

Summary
Utilizzando un Cas9 preassemblati complesso ribonucleoproteico (RNP) è un metodo potente per l'editing genomico precisa, efficiente. Qui, si evidenzia sua utilità in una vasta gamma delle cellule e degli organismi, tra cui cellule umane primarie e classico ed emergenti organismi modello.
Abstract
L'editing genomico eucariotica site-specific con CRISPR (cluster regolarmente interspaziati brevi ripetizioni palindromi)-sistemi di Cas (CRISPR-associata) è rapidamente diventato un luogo comune tra i ricercatori perseguire un'ampia varietà di domande biologiche. Gli utenti utilizzano più spesso la proteina Cas9 derivata da Streptococcus pyogenes in un complesso con una guida facilmente riprogrammata RNA (gRNA). Questi componenti vengono introdotti nelle cellule, e attraverso una base in abbinamento con una regione complementare del genoma doppia elica (dsDNA), l'enzima si unirà entrambi i fili per generare una rotture a doppio filamento (DSB). Riparazione successiva conduce a inserimento casuale o cancellazione eventi (indels) o l'incorporazione di DNA forniti da sperimentatore presso il sito della rottura.
L'uso di un singolo-guida RNA e Cas9 proteina purificata, preassemblati per formare un RNP e consegnati direttamente alle cellule, è un approccio potente per raggiungere gene altamente efficiente di editing. RNP editing particolarmente migliora il tasso di inserimento del gene, un risultato che è spesso difficile da raggiungere. Rispetto alla consegna tramite un plasmide, la più breve persistenza della RNP Cas9 all'interno della cellula conduce a meno eventi fuori bersaglio.
Nonostante i suoi vantaggi, molti utenti occasionali di CRISPR gene editing hanno meno familiari con questa tecnica. Per abbassare la barriera di ingresso, abbiamo delineare protocolli dettagliati per l'attuazione della strategia di RNP in una gamma di contesti, evidenziando i vantaggi distinti e le diverse applicazioni. Ci occupiamo di editing in due tipi di cellule umane primarie, le cellule T e le cellule staminali/progenitrici ematopoietiche (HSPCs). Mostriamo anche come Cas9 RNP editing consente la facile manipolazione genetica di interi organismi, tra cui il verme Caenorhabditis elegans di modello classico e più recentemente introdotto crostaceo di modello, Parhyale hawaiensis.
Introduction
Fil CRISPR-Cas9 sistema permette agli scienziati di alterare regioni mirate di qualsiasi genoma1. Questa tecnologia veloce e poco costoso ha rivoluzionato la ricerca di base e promette di rendere un profondo impatto sullo sviluppo di terapie personalizzate di malattia, l'agricoltura di precisione e oltre2. CRISPR editing è uno strumento di democratizzazione e implementazione del sistema in un nuovo laboratorio non richiede nessuna particolare esperienza nelle competenze di biologia molecolare del genoma engineering, solo di base. I ricercatori possono ora studiare organismi precedentemente insolubili con pochi mezzi alternativi per manipolazione genetica3,4. Negli ultimi cinque anni da solo, CRISPR editing genomico è stato usato per ingegnere oltre 200 differenti classi di vertebrati, invertebrati, piante e specie microbiche.
Adattato dal pathway di prokaryotic difesa CRISPR, gli elementi fondamentali necessari per l'editing di genoma site-specific sono le proteine Cas9, in genere da S. pyogenes e codone-ottimizzato con un segnale di aggiunta la localizzazione nucleare (NLS) e la sua specializzati RNA Guida5,6. Anche se non è discusso qui, altri Cas9 ortologhi o endonucleasi CRISPR possono anche essere utilizzati. Il gRNA natura è composto di due pezzi separatamente trascritti, il RNA CRISPR (crRNA) e il trans-attivazione crRNA (tracrRNA)7. Questi RNA possono essere fuse in una singola trascrizione, nota come il singolo-guida RNA (sgRNA)8. Maggior parte degli editor di genoma scegliere la snella sgRNA9, anche se il dual-guida è anche usato regolarmente10,11. Gli sperimentatori scegliere un bersaglio di DNA genomico 20 nucleotidi (nt), assicurando che si trova accanto a una breve firma licenza necessaria per il riconoscimento di Cas9, chiamato un motivo adiacente protospacer (PAM) e progettare una gRNA che contiene la sequenza complementare12 .
Una volta all'interno della cellula, il complesso RNP individua il bersaglio genomic, le coppie di basi gRNA con il DNA complementare per ogni fila, e quindi l'enzima idrolizza due filamenti di DNA per generare un doppio filo rompono2. Meccanismo di riparazione del cellulare consente di correggere il DSB da uno di almeno due vie: via pathway (NHEJ) errori non-omologo fine-assemblaggio o la riparazione di omologia-diretto (HDR), che incorpora perfettamente DNA contenente 'armi' di omologia su entrambi i lati della rottura. Il sentiero di riparazione ex in genere conduce alla indel formazione e rottura del gene conseguente, mentre quest'ultimo permette di sperimentatori inserire o modificare il DNA sequenze1.
L'efficienza e la precisione di editing dipendono i mezzi con cui Cas9 e gRNA immettere nella cella. Questi componenti possono essere consegnati a cellule coltivate, embrioni o organismi sotto forma di acidi nucleici o come un preassemblato RNP complesso13,14,15. Metodi basati su acido nucleico consegna comuni includono la trasduzione virale, transfezione o elettroporazione di mRNA o di DNA plasmidico. Guida RNA e proteina Cas9 vengono poi prodotti all'interno della cellula e si associano per formare un complesso.
La consegna diretta di RNP richiede la purificazione separata della Guida RNA e proteina Cas9. Questo può essere fatto in casa, o la proteina e sgRNA possono essere acquistati da uno dei diversi fornitori commerciali. Una volta acquisito, il Cas9 e gRNA sono mescolati per formare il complesso RNP enzimaticamente competente e presentare alle cellule di iniezione diretta in embrioni e le uova fecondati, trasfezione basata sui lipidi16o elettroporazione. Il primo rapporto di RNP editing compreso iniezione in c. elegans gonadi17. Microiniezione è ancora il mezzo preferito di introdurre RNP nel embrioni e interi organismi, anche se efficace elettroporazione è stata dimostrata in18,19 e ratto20 embrioni di topo. Descriviamo i protocolli per iniettare direttamente RNP gonadi di c. elegans e p. hawaiensis embrioni e consiglia un tipo specializzato di elettroporazione di consegnare RNP durante la modifica di cellule umane primarie. Questo metodo, il nucleofection, coinvolge ottimizzato elettroporazione programmi e soluzioni specifiche per tipo di cella e permette il RNP entrare sia il citoplasma e il nucleo21.
L'editing genomico con RNP offre diversi vantaggi distinti. Poiché i componenti di RNA e proteine sono pre-assemblati e qualità può essere assicurata prima della consegna, RNP editing evita molti trabocchetti connessi con la consegna dell'acido nucleico-basato. Vale a dire, non c'è alcun rischio di integrazione Cas9-codifica del DNA nel genoma ospite, mRNA non è mai esposto per degradazione ed elude problemi con in vivo gRNA o proteina espressione, pieghevole e associazione22,23. Inoltre, utilizzando RNP conduce per abbassare la tossicità e molti meno eventi fuori bersaglio rispetto l'espressione basata su plasmide, un risultato della più breve emivita di RNP dentro la cella24,25,26,27.
Infine, in modo dimostrabile RNP modifica porta ad alti tassi di editing in una varietà di linee cellulari umane, cellule primarie come fibroblasti, cellule staminali embrionali (ESCs), indotta da cellule staminali pluripotenti (iSPCs), HSPCs, e cellule di T16,24, 25,26,27,28,29; negli invertebrati, tra cui c. elegans, p. hawaiensise mosche della frutta3,17,30; in specie di vertebrati come zebrafish, topi e ratti31,32; nel piantare specie tra cui Arabidopsis, tabacco, lattuga, riso, vite, mela, mais e grano33,34,35,36; e in Chlamydomonas, Penicilliume Candida specie37,38,39. La frequenza di formazione di indel può essere più elevata quando si utilizza RNP rispetto alla consegna del plasmide e inserimento di HDR-mediato del DNA può essere più facile raggiungere25,27,29.
Il protocollo descritto qui utilizza il RNP Cas9 ed è una tecnica efficace, facilmente adattabile che è semplice da applicare a una vasta gamma di sistemi biologici40,41, soprattutto nelle cellule che sono altrimenti difficili da lavorare con e negli organismi senza sistemi affermati per la manipolazione genetica precisa. Iniziamo descrivendo come progettare, ottenere e assemblare il RNP Cas9 prima di coprire il suo utilizzo attraverso organismi e modello diversi tipi di cellule. Cellule staminali/progenitrici ematopoietiche (HSPCs) e cellule T vengono modificate utilizzando lo stesso metodo, il nucleofection, quindi sono coperti insieme nei passaggi 2 e 3 del presente protocollo. Procedure di editing per C. elegans sono descritte nei passaggi 4 e 5 e P. hawaiensis editing è coperto nei passaggi 6 e 7. Infine, poiché il successo di un esperimento di modifica del gene in qualsiasi organismo può essere valutato mediante sequenziamento di genotipo, sottopassaggi che descrive i metodi di analisi possibile per tutte le cellule e organismi descritti nel protocollo sono indicate nel passaggio 8.
Protocol
1. RNP Assembly
-
Progettare l'esperimento in anticipo, l'acquisizione di tutti i componenti di RNA, DNA e proteine prima del tempo. Come un primo passo, provare uno dei controlli positivi elencati nella tabella 1 e utilizzare i reagenti commerciali descritti nella tabella materiali per garantire un design sperimentale affidabile e l'integrità dei materiali. Per ulteriori suggerimenti sulla pianificazione di un nuovo esperimento di modifica del genoma, vedere documenti su questo argomento12,42,43.
Nota: Una volta montato come descritto nei passaggi successivi, RNP preparato in anticipo possono essere conservati a-80 ° C.- Dopo aver scelto quale gene bersaglio, utilizzare uno degli strumenti online gratuiti per progettare un'ottimale gRNA44,45,46,47,48. Essere sicuri di scegliere come target un esone se nella speranza di generare un ko.
Nota: Questi strumenti verranno aiuterà a identificare un sito di destinazione con un adiacente S. pyogenes PAM sequenza, il Punteggio di qualità e punteggio basso fuori bersaglio. - Purificare la proteina di Cas9 di S. pyogenes attraverso metodi pubblicati8, o acquistare da un fornitore commerciale.
- Preparare un tipico Cas9 tampone per la diluizione di RNA, RNP preparazione e deposito di proteine, che contiene 20 mM di pH HEPES 7.5, 150 mM KCl, glicerolo al 10% e 1 mM di TCEP. Utilizzare sempre acqua priva di nucleasi nei buffer che verrà utilizzato per risospendere o diluire RNA per prevenire il degrado.
- Produrre la Guida di RNA (tracrRNA e crRNA o sgRNA) attraverso una trascrizione in vitro utilizzando metodi pubblicati, o l'acquisto di un acido nucleico sintesi società17,21,49, 50 , 51.
- Se si inserisce un gene, sintetizzare o acquista un donatore del DNA template.
- Memorizzare le proteine e le aliquote di RNA a-80 ° C e scongelare su ghiaccio immediatamente prima dell'uso.
Nota: Ogni gelo-disgelo si abbassa leggermente l'efficienza. Protocolli dettagliati, ad accesso aperto per Cas9 purificazione52 e la trascrizione in vitro di sgRNAs53 sono disponibili altrove.
- Dopo aver scelto quale gene bersaglio, utilizzare uno degli strumenti online gratuiti per progettare un'ottimale gRNA44,45,46,47,48. Essere sicuri di scegliere come target un esone se nella speranza di generare un ko.
- Se lavora con c. elegans, passare al punto 1.5. Per il protocollo di p. hawaiensis , passare al punto 1.6. Se utilizzando sgRNA, passare al punto 1.4. Passate al paragrafo 1.3 per assemblare una gRNA per cellula primaria di editing.
-
Assemblare una gRNA mescolando quantità equimolari di tracrRNA e crRNA. Fare 100 µ l di 80 µM gRNA stock, circa 50 esperimenti di editing di genoma.
- Incubare il gRNA a 37 ° C per 30 min e poi lasciarlo raffreddare lentamente a temperatura ambiente.
-
Prep RNP per editing HSPC e T cellulare: assemblare un complesso RNP miscelando una quantità molare di x di 1-2 di gRNA a 200 pmol di proteina Cas9 in un volume totale di 10 µ l. molto lentamente, aggiungere Cas9 concentrato il gRNA (pre-diluito nel buffer Cas9) per circa 30 s , rendendo rapide Cerchi con la pipetta, portando la concentrazione finale di Cas9 a 20 µM.
- Preparare le provette di elettroporazione.
Nota: Questo protocollo è specifico per il sistema commerciale di cui alla Tabella materiali, ma RNP editing può essere ottenuta anche con altri dispositivi di elettroporazione. - Aggiungere 5 µ l (100 pmols, cellule di T) o 10 µ l (200 pmol, HSPCs) di RNP per ogni provetta.
- Se l'inserimento di nuovo DNA piuttosto che fare un knockout, Aggiungi 1 µ l di 100 µM (100 pmol) singolo-incagliato oligonucleotide donatore del DNA (ssODN)25,54,55 la cuvette o pozzetti della piastra.
- Procedere con il passo 2 per le istruzioni nel montaggio cellula primaria protocollo.
- Preparare le provette di elettroporazione.
-
Preparazione di RNP per l'editing di c. elegans : assemblare il complesso RNP aggiungendo i seguenti reagenti al fine di creare un volume finale di 20 µ l (le concentrazioni finali sono riportate tra parentesi): Cas9 (2 µM), pH HEPES 7.5 (10 µM), KCl (115 µM), crRNA (12 µM) , tracrRNA (40 µM) e la riparazione modelli se necessario (0,5 µM ssDNA o fino a 350 ng / µ l dsDNA).
Nota: L'efficienza di una riparazione Cas9-mediata DSB-basato su modelli è proporzionale alla concentrazione del dsDNA riparazione costrutto; quindi, maggiore è la concentrazione del modello riparazione, il più efficiente la riparazione basato su modelli. Tuttavia, un'iniezione di miscele contenenti superiore a 350 ng / µ l di dsDNA ha dimostrata di ridurre la vitalità dei vermi iniettati. Così, si consiglia di utilizzare fino a, ma non più di 350 ng / µ l di dsDNA nel mix per massimizzare l'efficienza di riparazione riducendo al minimo la letalità.- Aggiungere più crRNAs a target luoghi multipli simultaneamente, come necessario per l'approccio di screening co-CRISPR/co-conversion descritto al punto 5.4. Quando si aggiunge più di un crRNA, aggiungere ogni in sequenza per il mix master.
Nota: La quantità di ogni crRNA non deve necessariamente essere lo stesso, e anche raddoppiando la concentrazione totale di crRNAs nel mix master senza modificare la concentrazione di Cas9 non sembra interferire con la frequenza di mutagenesi in un locus specifico. Esempi sono descritti in dettaglio in Paix et al. 56. - Mescolare pipettando e girare la soluzione RNP a 16.000 x g per 5 s per assicurare che la soluzione è raccolto nella parte inferiore del tubo.
- Incubare la soluzione a 37 ° C per 15 m.
- Centrifugare il campione a 16.000 x g per 1 min a pellet eventuali particelle che potrebbero intasare l'ago sottile-annoiato microiniezione. Utilizzare il surnatante nei passaggi successivi.
- Andare al passaggio 4 per il resto del protocollo dei elegans del c .
- Aggiungere più crRNAs a target luoghi multipli simultaneamente, come necessario per l'approccio di screening co-CRISPR/co-conversion descritto al punto 5.4. Quando si aggiunge più di un crRNA, aggiungere ogni in sequenza per il mix master.
-
Prep RNP per p. hawaiensis editing: Preparare aliquote Cas9 monouso diluendo con acqua priva di nucleasi e rosso fenolo (per la visualizzazione di iniezioni) ad una concentrazione finale di 6.25 µM di rosso fenolo Cas9 e 0,15%.
- Assemblare il complesso RNP mescolando un 2-5 x eccesso molare di gRNA alla proteina Cas9 in un volume totale di 6 µ l. aggiungere 12 pmol di Cas9 a gRNA, portando la concentrazione finale di Cas9 a 2 µM, gRNA concentrazione di 4-8 µM e la concentrazione di rosso fenolo a 0,05%.
- Incubare la miscela a temperatura ambiente per 10 min al complesso RNP.
- Procedere con il passo 6 per le istruzioni nel montaggio hawaiensis p. protocollo.
2. cultura e preparazione delle cellule
Nota: Eseguire la procedura 2.1.1 a 3.3.3 in una cappa di sicurezza biologica.
-
Umani crioconservati acquisto mobilitato circolanti CD34+ HSPCs da un fornitore.
- Scongelare ~ 1 x106 HSPCs in un'acqua di 37 ° C bagno per 3 min e trasferirli in una provetta conica da 15 mL. Aggiungere 10 mL di mezzo di espansione senza siero da una fonte commerciale e girare il composto a 100 x g per 10 min. eliminare il surnatante e risospendere le cellule in 2 mL di SFEM completati. Le cellule in piastre da 6 pozzetti del piatto e cultura li in un incubatore a 37 ° C per 24-48 h prima l'elettroporazione RNP.
- Contare le celle con un emocitometro e trasferire il numero totale di HSPCs necessari (150.000-200.000 HSPCs per cuvette per essere individulamente) in una provetta da centrifuga.
- Girare il tubo a 100 x g per 10 min agglomerare le cellule.
-
Acquistare CD4 primario umano+ T cellule da un fornitore o isolarli da sangue umano intero dalla centrifugazione in gradiente di densità29.
- Prima dell'attivazione delle cellule T, pre-cappotto piastre di coltura 48 pozzetti con αCD3 (UCHT1) e αCD28 (CD28.2). Rivestire le piastre con 500 µ l di 10 µ g/mL αCD3 e 10 αCD28 µ g/mL in PBS per almeno 2 ore a 37 ° C.
Nota: Per alcuni loci, NHEJ può essere raggiunto senza pre-stimolazione, ma anche questo passaggio massimizza l'efficienza. - Cellule di cultura la T per 48 h a 37 ° C su αCD3/αCD28 legata all'anticorpo piastre in un medium RPMI completo [RPMI-1640 completate con 5 mM HEPES, 2 mM di alternativa commerciale a L-glutamina, 50 µ g/mL di penicillina/streptomicina, 50 µM di 2-mercaptoetanolo, 5 mM di non essenziali aminoacidi, 5 mM di piruvato di sodio e il 10% (vol/vol) FBS]. Cellule di cultura la T a una densità di 2.000.000 T cellule in 500 µ l di media per pozzetto di una piastra a 48 pozzetti.
- Conteggio del T cellule utilizzando un emocitometro e trasferimento il numero totale di cellule T necessari per l'elettroporazione esperimento (100.000-1.000.000 cellule T per cuvette per essere individulamente) in una provetta per centrifuga.
- Girare il tubo a 90 x g per 8 min agglomerare le cellule. Se le cellule sono state densità gradiente separati entro 2 giorni, li spin a 200 x g per 8 min.
- Prima dell'attivazione delle cellule T, pre-cappotto piastre di coltura 48 pozzetti con αCD3 (UCHT1) e αCD28 (CD28.2). Rivestire le piastre con 500 µ l di 10 µ g/mL αCD3 e 10 αCD28 µ g/mL in PBS per almeno 2 ore a 37 ° C.
-
Per entrambi i tipi cellulari, aspirare il surnatante con un pipetta/vuoto, rimuovere tutte le bolle.
- Risospendere delicatamente le cellule con 20 µ l di tampone di elettroporazione per cuvette.
- Aggiungere 20 µ l delle cellule (150.000-200.000 HSPCs o le cellule di T di 100.000-1.000.000) per ogni provetta, che già contiene 10 µ l di RNP e mescolare bene pipettando su e giù senza creare bolle.
3. RNP elettroporazione
- Electroporate le provette dopo la loro immissione in un Nucleofactor. Per la HSPCs, utilizzare il codice di impulso ER100. Per le cellule di T, utilizzare il codice di impulso EH-115.
-
Solo HSPCs: Aggiungere 100 µ l di un mezzo SFEM completato (riscaldato a 37 ° C) per ogni provetta immediatamente dopo l'elettroporazione e lasciare che le cellule recupero per 10-15 min.
- Trasferire le cellule in cultura li in un turno di 96 pozzetti-fondo piatto e aggiungere un ulteriore 100 µ l del mezzo SFEM completato per 24 h.
- Cambiarli in un mezzo fresco completato SFEM e li Incubare per altre 24-72 h.
- Rimuovere le cellule per la genotipizzazione li 48-96 h post-elettroporazione. Gira le cellule a 300 x g per 5 minuti e rimuovere il surnatante prima di iniziare l'estrazione del DNA (punto 8.2).
-
Solo le cellule di T: aggiungere 80 µ l di RPMI completare mezzi di coltura pre-riscaldato a 37 ° C dal serbatoio per ogni provetta o bene, usando una pipetta multicanale (se necessario).
- Li Incubare a 37 ° C per 15 min.
- Aggiungere il supporto appropriato, anticorpi, citochine, ecc per le piastre di destinazione e pre-riscaldare loro in un incubatore a 37 ° C.
- Trasferire 107 µ l delle cellule elettroporate dai pozzetti in una piastra a 96 pozzetti turno-fondo utilizzando una pipetta multicanale (se necessario).
- Per informazioni su come valutare i risultati di editing, andare al passaggio 8.
4. preparazione di c. elegans
-
1 giorno prima microiniezione: preparare le pastiglie di agarosio per la microiniezione.
- Fare una soluzione di agarosio 3% (p/v) in acqua aggiungendo agarosio all'acqua e portando la soluzione ad ebollizione su una piastra calda o nel forno a microonde.
- 24 x 50 mm x diapositive di vetro di copertura 1,5 mm su un tavolo di organizzare e utilizzare un pipetta Pasteur di vetro per mettere una goccia di piccolo (~ 15 µ l) di soluzione di agarosio sulla diapositiva. Rapidamente di appiattire la goccia di agarosio inserendo un altro vetrino coprioggetto sulla parte superiore. Consentire l'agarosio solidificare e quindi rimuovere una delle lamelle.
- Lasciare il vetrino coprioggetto rivestite su agarosio a faccia in su un tavolo ad asciugare durante la notte. Dopo 24 h, è possibile memorizzare le pastiglie di agarosio in un contenitore pulito e asciutto.
Nota: Questi possono essere utilizzati a tempo indeterminato.
- Tirare gli aghi di microinjection: utilizzo di tubi capillari in vetro borosilicato con filamenti (diametro 1,0 mm e interno diametro esterno 0,58 mm), tirare gli aghi basati su Mello e Fire57 e altre risorse58. Gli aghi possono essere utilizzati immediatamente o possono essere immagazzinati in un contenitore pulito e asciutto, rinforzato da supporti di argilla.
- Per la manutenzione dei vermi, preparare un agar di Nematode crescita Media (NGM) versata in piastre di Petri e macchiato con OP50 batteri (per protocolli standard di c. elegans manutenzione e ricette per mezzi di sviluppo, vedere Stiernagle59).
- Tappa i vermi per il microinjection: 12-24 h prima la microiniezione, pick ermafroditi L4-messa in scena di una piastra di agar a NG con batteri OP50 e incubare per una notte a 20 ° C. Per ogni mix di destinazione/iniezione Cas9, prendono ~ 30 vermi alla piastra.
-
Giorno di microiniezione: Caricare l'ago per microiniezione tirato con il surnatante soluzione RNP preparata al punto 1.5.
- Dispensare il surnatante dal punto 1.5.4 in una pipetta capillare tirato e recupero la soluzione dalla pipetta capillare nell'ago per microiniezione preparati (generalmente meno di 0.1 µ l di caricamento).
- Montare l'ago caricato sulla apparato microiniezione collegato a un micromanipolatore. Impostare la pressione di apparato di iniezione a 250 kPa e la pressione di equilibrio a 25 kPa.
-
Indietro rompere la punta dell'ago caricato per generare un bordo tagliente dell'ago. Posto un 15 mm x 15 mm x 1,5 mm quadrati coprioggetto sulla cima di un 24 x 50 mm x 1,5 mm vetrino coprioggetti.
- Overlay uno spigolo del quadrato coprivetrino con olio halocarbone 700.
- Posizionare l'ago nell'olio, al bordo del coprioggetto quadrati di 15 mm.
- Usando una mano per guidare il tavolino del microscopio e coprioggetto, spazzolare la diapositiva fino e lungo il bordo dell'ago mentre si preme il pedale/pulsante di iniezione. Rompere la punta dell'ago indietro, aumentando il flusso di liquido fuori l'ago. Ottenere una portata ottimale facendo l'iniezione mescolare flusso lungo il bordo dell'ago, formando ~ 1 bolla/s.
- Confermare che i vermi di L4 scelto 12-24h prima microiniezione sono inerente allo sviluppo in scena giovani adulti il giorno dell'iniezione. Scegli i vermi adulti giovani ad una piastra di agar a NG che manca OP50 batteri e consentire loro di strisciare per 5 min. Questo riduce la quantità di batteri trasferiti al pad iniezione, riducendo al minimo gli zoccoli dell'ago.
- Un'iniezione di agarosio pad/vetrino coprioggetti. mettere in un ambito di dissezione. Usando un plettro di verme, giaceva una piccola traccia di olio halocarbone lungo un bordo del pad.
-
Utilizzando il plettro di verme rivestito in olio, sollevare diversi worm fuori la piastra di agar a NG e nella traccia di olio. Con una peluria associata a una pipetta, come un soffio di ciglia o un gatto, posizionare i vermi in parallelo, delicatamente spingendo i vermi nel pad dell'agarosi. Fino a suo agio con la procedura di microiniezione, solo montare e iniettare un verme alla volta.
Nota: L'agarosio asciutto sarà stoppino l'umidità dai vermi, causando loro di aderire al pad. Di conseguenza, uno deve lavorare velocemente come i vermi possono essiccare.- Una volta in posizione e attaccato al pad, sovrapporre i vermi con un altro prendere alcune gocce di olio halocarbone (~ 20 µ l) dalla punta del worm.
5. c. elegans gonade microiniezione con RNP e cura post-iniezione
Nota: Il protocollo di microiniezione è adattato da Mello e Fire57e descritto dettagliatamente altrove60,61.
-
Porre il coprivetrino con i vermi montati sul microscopio iniezione. Con un basso ingrandimento (obiettivo 5 X, 10 X oculare), posizionare i vermi perpendicolari all'ago per iniezione.
- Interruttore di un alto ingrandimento (obiettivo 40x, oculare 10x), riposizionare l'ago adiacente al braccio della gonade corrispondente alla regione vicino i nuclei in metà - alla fine-pachitene.
- Utilizzando il micromanipolatore, spostare l'ago contro il worm, deprimente la cuticola leggermente. Quindi, con una sola mano, toccare il lato del palco microscopio per scuotere l'ago attraverso la cuticola. Premere il pedale/pulsante di iniezione e lentamente riempire il braccio della gonade con il mix di iniezione e rimuovere l'ago.
- Ripetere questo passaggio con l'altro braccio della gonade.
-
Una volta che i vermi sono iniettati, rimuovere il vetrino coprioggetto/agarosio pad e posizionarlo sotto un microscopio per dissezione.
- Utilizzando una pipetta capillare tirata, occorre spostare l'olio dai vermi pipettando un buffer M9 su di loro. Eseguire questo trattamento per rilasciare i vermi da agar.
- Dopo 10 min, quando i vermi sono thrashing intorno nel buffer, spostarli in una piastra di agar NG con batteri OP50 utilizzando la pipetta capillare tirata. Posizionare la piastra a 20 ° C per 2-3 h fino a quando i vermi hanno recuperato e si muovono.
- Una volta recuperato, individualmente trasferire i vermi piatti di NG-agar con OP50 e le piastre di trasferimento di un incubatore di 25 ° C.
-
Consentire la P0-iniettato vermi a crescere e a dare progenie per 3 giorni. Schermo la prole di1 F.
- Se si utilizza co-CRISPR o co-conversione62,63,64,65, quindi selezionare i vermi candidato per lo screening basato su se hanno il fenotipo mutante del gene di riferimento. Individualmente trasferire questi vermi contrassegnati a nuove piastre di agar a NG con OP50 e consentire loro di posare progenie2 F a 20 ° C.
Nota: Il fenotipo utilizzato per un co-CRISPR selezione o selezione deve fornire una stima iniziale per il successo di Cas9 editing. - Se non è presente il fenotipo di co-CRISPR, microinject un plasmide di controllo positivo per aiutare a migliorare l'efficienza di microiniezione.
Nota: per esempio, tra cui un plasmide del mix di iniezione che codifica mCherry-etichetta MYO-2 aiuterà a valutare l'efficienza di iniezione. Vermi correttamente iniettato con pCFJ90 avrà qualche prole con fluorescente pharynxes.
- Se si utilizza co-CRISPR o co-conversione62,63,64,65, quindi selezionare i vermi candidato per lo screening basato su se hanno il fenotipo mutante del gene di riferimento. Individualmente trasferire questi vermi contrassegnati a nuove piastre di agar a NG con OP50 e consentire loro di posare progenie2 F a 20 ° C.
- Esaminare i vermi di1 F per la presenza di modifiche desiderate. Scegliete la madre di1 F in un individuale pozzetto di una piastra a 96 pozzetti, lisare lei ed esaminare il suo DNA di amplificazione di PCR di inserto specifiche analisi di sequenza del DNA o, surveyor nuclease assay (CEL-1)66.
Nota: Queste analisi possono essere eseguite quando si utilizza un co-CRISPR/co-conversion o altri screening o selezione regimi65,66,67,68. - Per informazioni su come valutare i risultati di editing, andare al passaggio 8.
6. preparazione hawaiensis p.
- 1 giorno prima la microiniezione, arricchire per gli embrioni in anticipo mediante l'istituzione di un "serbatoio di coppia' la sera prima; le femmine appena separate conterrà embrioni appena fertilizzato. Vedere Rehm et al. 69 per i dettagli.
- Il giorno di microiniezione, raccogliere gli embrioni Parhyale unicellulare (0-4 h post-fertilizzazione) da anestetizzando le femmine gravide con 0,02% olio di chiodi di garofano in acqua di mare e raschiare delicatamente gli embrioni fuori suo marsupio coulors ventrale utilizzando una fiamma-tirato e pipetta di vetro arrotondato e una coppia noiosa di forcipe #3.
7. p. hawaiensis embrione microiniezione con RNP e cura post-iniezione
- Recupero informazioni un tubo capillare tirato con circa 1 µ l della miscela di iniezione RNP descritto sopra.
-
Utilizzare azoto compresso per microinject ogni embrione come descritto in Rehm et al. 69.
- Iniettare gli embrioni Parhyale sotto un microscopio per dissezione utilizzando un microinjector e un micromanipolatore. Carico di 1,5 µ l della miscela di iniezione nella parte posteriore di un tubo capillare tirato (4 pollici - 1.0 mm con filamenti, tirato usando una micropipetta tirando apparato) utilizzando un puntale di microloader.
- Impostare l'ago sull'apparato di iniezione e rompere la punta dell'ago (una piccola quantità) usando un paio di pinze per dissezione nell'ambito. Calibrare il volume consegnato iniettare olio halocarbone 700 e misurando il diametro della bolla.
- Tagliare un 'passare' fuori l'agente indurente utilizzando una lama di rasoio. Riempire a metà con acqua di mare filtro-sterilizzato e allineare gli embrioni Parhyale nel trogolo per stabilizzare.
- Iniettare gli embrioni utilizzando l'installazione di microiniezione, ogni embrione di stabilizzazione con un paio di pinze durante l'iniezione. Dopo l'iniezione, è possibile utilizzare una pipetta di vetro per trasferire gli embrioni su di una piastra di coltura fresco 60 mm riempita a metà con acqua di mare filtro-sterilizzato.
-
Se la prima divisione è già verificato per formare un embrione di 2 cellule (post-fertilizzazione di 4-6 h), è possibile generare gli animali completamente-mutante iniettando entrambi blastomeri. Per garantire una totale scissione della fase 2-cella, co-iniettare i blastomeri con FITC o TRITC destrano e osservare che il segnale è limitato a un singolo blastomero sotto un fluorescente ambito di dissezione dopo l'iniezione.
- In alternativa, è possibile generare 'metà-mutante' animali iniettando solo uno dei due blastomeres nella fase 2 celle (sinistra-destra approssimativamente diviso a seconda del tessuto e la posizione lungo l'asse A-P).
- Iniettare una cella in un embrione di 8 celle (post-fecondazione 7,5-9 h) per limitare la modifica a un singolo strato di germe. Vedere Gerberding et al. 70 per una mappa dei primi lignaggi di blastomeri.
-
Incubare gli embrioni in piastre di coltura di 60 mm (non più di 25 per ogni piatto), riempite a metà con acqua di mare filtro-sterilizzato, 'pre-ossigenato' utilizzando un gorgogliatore acquario o agitando vigorosamente il flacone.
- Posizionare i piatti degli embrioni in un plasticware vagamente-sigillato interno foderato con tovaglioli di carta bagnati per mantenere umidità e metterli in un'incubatrice di 26 ° C con un ciclo luce-buio di 12 h.
- Trasferire gli embrioni sopravvissuti per pulire i piatti con acqua di mare ogni pochi giorni.
Nota: Gli embrioni possono essere coltivati a temperatura ambiente, anche se si svilupperanno molto più lentamente.
-
Sezionare e correggere gli embrioni nelle varie fasi per un'analisi di espressione di in situ di ibridazione o anticorpo colorazione (Vedi Browne et al. 71 per una guida di gestione temporanea e ulteriori riferimenti per la dissezione e la fissazione72, in situ ibridazione73e74la macchiatura dell'anticorpo).
- Fare la dissezione aghi infilando un pezzo piegato di filamenti di tungsteno circa 0,5 a di lunghezza nell'estremità di un ago da insulina. Affinare l'ago in idrossido di sodio sotto una corrente. Utilizzare una siringa da 1 mL per la gestione dell'ago di dissezione.
- Riempire un pozzo di un piatto di vetro 3-pozzetti a metà con una soluzione di preparati di 9 parti PEM Buffer (0.1 M di pH di tubi 6.95, 2 mM di EGTA, 1 mM di MgSO4), 1-parte 10X PBS e 1 parte 32% PFA. Posizionare gli embrioni di 3-5 nel piatto e colpire un piccolo foro sul ogni embrione, utilizzando un ago di tungsteno tagliente a poke e uno leggermente offuscato per stabilizzare, permettendo il tuorlo di defluire e il fissativo per l'esecuzione in.
- Utilizzando una coppia di aghi affilati tungsteno, delicatamente civettuole distanza l'esterne due membrane che circondano l'embrione Parhyale . Li sezionava in fissativo per rendere più affidabile gli embrioni ma lavoro rapidamente per mantenere la membrana da diventare fisso per l'embrione, che rende più difficile la rimozione della membrana. Consentire gli embrioni alla difficoltà per un totale di 15-20 minuti per la macchiatura dell'anticorpo o 40-50 min per ibridazione in situ .
- Immagine dal vivo hatchlings e analizzarli per fenotipi morfologici e comportamentali o difficoltà e li macchia per analisi più dettagliate. Sollevare le hatchlings alla maturità sessuale a 2-3 mesi per stabilire knockout e linee transgeniche (Vedi Kontarakis e Pavlopoulos75 per la cura del cucciolo e altri dettagli utili).
8. valutare i risultati di Editing
- Se del caso, cercare un fenotipo visual o funzionale in celle modificate o organismi.
Nota: Questo processo varia ampiamente da applicazione e alcuni esempi sono descritti alla fine della loro procedura pertinente protocollo precedente. Dopo aver corretto la mutazione delle cellule di falce in HSPCs, è possibile analizzare la produzione di emoglobina di eritroblasti differenziati mediante HPLC (Figura 1A). Un knockout del gene del ricevitore di IL-2 in cellule di T può essere confermato dalla macchiatura superficiale e flusso cytometry (Figura 1B). Per valutare i fenotipi di c. elegans e hawaiensis p. , osservare la morfologia degli animali e il comportamento sotto un microscopio chiaro o fluorescente (figure 1 e 1 D). - Per determinare l'efficienza e il tipo delle modifiche genomiche generato, lisare le piscine di celle modificate ed estrarre il loro DNA genomico utilizzando un kit di estrazione commerciale21.
-
Per una rapida stima della formazione indel, PCR-amplificare almeno 200 paia di basi intorno il taglio del sito ed eseguire un endonuclease1 T7 (T7E1)76 o Geometra (nucleasi CEL-1) dosaggio77.
- Se una formazione di indel sito Cas9-taglio o successo HDR sarà creare o rimuovere un sito di restrizione nota, considerare l'utilizzo di una digestione degli enzimi di limitazione per stimare l'editing di efficienza6. L'analisi di polimorfismo (RFLP) restrizione lunghezza frammento può essere un modo pratico per verificare l'efficienza, se capita di essere disponibile.
- Per una quantificazione accurata della efficienza di editing e la determinazione dei risultati dell'editing predominante, inviare l'amplicone PCR per una standard Sanger sequenziamento con primer sia avanti e indietro.
Nota: Se l'analisi di un singolo clone o organismo, l'analisi dei risultati di Sanger è semplice, come illustrato nella Figura 2A. Se l'analisi di un pool di cellule, quindi analizzare i cromatogrammi con il tool online78, come illustrato nella Figura 2B. - Per una completa quantificazione e sequenze di editing dei risultati, è necessario eseguire sequenziamento profondo27,54, come raffigurato in Figura 2C.
- Per valutare un particolare insieme di modifiche fuori bersaglio, PCR-amplificare i predetti siti fuori bersaglio e mandarli per NGS. Per abilitare il rilevamento di traslocazioni cromosomiche, eseguire Guida-seq79 o ad alta velocità, spostamento di genoma sequenziamento (HTGTS)80. Per un quadro completo delle modifiche fuori bersaglio in una popolazione clonale, eseguire sequenziamento intero genoma (WGS)81,82,83.
Nota: Ci sono una varietà di metodi per quantificare le modifiche del genoma e fuori bersaglio, ha spiegate ulteriormente in rassegna vari articoli84,85,86.
Representative Results
Questi esperimenti Visualizza come pre-assemblati Cas9 RNP può essere utilizzato per manipolare il genoma di cellule primarie e interi organismi. I ricercatori purificano o acquistare sgRNA e proteina Cas9, combinano i due componenti per la pre-formare il complesso e introducono il RNP in loro cellule o organismo di interesse. Dopo aver lasciato abbastanza tempo per l'editing per verificarsi e per la prole di ultima generazione per essere Nato (se applicabile), controllare i fenotipi e/o raccogliere le cellule per la genotipizzazione. Fenotipi possono essere osservati tramite saggi funzionali, analisi di espressione, visualizzazione (ad occhio o con microscopia) o altri metodi, a seconda dell'esperimento.
Ad esempio, HSPCs che sono stati modificati per correggere la mutazione di β-globina che causa anemia falciforme può essere differenziato in eritrociti e analizzati per la produzione di sano o la falce dell'emoglobina27,87 (Figura 1 A). T le celle modificate a knock out del gene del ricevitore di IL-2 ad alta affinità, CD25 (IL2RA), possono essere analizzati dalla macchiatura superficiale e flusso cytometry88e funzionalmente analizzati per rilevare una segnalazione risposta a stimolo del-2 (Figura 1B ). L'efficacia antitumorale in vivo di auto-T cellule11e cellule T possono anche essere riprogrammate in molti modi clinicamente importanti che richiedono la valutazione di diversi fenotipi, tra cui l'efficacia di HIV infezione89 .
Utilizzando un approccio di screening co-CRISPR/co-conversion, c. elegans worm vengono modificati contemporaneamente a due loci62. HDR al gene di riferimento dpy-10 utilizzando un ssODN riparazione risultati di modello in una mutazione di guadagnare-de-funzione facilmente segnato dominante dpy-10 . Eterozigoti F1dpy-10(gof) animali sono rullo (Rol) e gli animali omozigoti dpy-10(gof) sono losca (Dpy). La presenza del fenotipo indica che Cas9 l'editing si è verificato in questi animali e migliora le probabilità di individuare un evento editing presso il secondo locus negli animali1 Rol o Dpy F. 33-50% di iniettato P0 vermi producendo 20 o più prole1 F che sono Rol o Dpy90dovrebbe provocare un esperimento riuscito di editing. Quindi è possibile scegliere gli animali non-Rol tornare dpy-10 wildtype e selezionare per la modifica omozigotica di interesse. Come regola generale, la concentrazione di crRNA del gene di riferimento co-CRISPR di targeting dovrebbe essere metà che del crRNA di mira il gene di interesse. Se una modifica del gene di interesse non viene recuperata, i rapporti dei due CRISPR RNA possono essere regolati per aumentare le probabilità di recuperare la mutazione desiderata. Per esempio, aumentando la quantità di crRNA per il gene di interesse riguardante la crRNA del gene di riferimento aumenterà la percentuale di vermi che possiedono modifiche nel gene di interesse all'interno della popolazione di vermi che possiedono anche modifiche al luogo del gene di riferimento. Frequenze di co-conversione variano, ma le tariffe sono in genere 20-60%, spesso producendo omozigotiche modifiche nella generazione1 F (Figura 1C).
P. hawaiensis hatchlings che sono state omesse per buttare giù il gene Addominale-B (Abd-B) visualizzare anomalie morfologiche chiaro3 (Figura 1D). Questo gene è necessario per la corretta addominale modellatura, e relativi risultati di rottura in toracica tipo saltando e zampe sostituendo le gambe nuoto e ancoraggio che sono di solito presentano sull'addome.
Determinare esiti a livello genotipico di editing genomico richiede sequenziamento o un'analisi in vitro che rileva le variazioni di sequenza. Qui, indichiamo dati di sequenziamento rappresentante dai nostri tipi cellulari modello e organismi, evidenziando i diversi approcci alla quantificazione di editing. Si noti che le etichette di figura sono generalizzate perché tutti i metodi mostrati qui possono essere applicati a qualsiasi sistema biologico.
Approcci basati sul sequenziamento variano in complessità tecnica e profondità dei risultati. Per popolazioni clonali modificate o organismi individuali facilmente separabili, possono essere sequenziati individui modificati dopo estrazione del DNA genomico. Risultati di sequenziamento Sanger standard rivelerà il cambiamento di sequenza nel sito Cas9-taglio in un dato individuo, con ipotetici frameshift che sconvolgerebbe la sua funzione (Figura 2A). Lo strumento online utilizzato per il sequenziamento è un altro approccio di base di sequenziamento Sanger che può essere applicato a popolazioni miste, piuttosto che singoli mutanti78. Sequenze sono analizzate con uno strumento online che può approssimare complessiva efficienza di editing come pure i risultati sequenza predominante. I dati rappresentativi sono illustrati nella Figura 2B.
Il metodo di sequenziamento più approfondito qui descritto è sequenziamento profondo (a volte denominato sequenziamento high throughput o generazione). Questo metodo fornisce sequenze di DNA da singoli genomi in una popolazione mista. Tali dati possono essere illustrati in una varietà di modi. Qui, abbiamo classificato individuale sequenziamento letture da celle modificate sulla base del risultato di editing (Figura 2C). Maggior parte delle cellule vengono modificata tramite la via NHEJ, che in genere si traduce in rottura del gene. In altri casi, il gene dell'obiettivo è stato scambiato per una versione alternativa via HDR27.
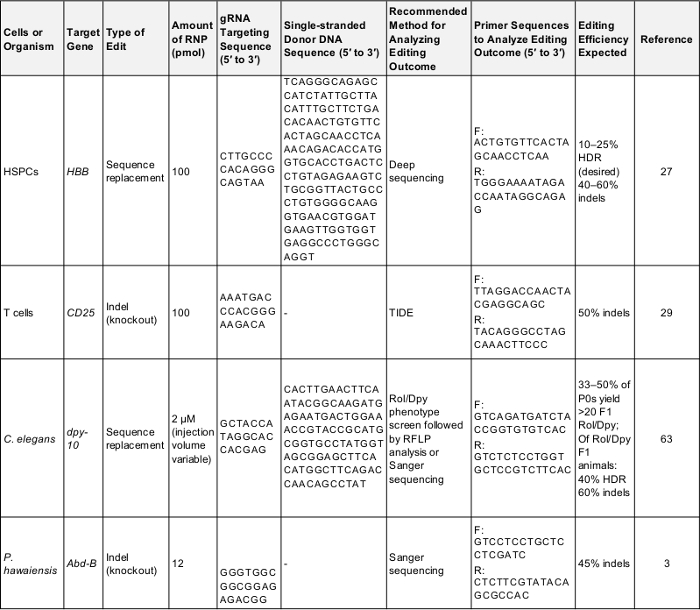
Tabella 1: positivo controlla per genoma preliminare esperimenti di editing. Questa tabella mostra le informazioni chiave necessarie per eseguire un genoma di prima volta editing esperimento in ciascuna delle cellule e degli organismi descritti nel presente protocollo. Seguendo questi parametri è probabile per produrre un risultato positivo che può essere utilizzato per verificare il protocollo o come base per il confronto una volta lo sperimentatore prende di mira un gene di proprio interesse. F: avanti, retromarcia r:, HDR: riparazione omologia-diretto. Per favore clicca qui per scaricare questa tabella.

Figura 1 : Rappresentante fenotipica risultati da Cas9 RNP editing di cellule umane primarie e organismi. (A) si tratta di un'analisi HPLC risultati che dopo successo genoma di editing, HSPCs che sono differenziati in ritardo-fase eritroblasti produrrà più funzionale dell'emoglobina di emoglobina di falce. Eritrociti mutanti producono emoglobina falciforme (HbS), mentre cellule modificato correttamente produrrà emoglobina sano (HbA e HbA2) così come l'emoglobina fetale (HbF). L'assorbanza è rappresentato graficamente in unità arbitrarie (au). Questo pannello è stato pubblicato in DeWitt eal t 27. è ristampato con il permesso dell'associazione americana per l'avanzamento della scienza. (B) sulla sinistra, per ogni condizione, questo pannello mostra flusso cytometry dati mostrando che superficie-macchiato T cellule non esprimono CD25 dopo il gene di CD25 è stato buttato con RNP. L'abbondanza di CD25 viene tracciata sull'asse x con le dimensioni della cella sull'asse y. A destra, per ogni condizione, questo pannello mostra la quantificazione di fosfo-Stat5 (pStat5) dopo un'induzione con il-2. La segnalazione è ridotta quando il recettore il-2 è assente (CD25 KO). L'abbondanza di pStat5 viene tracciata sull'asse x e i dati risultanti da tre diversi livelli di IL-2 input vengono confrontati in verticale. (C) questo pannello mostra una Caenorhabditis elegans co-CRISPR/co-conversion schermata dpy-10 come l'indicatore di CO-Conversione di targeting. Due guida RNA bersaglio due loci, dpy-10 e il tuo preferito gene (yfg), in stesso P0-animale iniettato. HDR a dpy-10 provoca un fenotipo Rol o Dpy. La selezione di Rol - o Dpy-F1 animali aumenta le probabilità di individuare modifiche presso il secondo locus. (D) questo pannello mostra che wildtype hawaiensis Parhyale hatchlings normale addomi con gambe nuoto e ancoraggio. Le covate di knock-out di Abd-B (individui di0 F) sviluppano un addome trasformato verso il torace. Così, il nuoto e le gambe ancore sono scomparsi e sostituite da saltare e a piedi gambe connesse con un torace normale. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2 : I risultati tipici di modificare i metodi di analisi del risultato. (A) questo pannello mostra esempi del Sanger sequenziamento risultati da diversi organismi di1 hawaiensis p. F, inclusi la sequenza di wildtype e tre diversi indels che disturba la funzione genica spostando l'open reading frame. (B) marea questi risultati mostrano la gamma di operazioni di inserimento e gli eventi di eliminazione che si è verificato in un sito di Cas9-destinazione in un pool di cellule T in sequenza. L'asse x indica la lunghezza di una determinata inserzione o omissione in nucleotidi. (C) questi risultati di sequenziamento profondo non mostrano nessun editing genomico senza nucleofection o gRNA e successo editing con intatta Cas9 RNP, raggruppati per risultato di riparazione del DNA in HSPCs. Clicca qui per visualizzare una versione più grande di questa figura.
Discussion
Che istituisce un genoma robusto protocollo di modifica in una cella, riga o organismo di interesse richiede l'ottimizzazione ed empirica test di vari parametri chiave, discussi in questa sezione. Cercando alcune variazioni degli approcci generali qui presentati è fortemente incoraggiata. La limitazione di chiave di questo protocollo è che applicando questi metodi ad altre cellule o organismi possono condurre ad un risultato diverso a seconda della specie studiata, e un disegno sperimentale che conduce a un knockout del gene ad alta efficienza non può promuovere l'inserimento di DNA. Pertanto, si consiglia di iniziare con i metodi qui presentati e risoluzione dei problemi come descritto di seguito.
Risoluzione dei problemi di qualità di reagente di editing genomico:
Generazione o l'acquisto di reagenti di alta qualità è un passo fondamentale in qualsiasi protocollo di modifica di genoma. Cas9 proteina possa essere purificata in laboratorio o acquistata in commercio. Molti protocolli di notare una concentrazione finale per Cas9 nelle ricette di RNP, ma il gene ottimo attività di editing dipenderà l'attività specifica di qualsiasi preparazione individuale per la proteina Cas9, che varia a seconda della fonte. Una volta che il protocollo presentato qui sta lavorando, considera l'ottimizzazione della quantità di RNP utilizzato da titolazione Cas9 livelli per stabilire una concentrazione ottimale: uno che fornisce la fenditura del DNA target altamente specifico senza inutili fuori bersaglio scissione causata da eccessivo Cas940.
Guida RNA purezza e omogeneità può anche essere fattori determinanti del successo22di editing genomico. SgRNAs acquistati o componenti separati di crRNA e tracrRNA sono generalmente alta qualità reagenti e una varietà di modificazioni chimiche sono disponibili per combattere i problemi con degradazione di RNA o di infondere caratteristiche aggiuntive a RNP91. Mentre modificati chimicamente gRNAs potrebbe non essere necessario per standard genoma esperimenti di editing, alcuni gruppi hanno osservato molto più alti di editing efficienze con tali reagenti, quindi potrebbero essere la pena di provare dopo il processo di mastering e/o quando gRNA degradazione sembra essere un problema22,91. Trascrizione in vitro e gel successiva purificazione è un'alternativa economica, che può essere sufficiente per routine genoma esperimenti17,21,49,50di editing. Ulteriormente, i diversi approcci che sono comunemente applicato per produrre omogenea gRNA popolazioni in vivo, compreso l'asportazione ribozima e tRNA-based di singole guide, può essere esteso a in vitro preparazione di RNA per generare pulitore prodotti92.
Donatore del DNA e RNA guida progettare suggerimenti:
Selezione di RNA di guida è un fattore critico nel raggiungimento di editing on target altamente efficiente, riducendo al minimo le probabilità di fenditura fuori bersaglio. Per facilitare la selezione della guida, parecchi studi hanno utilizzato schermi di alto-rendimento accoppiati con sequenziamento di nuova generazione per compilare caratteristiche di sequenza di successo guide47,79,93,94, 95,,96. Queste caratteristiche sono state utilizzate per sviluppare algoritmi predittivi e strumenti online per aiutare nella guida selezione44,45,46,47,48. Tali algoritmi sono fondati su schermi utilizzando sistemi basati su DNA per guida espressione del RNA. Guide sono espresse utilizzando un promotore Pol III, e pertanto la loro espressione è soggetta alle limitazioni associate trascrizione Pol III, ad esempio la terminazione prematura quando rileva tracce di uracile97,98, 99. Tuttavia, l'uso di RNP fatto con in vitro-guida sintetizzato RNAs ignora tali preoccupazioni e semplifica i vincoli sul design di guida. Una caratteristica comune che è emerso da questi algoritmi ed è stata confermata in numerosi studi con l'editing genomico altamente efficace, è la presenza di una purina, particolarmente una guanina, all'estremità 3 ' della sequenza target-specifici della guida. Questa funzionalità di guida ha avuto molto successo tra gli organismi che vanno dai mammiferi di c. elegans, moscerini della frutta e zebrafish65,100,101. Inoltre, per c. elegans, progettazione di guide con un dinucleotide GG all'estremità 3 ' della regione di destinazione della guida è una strategia efficace per la predizione di guida altamente efficace RNAs65. Idealmente, è possibile testare più guide in parallelo per determinare quale è il maggior successo per una determinata applicazione.
Quando si tenta di introdurre una sequenza di DNA nel genoma, il design del donatore o del modello del DNA è anche cruciale. Donatori di singolo-incagliato oligonucleotide (ssODNs) vengono inseriti in modo più affidabile rispetto a altri modelli tipici riparazione, lineare double-stranded e plasmide DNA54,55,102. Alcuni loci, HDR efficienza può essere migliorata con ssODNs che sono complementari alle non-bersaglio o sfollati filo del DNA e possedere armi di omologia che sono asimmetriche in lunghezza27,55. Poiché il modello di riparazione viene inserito nel sito di taglio e comprende la sequenza mirata, deve intraprendere per impedire che Cas9 fendendo il donatore del DNA prima o dopo l'inserimento di genomica. Questa operazione viene eseguita facendo mutazioni silenti per la sequenza di PAM o la regione di seme, evitando il riconoscimento di Cas9 pur mantenendo la funzione del gene inserito21,103. Se anche singolo nucleotide cambia il PAM hanno probabilità di abolire associazione104, provare a cambiare almeno quattro nucleotidi per essere sicuri.
Significato e applicazioni future:
Editing con CRISPR-Cas9 genomico è emerso come un potente metodo che consente di facile manipolazione genetica di qualsiasi organismo. Editing con il RNP Cas9 prende un po' più sforzo in un primo momento, ma è semplice da usare una volta reagenti e protocolli sono stabiliti in un laboratorio. Modifica delle celle con pre-assemblati RNP invece di DNA del plasmide conduce a una maggiore efficienza di editing generale, compreso l'inserimento del gene difficile da raggiungere via HDR, con meno effetti fuori bersaglio24,25,26 , 27 , 29. Inoltre, gli sperimentatori evitare problemi con l'espressione genica, RNA degradazione, piegatura della proteina e l'associazione tra gRNA e Cas9 molecole sintetizzate separatamente all'interno della cella22,23. Modifica di RNP elude anche preoccupazioni di sicurezza per mutagenesi inserzionale e sostenuta espressione che può sorgere quando i metodi di consegna virali sono usati clinicamente14. A causa di questi vantaggi, molti scienziati conducendo pre-clinici, esperimenti di proof-of-concept bomboniera RNP editing per applicazioni terapeutiche umane. In vivo ed ex vivo basato su RNP genoma editing approcci sono in sviluppo a trattare o curare anche una varietà di condizioni, da malattie genetiche come Duchenne distrofia muscolare105 e cellule falciformi27 a L'HIV1129 e cancro. Interessante, Cas9 RNP viene impiegato sempre più come un metodo di consegna per ingegneria agricola perché permette di 'DNA-free' editing delle piante33,34,36.
Disclosures
Gli autori Alexander Marson e Jacob E. Corn sono co-fondatori di Spotlight terapeutica. Jacob E. Corn è un consulente missione terapeutica e suo laboratorio ha ricevuto il sostegno di ricerca sponsorizzata da AstraZeneca e Pfizer. Alexander Marson è un consigliere di Juno Therapeutics e patto Therapeutics, e il suo laboratorio ha ricevuto il sostegno di ricerca sponsorizzata da Juno Therapeutics, Epinomics e Sanofi. Suo laboratorio ha anche applicato per brevetti relazionati alla tecnologia Cas9 RNP.
Acknowledgments
Ringraziamo il precedenti molti membri dei nostri laboratori e la comunità di editing di genoma Bay Area per il loro contributo allo sviluppo di questi metodi. Ringraziamo Ross Wilson per la lettura critica di questo manoscritto.
Ricerca di Alexander Marson è supportato da un regalo dal Jake Aronov e concedere un National Multiple Sclerosis Society (CA 1074-A-21). Alexander Marson detiene un premio alla carriera per gli scienziati medici del Burroughs Wellcome fondo ed è un investigatore di Biohub Zuckerberg Chan. Ricerca di Jacob E. Corn è supportato dai Li Ka Shing Foundation, il patrimonio Medical Research Institute Medical e il California Institute for Regenerative Medicine. Behnom Farboud e di Barbara J. Meyer ricerca è finanziata in parte dalla concessione NIGMS R01 GM030702 a Barbara J. Meyer, che è un ricercatore dell'Howard Hughes Medical Institute. Ricerca Erin Jarvis e Nipam H. Patel è stato in parte finanziata dalla sovvenzione NSF IOS-1257379 ed Erin Jarvis riconosce sostegno da un GRFP NSF e una borsa di studio Philomathia.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
