS]. Kultur T celler på en tetthet av 2.000.000 T-celler i 500 µL av media per brønn av en 48-vel plate.
- Telle T celler ved hjelp av en hemocytometer og overføre eksperimenter antall T celler nødvendig for electroporation (100.000-1,000,000 T celler per cuvette skal electroporated) slik sentrifuge.
- Spinne røret på 90 x g i 8 min til pellets cellene. Hvis cellene har vært tetthet gradert atskilt innen 2 dager, spinne dem på 200 x g i 8 min.
- Før T celle aktivering, pre pels 48-brønnen plater med αCD3 (UCHT1) og αCD28 (CD28.2). Pels platene med 500 µL av 10 µg/mL αCD3 og 10 µg/mL αCD28 i PBS minst 2 h på 37 ° C.
-
Sug opp nedbryting med en pipette/vakuum, fjerne bobler for begge celletyper.
- Forsiktig resuspend cellene med 20 µL av electroporation buffer per cuvette.
- Legge til 20 µL av cellene (150.000-200.000 HSPCs eller 100.000-1,000,000 T celler) hver cuvette, som allerede inneholder 10 µL av RNP og bland godt av pipettering opp og ned uten bobler.
3. RNP Electroporation
- Electroporate cuvettes etter å plassere dem i en nucleofector. For HSPCs, kan du bruke puls koden ER100. For T-celler, bruke puls EH-115.
-
HSPCs bare: Tilsett 100 µL av en supplert SFEM medium (oppvarmet til 37 ° C) til hver cuvette umiddelbart etter electroporation og la cellene gjenopprette for 10-15 min.
- Overføre cellene til kultur i en 96-brønns runde bunn plate og legge en ekstra 100 µL av supplert SFEM mediet for 24 timer.
- Endre dem til en frisk supplert SFEM medium og ruge dem for en ekstra 24-72 h.
- Fjerne celler for genotyperingteknologi dem 48-96 h innlegg-electroporation. Spinn cellene på 300 x g i 5 min og fjerne nedbryting før du begynner DNA utvinning (trinn 8.2).
-
T celler bare: legge til 80 µL av RPMI fullføre kultur medier pre varmet til 37 ° C fra reservoaret hver cuvette eller vel, med en flerkanals pipette (hvis nødvendig).
- Inkuber dem på 37 ° C i 15 min.
- Tilsett mediefil, antistoffer, cytokiner, etc. til målet plate(s) og pre varme dem i en 37 ° C inkubator.
- Overføre 107 µL electroporated cellene fra brønnene til en runde bunn 96-brønns plate med en flerkanals pipette (hvis nødvendig).
- For informasjon om vurdere redigering resultatene, hopper du til trinn 8.
4. C. elegans forberedelse
-
1 dag før microinjection: forberede agarose pads for microinjection.
- Lage en 3% (w/v) agarose løsning i vann ved å legge agarose til vann og bringe løsningen til en byll på en kokeplate eller i en mikrobølgeovn.
- Ordne 24 x 50 mm x 1,5 mm cover glass lysbilder på et bord og bruke et glass Pasteur Pipetter til å plassere en liten (~ 15 µL) dråpe agarose løsning på lysbildet. Raskt flat agarose fall ved å plassere en dekkglassvæske på toppen. Tillate agarose å stivne og fjern deretter en av coverslips.
- La den agarose-belagt dekkglassvæske opp på en tabletop overnatting tørke. Etter 24 timer, kan du lagre agarose pads i en ren, tørr beholder.
Merk: Disse kan brukes på ubestemt tid.
- Trekk microinjection nålene: Borosilikatglass kapillærene med filamenter (ytre diameter 1.0 mm og indre diameter 0.58 mm), trekke nålene basert på Mello og brann57 og andre ressurser58. Nålene kan brukes umiddelbart eller lagres i en ren, tørr beholder, avstivet av leire støtter.
- For vedlikehold av ormer, forberede en Rundormer vekst medier (NGM) agar helles i Petri plater og oppdaget med OP50 bakterier (for protokoller på standard C. elegans vedlikehold og oppskrifter for vekst medier, se Stiernagle59).
- Scenen ormer for microinjection: 12-24 h før microinjection, plukke L4-trinnvis hermafroditter til en ny NG-agar plate med OP50 bakterier og ruge dem over natten på 20 ° C. For hver Cas9 mål/injeksjon blanding, plukke ~ 30 ormer til platen.
-
Dag med microinjection: Laste trakk microinjection nålen med RNP løsning supernatant i trinn 1.5.
- Pipetter nedbryting fra trinn 1.5.4 i en trakk kapillær pipette og etterfylle løsningen fra kapillær Pipetter i forberedt microinjection nålen (vanligvis lasting mindre enn 0,1 µL).
- Montere lastet nålen på microinjection apparatet knyttet til en micromanipulator. Angi injeksjon apparater presset til 250 kPa og balanse presset til 25 kPa.
-
Bryte tilbake lastet pinne-spissen generere en skarp nål kanten. Plass en 15 x 15 mm x 1,5 mm firkantet dekkglassvæske på en 24 x 50 mm x 1,5 mm dekkglassvæske.
- Overlappe en kant av den firkantede dekkglassvæske med halocarbon olje 700.
- Plasser nålen i olje, på kanten av 15 mm firkantet dekkglassvæske.
- Bruker en hånd å guide mikroskop scenen og dekkglassvæske, børste lysbildet opp og langs kanten av nålen mens deprimerende injeksjon pedal/knapp. Bryte pinne-spissen på tilbake, øke flyten av væske ut av nålen. Oppnå en optimal gjennomstrømning ved injeksjon blande strømmen langs kanten av nålen, danner ~ 1 boble/s.
- Bekreft at L4 ormer plukket 12-24 h før microinjection er developmentally trinnvis unge voksne ved injeksjon. Velg de unge voksne ormene en NG-agar platen som mangler OP50 bakterier og tillate dem å gjennomsøke rundt for 5 min. Dette reduserer mengden av bakterier overføres til injeksjon pad, minimere nål tresko.
- Plass en agarose injeksjon pad/dekkglassvæske på et disseksjon omfang. Bruke en orm hakke, lå et lite spor halocarbon olje langs en av kantene på puten.
-
Bruker ormen plukke belagt i olje, løft flere ormer av NG-agar platen og inn i sporet av olje. En fint hår festet til en pipette, for eksempel en øyenvippe eller katten whisker, plasser ormer i parallell, forsiktig skyve ormer i agarose puten. Til komfortable microinjection fremgangsmåten, bare montere og injisere ettall orm samtidig.
Merk: Den tørre agarose veken fuktighet fra ormer, forårsaker dem til å overholde puten. Derfor må man jobber raskt som ormer kan utenå tørrlegge overflaten.- En gang i posisjon og knyttet til puten, overlegg ormer med en noen dråper halocarbon olje (~ 20 µL) fra spissen av ormen plukke.
5. C. elegans Gonad Microinjection med RNPs og etter injeksjon vare
Merk: Microinjection protokollen er tilpasset fra Mello og brann57og beskrevet i detalj andre steder60,61.
-
Plass dekkglassvæske med ormene montert på injeksjon mikroskopet. Plasser ormer vinkelrett injeksjon nålen under en lav forstørrelse (5 X mål, 10 X okulær).
- Bytt til en høy forstørrelse (40 X mål, 10 X okulær), flytte nålen tilstøtende gonad armen tilsvarer regionen nær kjerner i midt - å sen-pachytene.
- Bruker micromanipulator, Flytt nålen mot ormen deprimerende cuticle litt. Tapp deretter siden av mikroskopet scenen å støte nålen gjennom cuticle med én hånd. Trykke ned injeksjon pedal/knapp og sakte fyll gonad armen med injeksjon mix og fjerne nålen.
- Gjenta dette trinnet med andre gonad armen.
-
Når ormer er injisert, Fjern dekkglassvæske/agarose pad og plassere den under dissecting mikroskop.
- Bruker en trakk kapillær pipette, fortrenge olje fra ormer av pipettering en M9 buffer over dem. Utføre denne behandlingen å løslate ormer fra agar.
- Etter 10 min, når ormer er juling rundt i bufferen, flytte dem til en NG-agar plate med OP50 bakterier bruker den trakk kapillær pipette. Plass platen ved 20 ° C i 2-3 h før ormer har gjenopprettet og går rundt.
- En gang gjenopprettet, individuelt overføre ormer til NG-agar plater med OP50 og overføre platene til en 25 ° C inkubator.
-
Tillate P0-injisert ormer å vokse og legge avkom for 3 dager. Skjermen F1 avkom.
- Hvis bruker co-CRISPR eller co konvertering62,Velg63,64,65, ormene kandidat for screening basert på om de har mutert fenotypen av referanse genet. Individuelt overføre disse merket ormer til nye NG-agar plater med OP50 og tillate dem å legge F2 avkom på 20 ° C.
Merk: Fenotypen brukes for co-CRISPR screening eller valget bør gi tidlig anslaget for suksess Cas9 redigering. - Hvis co-CRISPR fenotypen ikke finnes, microinject en positiv kontroll plasmider å bistå i å forbedre microinjection.
Merk: For eksempel, inkludert en plasmider injeksjon blandingen som koder mCherry-merket MYO-2 vil hjelpe vurdere Injeksjonseffektiviteten. Ormer ble injisert med pCFJ90 har noen avkom med fluorescerende pharynxes.
- Hvis bruker co-CRISPR eller co konvertering62,Velg63,64,65, ormene kandidat for screening basert på om de har mutert fenotypen av referanse genet. Individuelt overføre disse merket ormer til nye NG-agar plater med OP50 og tillate dem å legge F2 avkom på 20 ° C.
- Undersøke F1 ormer for tilstedeværelsen av de ønskede endringene. Velg F1 mor til en individuell godt av en 96-brønns plate, lyse henne og undersøke hennes DNA av enten insert-spesifikke PCR forsterkning, DNA sekvens analyse eller takstmann nuclease analysen (CEL-1)66.
Merk: Disse analyser kan utføres når du bruker en co-CRISPR/co-conversion eller andre screening eller utvalg regimer65,66,67,68. - For informasjon om vurdere redigering resultatene, hopper du til trinn 8.
6. P. hawaiensis forberedelse
- 1 dag før microinjection, berike for tidlig embryo ved å sette opp en 'par tank' kvelden før; nylig separerte kvinner vil inneholde ferske befruktet embryoer. Se Rehm et al. 69 for mer informasjon.
- Ved microinjection, samle encellede Parhyale embryo (0-4 h etter befruktning) av anesthetizing gravid kvinner med 0,02% fedd olje i sjøvann og forsiktig skrape embryo av hennes ventrale Foreldreomsorg og yngelens pose med en flamme, trakk og avrundet glass pipette og noen kjedelig av #3 tang.
7. P. hawaiensis Embryo Microinjection med RNPs og etter injeksjon vare
- Etterfylle en trakk kapillær rør med ca 1 µL av RNP injeksjon miksen beskrevet ovenfor.
-
Bruke komprimert nitrogen til microinject hver embryo som beskrevet i Rehm et al. 69.
- Injiser Parhyale embryo under dissecting mikroskop med en microinjector og en micromanipulator. Laste 1,5 µL av injeksjon miksen på baksiden av en trakk kapillær tube 4 inches - 1.0 mm med filamenter, trakk bruker brønnene trekke apparater bruker en microloader pipette tips.
- Definer nålen på injeksjon apparatet og bryte spissen av nålen (en svært liten mengde) med en tang under dissecting omfanget. Kalibrere volumet av sprøytebruk i halocarbon olje 700 og måle diameteren på boblen.
- Skjær et trau av herding agent med et barberblad. Fyll den halvveis med filter-steriliseres sjøvann, og linje Parhyale embryo opp gjennom å stabilisere.
- Injisere embryo bruk microinjection, stabilisere hver embryoet med en tang under injeksjon. Etter injeksjon, bruk en glass overføring pipette overføre embryoene til en frisk 60 mm kultur parabol halvveis fylt med filter-steriliseres sjøvann.
-
Hvis de første divisjonen har allerede skjedd å danne en 2-celle embryo (4-6 h etter befruktning), generere fullt-mutant dyr ved å injisere begge blastomeres. For å sikre en total cleavage av 2-celle scenen, co injisere blastomeres med FITC eller TRITC dekstran og observere at signalet er begrenset til en enkelt blastomere under en fluorescerende dissekere omfang etter injeksjon.
- Eventuelt generere 'halv-mutant' dyr ved å injisere bare ett av de to blastomeres på 2-celle scenen (omtrent delt venstre-høyre avhengig av vev og plasseringen langs aksen A-P).
- Sette inn en celle i et 8-cellers embryo (7.5-9 h etter befruktning) å begrense redigering til et enkelt bakterie lag. Se Gerberding et al. 70 kartet tidlig blastomere linjene.
-
Inkuber embryo 60 mm kultur retter (ikke mer enn 25 per fat), halvveis fylt med filter-steriliseres sjøvann 'pre oksygenert' bruker en akvariet bubbler eller ved å riste kraftig.
- Plass retter av embryo i en løst lukket plasticware med våt papirhåndklær å opprettholde fuktighet og plassere dem i en 26 ° C inkubator med en 12 h lys og mørke syklus.
- Overføre de overlevende embryoene for å rense sjøvann retter hver par dager.
Merk: Embryo kan kultivert i romtemperatur, selv om de utvikler mye saktere.
-
Analysere og løse embryoene på ulike stadier for en uttrykk analyse av i situ hybridisering eller antistoff flekker (se Browne et al. 71 for en oppsetning guide og flere referanser for disseksjon og fiksering72, i situ hybridisering73og antistoff flekker74).
- Gjøre disseksjon nåler av threading en bøyd stykke tungsten wire ca 0,5 i lengde i slutten av en insulin-nål. Skjerpe nålen i natriumhydroksid under en strøm. Bruk en 1 mL sprøyte som håndtaket disseksjon nålen.
- Fylle en brønn på en 3-vel glass rett halvveis en nylaget løsning 9 deler PEM Buffer (0.1 M av rør pH 6.95, 2 mM EGTA, 1 mM MgSO4), 1-del 10 x PBS og 1 del 32% PFA. Plasser 3-5 embryo i retten og dytte et lite hull i hver embryo, bruker en skarp tungsten nål poke og en litt sløv for å stabilisere, slik at eggeplomme å strømme ut og bindemiddel i.
- Med en skjerpet tungsten nåler, forsiktig erte unna ytre to membraner rundt Parhyale fosteret. Analysere dem i bindemiddel gjøre embryoene mer robust, men arbeidet raskt å holde membranen blir festet til fosteret, som gjør membran fjerning vanskeligere. Kan embryoene til rette for totalt 15-20 min. til antistoff farging eller 40-50 min for i situ hybridisering.
- Bilde live skilpadder og analysere dem for morfologiske og atferdsmessige fenotyper eller fikse og stain dem for mer detaljerte analyser. Øke hatchlings til seksuell modenhet på 2-3 måneder å etablere knockout og transgene linjer (se Kontarakis og Pavlopoulos75 for hatchling omsorg og andre nyttige detaljer).
8. vurdere redigering resultater
- Eventuelt kan du se etter en visuell eller funksjonelle fenotypen i den redigerte celler og organismer.
Merk: Denne prosessen varierer mye av programmet, og noen eksempler er beskrevet på slutten av deres aktuelle protokollen trinnene ovenfor. Etter korrigering sigd celle mutasjon i HSPCs, analysere hemoglobin produksjon ved differensiert erythroblasts bruker mye HPLC (figur 1A). En knockout av IL-2 reseptor genet i T celler kan bekreftes av overflaten flekker og flow cytometri (figur 1B). For å vurdere C. elegans og P. hawaiensis fenotyper, observere dyr morfologi og oppførsel under et lys- eller fluorescerende mikroskop (tall 1 c og 1 D). - For å fastslå effektiviteten og type genomisk redigeringene generert, lyse bassengene redigerte celler og ekstra genomisk DNA ved hjelp av en kommersiell utvinning kit21.
-
For en rask estimering av indel formasjon, PCR-forsterke minst 200 base parene rundt kuttet nettstedet og utføre en T7 endonuclease1 (T7E1)76 eller takstmann (CEL-1 nuclease) analysen77.
- Hvis en indel formasjon på webområdet Cas9-kutt eller vellykkede HDR vil opprette eller fjerne en kjent begrensning-område, kan du vurdere å bruke en begrensning enzym fordøyelsen for å anslå redigering effektivitet6. Begrensning fragment lengde polymorfisme (RFLP) analysen kan enkelt kontrollere effektiviteten hvis det skjer være tilgjengelig.
- For en nøyaktig kvantifisering av redigering effektiviteten og fastsettelse av dominerende redigering utfall, sende PCR-amplicon for en standard Sanger sekvensering med både forover og bakover grunning.
Merk: Hvis analysere en enkel klone eller organisme, analyse av Sanger resultatene er enkel, som vist i figur 2A. Hvis analysere en pool av celler, deretter analysere chromatograms med online verktøyet78, som vist i figur 2B. - For en full kvantifisering og sekvenser av resultater, utføre dype sekvensering27,54, som vist i figur 2C.
- Å vurdere et bestemt sett av off-målet endres, PCR-forsterke spådd off-målet nettsteder og sende dem til NGS. For å aktivere gjenkjenning av chromosomal translocations, utføre GUIDE-seq79 eller høy gjennomstrømning og genom hele translokasjon sekvensering (HTGTS)80. Et komplett bilde av off-målet redigeringene i en klonal befolkning, kan du utføre hele-genome sekvensering (WGS)81,82,83.
Merk: Det er en rekke metoder for kvantifisering på - og off-målet Genova endringer, forklarte videre i ulike gjennomgang artikler84,85,86.
Representative Results
Disse eksperimentene viser hvordan ferdigmontert Cas9 RNP kan brukes til å manipulere genomer primære celler og hele organismer. Forskere rense eller kjøpe Cas9 protein og sgRNA, kombinerer to komponentene pre-danne komplekset, og innføre RNP i sine celler eller organisme rundt. Etter tillater nok tid for redigering for å oppstå og for avkom av neste generasjon å bli født (hvis aktuelt), sjekk for fenotyper og/eller samle celler for genotyperingteknologi. Fenotyper kan sees via funksjonelle analyser, uttrykk analyser, visualisering (av øye eller med mikroskopi) eller andre metoder, avhengig av eksperimentet.
For eksempel kan HSPCs som er endret for å løse β-globin mutasjon som forårsaker sigdcelleanemi være differensiert i erytrocytter og assayed for produksjon av sunn og sigd hemoglobin27,87 (figur 1 A). T celler endret for å slå ut høy affinitet IL-2 reseptor genet, CD25 (IL2RA), kan analyseres av overflaten flekker og flyt cytometri88, og funksjonelt analyseres for å oppdage en signalnettverk svar IL-2 stimulering (figur 1B ). T celler kan også omprogrammeres i mange klinisk viktige måter som krever vurdering av Norge, inkludert effekten av HIV infeksjon89 og i vivo antitumor effekten av bil-T celler11.
Med en co-CRISPR/co-conversion screening tilnærming, er C. elegans ormer endret samtidig på to loci62. HDR på dpy-10 referanse genet bruker en ssODN reparasjon mal resultater i en enkelt scoret dominerende dpy-10 gevinst-av-funksjon mutasjon. Heterozygote F1dpy-10(gof) dyrene er Berg (Rol) og homozygous dpy-10(gof) dyr er Munan (Dpy). Tilstedeværelsen av fenotypen angir at Cas9 redigering i disse dyrene og forbedrer oddsen for å identifisere en redigering hendelse på andre locus i Rol eller Dpy F1 dyr. En vellykket redigering eksperiment bør resultere i 33-50% av injisert P0 ormer gir 20 eller flere F1 avkom som Rol eller Dpy90. Det er da mulig å velge ikke-Rol dyr returnere dpy-10 til wildtype og velger homozygous Rediger rundt. Som en tommelfingerregel, bør konsentrasjonen av crRNA målretting co-CRISPR referanse genet være halvparten av crRNA rettet mot genet av interesse. Hvis en endring i genet av interesse ikke er gjenopprettet, kan prosenter av de to CRISPR RNAs justeres for å øke sjansene for å utvinne ønsket mutasjon. For eksempel vil øker mengden av crRNA for genet av interesse i forhold til referanse genet crRNA øke prosentandelen av ormer besitter redigeringer i genet av interesse i befolkningen i ormer som har redigeringene referanse genet locus. Co konvertering frekvenser varierer, men prisene er vanligvis 20-60%, ofte gir homozygous endringer i F1 generasjon (figur 1C).
P. hawaiensis skilpadder som er endret for å slå ut Magesmerter-B genet (Abd-B) vise klart morfologiske avvik3 (figur 1D). Dette genet er nødvendig for riktig abdominal mønstre avbrudd resultatene i thorax-type hoppe og gå Ben erstatte svømming og anker bena som vanligvis tilstede på magen.
Bestemme genomet redigering resultater på genotypic nivå krever sekvensering eller i vitro analysen som oppdager endringene til oppstartsekvensen. Her viser vi representant sekvensering data fra vår modell celletyper og organismer, fremheve ulike tilnærminger til redigering kvantifisering. Merk at figur etikettene er generalisert fordi alle metodene vises her kan brukes på alle biologiske systemer.
Sekvensering tilnærminger varierer i tekniske kompleksiteten og dybden av resultater. Klonal redigerte populasjoner eller lett delbar individuelle organismer, kan redigert enkeltpersoner rekkefølge etter genomisk DNA utvinning. Standard Sanger sekvensering resultater vil avdekke sekvens endringen på webområdet Cas9-kutt i en gitt person, med hypotetisk frameshifts som ville forstyrre funksjonen (figur 2A). Online verktøyet brukes for sekvensering er en annen Sanger sekvensering tilnærming som kan brukes til blandet populasjoner i stedet for individuelle mutanter78. Sekvenser analyseres med et nettbasert verktøy som kan tilnærme redigering virkningsgraden samt dominerende sekvens resultater. Representant dataene vises i figur 2B.
Den mest grundige sekvensering metoden beskrevet her er dyp sekvensering (noen ganger referert til som høy gjennomstrømming eller neste generasjons sekvensering). Denne metoden gir DNA-sekvenser fra personlige genomer i en blandet befolkning. Slike data kan illustreres i en rekke måter. Her har vi klassifisert personlige sekvensering leser fra redigerte cellene basert på redigering resultatet (figur 2C). De fleste cellene redigeres via NHEJ veien, som vanligvis resulterer i genet avbrudd. I andre har målet genet blitt byttet ut en alternativ versjon via HDR27.
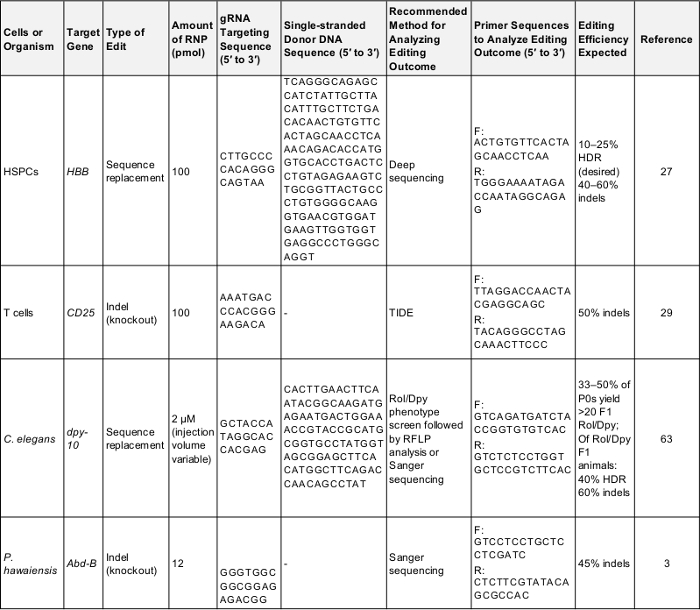
Tabell 1: positiv kontrollerer for foreløpige genomet redigering eksperimenter. Denne tabellen viser viktige informasjonen du trenger for å utføre en første gang genomet redigering eksperiment i alle celler og organismer som er beskrevet i denne protokollen. Etter disse parameterne er sannsynlig å gi et vellykket resultat som kan brukes til å teste protokollen eller som en baseline for sammenligning når eksperimentator er rettet mot et gen av deres egen interesse. F: fremover, R: bakover, HDR: homologi-rettet reparasjon. Klikk her for å laste ned denne tabell.

Figur 1 : Representant fenotypiske skyldes Cas9 RNP redigering av primære menneskelige celler og organismer. (A) dette er en såkalt HPLC spor viser at etter vellykket genomet redigering, HSPCs som skilles i sent erythroblasts vil produsere mer funksjonelle hemoglobin enn sickle hemoglobin. Mutant erytrocytter produserer sigd hemoglobin (HbS), mens-endret celler vil produsere friske hemoglobin (HbA og HbA2) samt fetal hemoglobin (absloutt). Absorbansen er grafisk i vilkårlig enheter (au). Dette panelet ble først publisert i DeWitt et al. 27. det er gjengitt med tillatelse fra American Association for Advancement of Science. (B) til venstre for hver tilstand, dette panelet viser flyt cytometri data som viser at overflaten-farget T celler ikke uttrykke CD25 etter CD25 genet har blitt slått ut med RNP. CD25 overflod tegnes inn på x-aksen med cellestørrelsen på y-aksen. Til høyre for hvert vilkår, viser dette panelet Phospho-Stat5 (pStat5) kvantifiseringen etter en induksjon med IL-2. Signalene reduseres når IL-2 reseptoren er fraværende (CD25 KO). PStat5 overflod tegnes inn på x-aksen og dataene fra tre forskjellige nivåer av IL-2 input sammenlignes loddrett. (C) dette panelet viser en Caenorhabditis elegans co-CRISPR/co-conversion skjerm målretting dpy-10 som co konvertering markøren. To guide RNAs målet to loci, dpy-10 og din favoritt genet (yfg), i samme P0-injisert dyr. HDR på dpy-10 resultater en Rol eller Dpy fenotypen. Valg av Rol - eller Dpy-F1 dyr øker sjansene for identifisere endringer på andre locus. (D) dette panelet viser at wildtype Parhyale hawaiensis skilpadder har normal sklerotisert med svømming og anker ben. Abd-B knock-out hatchlings (F0 personer) utvikle en mage forvandlet mot thorax. Dermed er svømming og ankere Ben borte og erstattet av hopp og gå bena forbundet med et vanlig syn. Klikk her for å se en større versjon av dette tallet.

Figur 2 : Typiske resultater fra redigering utfall analyse metoder. (A) dette panelet viser eksempler på Sanger sekvensering resultater fra individuelle F1 P. hawaiensis organismer, inkludert wildtype sekvensen og tre forskjellige indeler som forstyrrer funksjonen genet av skiftende åpent lesing rammen. (B) disse TIDEVANNET resultatene vise innsettinger og sletting hendelser som fant sted på et Cas9 målområde i et basseng av sekvensert T-celler. X-aksen angir lengden på en gitt innsetting eller sletting i nukleotider. (C) disse dype sekvensering resultatene viser ingen genomet redigering uten nucleofection eller gRNA og vellykket redigering med intakt Cas9-RNP, gruppert etter DNA reparasjon utfallet i HSPCs. Klikk her for å se en større versjon av dette tallet.
Discussion
Etablere et robust genom redigering protokollen i en celle linje eller organisme rundt krever optimalisering og empirisk testing av flere viktige parametere, drøftes i dette avsnittet. Prøver noen varianter av de generelle metodene som presenteres her er svært oppmuntret. Nøkkel begrensning av denne protokollen er at å bruke disse metodene til andre celler eller organismer kan føre til en ulike utfall avhengig av arten studerte, og en eksperimentell design som fører til en høy effektivitet genet knockout kan ikke promotere DNA innsetting. Derfor anbefaler vi starter med metodene presenteres her og feilsøking som beskrevet nedenfor.
Feilsøking genomet redigering reagens kvalitet:
Generere eller kjøpe høykvalitets reagenser er en avgjørende skritt i noen genomet redigering protokollen. Cas9 protein kan renset i laboratoriet eller kjøpt kommersielt. Mange protokoller Merk en siste konsentrasjon for Cas9 i RNP oppskrifter, men optimal genet redigering aktivitet avhenger til aktiviteten med noen personlige Cas9 protein forberedelser, som varierer avhengig av kilden. Når protokollen presenteres her fungerer, kan du vurdere å optimalisere mengden RNP brukt av titrating Cas9 for å etablere en optimal konsentrasjon: en som gir svært spesifikke mål DNA cleavage uten unødvendige off-målet cleavage skyldes overdreven Cas940.
Guide RNA renhet og homogenitet kan også være determinanter av genomet redigering suksess22. Kjøpte sgRNAs eller separate crRNA og tracrRNA komponenter er generelt høy kvalitet reagenser og en rekke kjemiske modifikasjoner er tilgjengelige mot problemer med RNA degradering eller tilegner funksjoner RNP91. Mens kjemisk endret gRNAs ikke kan være nødvendig for standard genomet redigering eksperimenter, noen grupper har observert mye høyere redigering effektiviteten med slike reagenser, så de kan være verdt å prøve etter mestring prosessen og/eller når gRNA fornedrelse synes å være et problem22,91. In vitro transkripsjon og påfølgende gel rensing er et billig alternativ, som kan være tilstrekkelig for rutinemessig genomet redigering eksperimenter17,21,49,50. Videre, flere tilnærminger som brukes vanligvis for å produsere homogen gRNA bestander i vivo, inkludert ribozyme og tRNA-baserte excision av enkelte hjelpelinjene, kan utvides til in vitro RNA forberedelse til å generere renere produkter92.
Guide RNA og donor DNA design tips:
Guide RNA utvalg er en kritisk faktor i å oppnå svært effektiv på målet redigering samtidig minimere sjansene for off-målet cleavage. For å hjelpe guide valg, har flere studier brukt høy gjennomstrømming skjermer sammen med neste generasjons sekvensering for å kompilere sekvens funksjoner av vellykket guider47,79,93,94, 95,96. Disse funksjonene har vært brukt til å utvikle prediktiv algoritmer og online verktøy for å hjelpe guide utvalg44,45,46,47,48. Slike algoritmer er jordet på skjermer ved hjelp av DNA-baserte systemer for guide RNA uttrykk. Guider er uttrykt ved hjelp av en Pol III promoter, og deres uttrykk er derfor utsatt for begrensningene knyttet til Pol III transkripsjon, som tidlig avslutning når møter spor av uracil97,98, 99. Bruk av RNPs gjort med in vitro-syntetisert guide RNAs utenom disse bekymringene og forenkler begrensningene i guide design. En vanlig funksjon som fremkom fra disse algoritmene og er bekreftet i flere studier med effektive genomet redigering, er tilstedeværelsen av en purine, spesielt en guanine, på 3 slutten av guiden mål-spesifikke. Denne guide funksjonen har vært svært vellykket blant organismer spenner fra pattedyr C. elegans, frukt fluer og sebrafisk65,100,101. I tillegg for C. eleganser designe guider med en GG dinucleotide på 3 slutten av guiden målretting regionen en effektiv strategi for å forutsi effektive guide RNAs65. Ideelt sett teste flere guider parallelt hva som er mest vellykkede for et gitt program.
Når du prøver å innføre en DNA sekvens i genomet, er utformingen av donor eller mal DNA også avgjørende. Single-strandet oligonucleotide givere (ssODNs) settes mer pålitelig enn andre typiske reparasjon maler, lineær double-strandet og plasmider DNA54,55,102. På noen loci, kan HDR effektivitet forbedres med ssODNs som er komplementære til ikke-målet eller fordrevet DNA strand og har homologi våpen som er asymmetriske i lengde27,55. Siden malen reparasjon settes på webområdet kutt og inkluderer målrettet sekvensen, tas skritt å hindre Cas9 i å spalte donor DNA før eller etter genomisk innsetting. Dette gjøres ved å stille mutasjoner PAM rekkefølgen eller frø regionen, unngå anerkjennelse av Cas9 samtidig beholde funksjonen til innsatte genet21,103. Selv single nukleotid endringer av PAM er sannsynlig å avskaffe bindende104, prøv å endre minst fire nukleotider for å være sikker.
Betydning og framtidige applikasjoner:
Genomet redigering med CRISPR-Cas9 har dukket opp som en kraftfull metode aktivere lettvinte genetisk manipulering av noen organismer. Redigering med Cas9 RNP tar litt mer innsats først, men er enkel å bruke når reagenser og protokoller er etablert i et laboratorium. Redigerer du celler med pre-monterte RNP i stedet for plasmider DNA fører til høyere samlede redigering effektivitet, inkludert vanskelig å oppnå genet innsetting via HDR, med færre off-målet effekter24,25,26 , 27 , 29. videre eksperimentelle unngå problemer med genuttrykk RNA fornedrelse, proteinfolding og tilknytningen mellom gRNA og Cas9 molekyler syntetisert separat i cellen22,23. RNP redigering også omgår sikkerhet bekymringer om insertional mutagenese og vedvarende uttrykk som kan oppstå når viral leveringsmetoder er brukt klinisk14. På grunn av disse fordelene favoriserer mange forskere gjennomfører pre-klinisk, proof-of-concept eksperimenter RNP redigering for menneskelig terapeutiske programmer. Både i vivo og ex vivo RNP-baserte genomet redigering tilnærminger er i utvikling å behandle eller selv kurere en rekke forhold, genetiske sykdommer som Duchenne muskeldystrofi105 og sigdcelleanemi27 HIV29 og kreft11. Interessant, er Cas9 RNP stadig ansatt som en leveringsmetode for landbruket tekniske fordi 'DNA-ledig' redigering av planter33,34,36.
Disclosures
Forfatterne Alexander Marson og Jacob E. Corn er grunnleggerne av Spotlight Therapeutics. Jacob E. Corn er rådgiver for oppdraget Therapeutics hans laboratoriet har fått sponset forskningsstøtte fra AstraZeneca og Pfizer. Alexander Marson er rådgiver Juno Therapeutics og pakt Therapeutics, og hans laboratoriet har fått sponset forskningsstøtte fra Juno Therapeutics, Epinomics og Sanofi. Hans laboratorium har også søkt om patenter relatert til Cas9 RNP teknologi.
Acknowledgments
Vi takker mange tidligere medlemmer av våre laboratorier og Bay Area genomet redigering samfunnet for sine bidrag til utviklingen av disse metodene. Vi takker Ross Wilson for kritisk lesing dette manuskriptet.
Alexander Marson forskning støttes av en gave fra Jake Aronov og en nasjonal multippel sklerose samfunnet gir (CA 1074-A-21). Alexander Marson holder en Career-pris for medisinske forskere fra Burroughs Wellcome fondet og er en Chan Zuckerberg Biohub etterforsker. Jacob E. Corn forskning støttes av Li Ka Shing Foundation, arv medisinsk forskning Medical Institute og California Institutt for regenerativ medisin. Behnom Farboud og Barbara J. Meyers forskning er finansiert delvis av NIGMS tilskudd R01 GM030702 til Barbara J. Meyer, som er en etterforsker av Howard Hughes Medical Institute. Erin Jarvis og Nipam H. Patels forskning er finansiert delvis av NSF stipendet IOS-1257379 og Erin Jarvis anerkjenner støtte fra en NSF-GRFP og en Philomathia Graduate Fellowship.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
