

Summary
Utilizando un Cas9 Confecciones complejo de ribonucleoproteína (RNP) es un método eficaz para la edición del genoma exacto, eficiente. Aquí, podemos destacar su utilidad en una amplia gama de células y organismos, incluyendo las células humanas primarias y ambos clásicos y emergentes en organismos modelo.
Abstract
Genoma eucariótico específico edición con CRISPR (clusters regularmente otro corto palindrómico repeticiones)-sistemas de Cas (CRISPR-asociado) se ha convertido rápidamente en un lugar común entre los investigadores persiguen una amplia variedad de cuestiones biológicas. Los usuarios emplean más a menudo la proteína Cas9 derivada de Streptococcus pyogenes en un complejo con una guía fácilmente reprogramada RNA (gRNA). Estos componentes se introducen en las células, y a través de una base que se aparea con una región complementaria del genoma double-stranded DNA (dsDNA), la enzima separa ambas hebras para generar una rotura de doble cadena (DSB). Reparación posterior conduce a la inserción al azar o eventos de eliminación (indels) o la incorporación de ADN proporcionadas por el experimentador en el sitio de la rotura.
El uso de una guía solo ARN y Cas9 proteína purificada, premontada, para formar una RNP y entregados directamente a las células, es un enfoque potente para lograr la edición altamente eficaz de genes. RNP edición especial mejora la tasa de inserción génica, un resultado que a menudo es difícil lograr. En comparación con la entrega a través de un plásmido, la menor persistencia de la RNP Cas9 dentro de la célula conduce a menos eventos de fuera de objetivo.
A pesar de sus ventajas, muchos usuarios casuales de CRISPR gene edición están menos familiarizados con esta técnica. Para bajar la barrera de entrada, Contorneamos protocolos detallados para la implementación de la estrategia de RNP en una variedad de contextos, destacando sus beneficios distintos y diversas aplicaciones. Cubrimos la edición en dos tipos de células humanas primarias, células T y las células progenitoras hematopoyéticas (HSPCs). También mostramos cómo Cas9 RNP edición permite la fácil manipulación genética de organismos enteros, incluyendo el ascáride elegans de Caenorhabditis del modelo clásico y el más recientemente había introducido crustáceo modelo Parhyale hawaiensis.
Introduction
fThe sistema CRISPR Cas9 permite a los científicos alterar las regiones específicas de cualquier genoma1. Esta rápida y barata la tecnología ha revolucionado la investigación básica y promete hacer un profundo impacto en el desarrollo de terapias personalizadas de la enfermedad, agricultura de precisión y más allá de2. CRISPR edición es una herramienta democratizadora y aplicación del sistema en un nuevo laboratorio no requiere ninguna especial experiencia en técnicas de biología molecular ingeniería, apenas básicos genoma. Los investigadores ahora pueden estudiar organismos previamente intratables con unos medios alternativos para manipulación genética3,4. En los últimos cinco años, CRISPR edición del genoma se ha utilizado para más de 200 diferentes vertebrados, invertebrados, plantas y especies microbianas.
Adaptado CRISPR defensa procariotas del camino de la, los elementos de base necesarios para la edición del genoma específico son la proteína Cas9, típicamente de S. pyogenes y codón-optimización con una señal añadida localización nuclear (NLS) y su especializada RNA guía5,6. Aunque no se discute aquí, otros Cas9 orthologues o endonucleasas CRISPR pueden usarse también. El gRNA natural se compone de dos piezas por separado transcritos, CRISPR ARN (crRNA) y la activación de trans crRNA (tracrRNA)7. Estos RNAs se pueden fusionar en una sola transcripción, conocida como la guía solo RNA (sgRNA)8. Editores de genoma más elegir el sgRNA aerodinámico9, aunque la guía doble también es utilizado regularmente10,11. Experimentadores elegir destino un ADN genómico 20 nucleótidos (nt), asegurando que se encuentra al lado de una corta licencia firma Cas9 reconocimiento, llamado un motivo adyacente protospacer (PAM) y diseñar un gRNA que contiene la secuencia complementaria12 .
Una vez dentro de la célula, el complejo RNP localiza su objetivo genomic, los gRNA de pares de bases con el ADN complementario del filamento, y entonces la enzima separa ambos filamentos de la DNA para generar un doble filamento romperán2. Maquinaria de reparación de la célula fija el OSD por uno de al menos dos rutas: vía el camino de (NHEJ) end-joining propenso no-homólogas o la reparación dirigida de homología (HDR), que incorpora ADN con 'armas' de homología a ambos lados de la rotura. La vía de reparación anterior típicamente conduce a formación de indel e interrupción consecuente gene, mientras que este último permite experimentadores colocar o cambiar las secuencias de ADN1.
La edición eficiencia y precisión dependen de los medios por los cuales Cas9 y gRNA entran en la célula. Estos componentes pueden entregarse a cultivos de células, embriones u organismos en forma de ácidos nucleicos o como Confecciones RNP complejo13,14,15. Métodos de entrega basados en ácidos nucleicos comunes incluyen la transducción viral, transfección o electroporación de mRNA o ADN plásmido. Guía RNA y proteína Cas9 entonces se producen dentro de la célula y asocian para formar un complejo.
La entrega directa de RNP requiere la purificación separada de la guía de RNA y proteína Cas9. Esto se puede hacer en casa, o la proteína y el sgRNA pueden comprarse uno de varios proveedores comerciales. Una vez adquirido, el Cas9 y gRNA son mezclados para formar el complejo RNP enzimáticamente competentes y a las células por inyección directa en embriones de huevos fertilizados, transfección basada en lípidos16o electroporación. El primer informe de RNP edición implicó inyección en C. elegans gónadas17. Microinyección sigue siendo el medio preferido de la introducción de la RNP en embriones y organismos enteros, aunque electroporación eficaz se ha demostrado en rata y19 20 embriones de ratón18,. Describir los protocolos para inyectar directamente el RNP en gónadas de C. elegans y p. hawaiensis embriones y recomendar un tipo especializado de la electroporación para entregar RNP al editar las células humanas primarias. Este método, nucleofection, implica programas de electroporación optimizado y las soluciones específicas del tipo celular y permite la RNP entrar en el citoplasma y el núcleo21.
La edición del genoma con RNP ofrece varias ventajas. Porque los componentes de RNA y proteínas son pre-ensamblados y calidad puede ser asegurada antes de la entrega, RNP edición evita muchos riesgos asociados con la entrega basados en ácidos nucleicos. Es decir, no hay riesgo de integración Cas9 codificación del ADN en el genoma del anfitrión mRNA nunca está expuesto para la degradación y evita problemas con en vivo gRNA o proteína expresión, plegamiento y asociación22,23. Además, usando RNP conduce a menor toxicidad y menos eventos de fuera del objetivo de la expresión basada en el plásmido, consecuencia de vida media más corta de la RNP dentro de la célula24,25,26,27.
Por último, RNP edición demostrable conduce a altas tasas de edición en una variedad de líneas celulares humanas, células primarias tales como fibroblastos, las células madre embrionarias (ESCs), inducida por células pluripotentes (iSPCs), HSPCs, y T de las células16,24, 25,26,27,28,29; en invertebrados como Caenorhabditis elegans, hawaiensis p.,17,de3,30de moscas de la fruta; en especies de vertebrados como peces cebra, ratones y ratas31,32; en la planta especies incluyendo Arabidopsis, tabaco, lechuga, arroz, vid, manzana, maíz y trigo33,34,35,36; y en Chlamydomonas, Penicilliumy Candida especies37,38,39. La frecuencia de formación de indel puede ser mayor cuando se utiliza el RNP en comparación con la entrega de plásmido e inserción de ADN mediada por el HDR puede ser más fácil alcanzar25,27,29.
El protocolo descrito aquí utiliza el RNP Cas9 y es una técnica eficaz, fácilmente adaptable que es fácil aplicar a una amplia variedad de sistemas biológicos40,41, especialmente en las células que de otra manera difícil de trabajar y en las organismos sin sistemas bien establecidos para la manipulación genética precisa. Empezamos describiendo cómo diseñar, obtener y montar el RNP Cas9 antes que cubre su uso a través de organismos y tipos celulares de diferentes modelos. (HSPCs) de las células progenitoras hematopoyéticas y las células T se editan siguiendo el mismo método, nucleofection, así que juntos están cubiertas en los pasos 2 y 3 del presente Protocolo. Editar procedimientos para C. elegans se describen en los pasos 4 y 5 y P. hawaiensis edición se describe en los pasos 6 y 7. Finalmente, puesto que el éxito de un experimento de edición de genes en cualquier organismo puede evaluarse por la secuencia de genotipo, pasos que describe métodos de análisis posible para todas las células y los organismos descritos en el protocolo se describen en el paso 8.
Protocol
1. RNP Asamblea
-
Diseñar el experimento con suficiente antelación, adquirir todos los componentes de RNA, DNA y proteínas antes de tiempo. Como un primer paso, probar uno de los controles positivos enumerados en la tabla 1 y uso de los reactivos comerciales descritos en la tabla de materiales para un diseño experimental confiable y la integridad de los materiales. Para sugerencias adicionales sobre la planificación de un nuevo experimento de edición del genoma, ver documentos en este tema12,42,43.
Nota: Una vez montado como se describe en los pasos posteriores, RNPs preparados por adelantado pueden ser almacenadas a-80 ° C.- Después de elegir que gen para blanco, utilice uno de las herramientas en línea gratuitas para diseñar un gRNA óptima44,45,46,47,48. Asegúrese de apuntar un exón si con la esperanza de generar un golpe de gracia.
Nota: Estas herramientas ayudarán a identificar un sitio de destino con un adyacente S. pyogenes PAM secuencia, baja puntuación objetivo y resultado de alta calidad. - Purificar la proteína Cas9 de S. pyogenes a través de métodos publicados8, o adquirir de un vendedor comercial.
- Preparar una memoria intermedia de Cas9 típico para la dilución de RNA, RNP preparación y almacenamiento de proteínas, que contiene 20 mM de HEPES pH 7.5, 150 mM KCl, glicerol al 10% y 1 mM de TCEP. Siempre usar agua libre de nucleasas en búferes que se utilizará para resuspender o Diluya el RNA para evitar la degradación.
- Producir a la guía RNA (tracrRNA y crRNA o sgRNA) a través de una transcripción en vitro usando métodos publicados, o adquirir de un ácido nucleic síntesis empresa17,21,49, 50 , 51.
- Si insertar un gen, sintetizar o comprar a un donante de plantilla de la DNA.
- Almacenar la proteína y alícuotas de RNA a-80 ° C y descongelar el hielo inmediatamente antes del uso.
Nota: Cada una congelación y descongelación disminuye ligeramente la eficiencia. Protocolos detallados, de libre acceso para Cas9 purificación52 y la transcripción en vitro de sgRNAs53 están disponibles en otros lugares.
- Después de elegir que gen para blanco, utilice uno de las herramientas en línea gratuitas para diseñar un gRNA óptima44,45,46,47,48. Asegúrese de apuntar un exón si con la esperanza de generar un golpe de gracia.
- Si trabaja con C. elegans, vaya al paso 1.5. Para el protocolo p. hawaiensis , vaya al paso 1.6. Si utiliza sgRNA, vaya al paso 1.4. Proceda al paso 1.3 para montar un gRNA para modificar las celdas primarias.
-
Montar un gRNA mezclando cantidades equimolares de tracrRNA y crRNA. Hacer 100 μl del stock de gRNA 80 μm, de unos 50 experimentos edición del genoma.
- Incubar el gRNA a 37 ° C durante 30 minutos y luego deje que se enfríe lentamente a la temperatura ambiente.
-
Preparación de RNP para HSPC y modificar las celdas: montar una complejo RNP mezclando una cantidad molar de 1-2 x de gRNA a 200 pmol de Cas9 proteína en un volumen total de 10 μl. muy lentamente, añadir Cas9 concentrado a la gRNA (previamente diluida en el Cas9 buffer) para cerca de 30 s , haciendo círculos rápidos con la pipeta, lo que la concentración final de Cas9 a 20 μm.
- Preparar las cubetas de electroporación.
Nota: Este protocolo es específico para el sistema comercial contemplado en la Tabla de materiales, pero RNP edición puede conseguirse también con otros dispositivos de electroporación. - Añadir 5 μl (100 pmols, las células de T) o 10 μl (200 pmol, HSPCs) de la RNP a cada cubeta.
- Si insertar nuevo ADN en lugar de hacer un golpe de gracia, añadir 1 μl de 100 μm (100 pmol) monocatenario oligonucleótidos donantes ADN (ssODN)25,54,55 a las cubetas o pocillos de la placa.
- Saltar al paso 2 para las siguientes instrucciones en el montaje de la célula primaria Protocolo.
- Preparar las cubetas de electroporación.
-
Preparación de la RNP para la edición de C. elegans : montar el RNP compleja mediante la adición de los reactivos siguientes para crear un volumen final de 20 μl (concentración final se observa en paréntesis): Cas9 (2 μm), pH HEPES 7.5 (10 μm), KCl (115 μm), crRNA (12 μm) , tracrRNA (40 μm) y la reparación plantillas si es necesario (0,5 μm ssDNA o hasta 350 dsDNA ng/μL).
Nota: La eficacia de una reparación mediaron Cas9 DSB-con plantillas es proporcional a la concentración de la construcción reparación de dsDNA; por lo tanto, cuanto mayor sea la concentración de la plantilla de reparación más eficiente la reparación con plantilla. Sin embargo, la inyección de mezclas que contienen más de 350 ng/μl de dsDNA ha demostrado reducir la viabilidad de los gusanos inyectados. Por lo tanto, es mejor usar, pero no más de 350 ng/μl de dsDNA en la mezcla para maximizar la eficiencia de la reparación y reducir al mínimo su letalidad.- Añadir múltiples crRNAs para atacar simultáneamente múltiples loci, según sea necesario para el enfoque de proyección co-CRISPR/co-conversion que se describe en el paso 5.4. Al agregar más de un crRNA, añadir cada uno secuencialmente a la mezcla principal.
Nota: La cantidad de cada crRNA no necesita ser el mismo, y aun duplicando la concentración total de crRNAs en la mezcla principal sin cambiar la concentración de Cas9 no parece interferir la frecuencia de mutagénesis en un locus específico. Ejemplos se describen en detalle en Paix et al. 56. - Mezclar mediante pipeteo y exprimir la solución RNP a 16.000 x g durante 5 s para asegurar que la solución se recoge en la parte inferior del tubo.
- Incubar la solución a 37 ° C durante 15 m.
- Centrifugar la muestra a 16.000 x g durante 1 min a cualquier partículas que podrían obstruir la aguja fina-aburrido microinyección de pellets. Utilizar el sobrenadante en los pasos posteriores.
- Vaya al paso 4 para el resto del Protocolo de C. elegans .
- Añadir múltiples crRNAs para atacar simultáneamente múltiples loci, según sea necesario para el enfoque de proyección co-CRISPR/co-conversion que se describe en el paso 5.4. Al agregar más de un crRNA, añadir cada uno secuencialmente a la mezcla principal.
-
Preparación de la RNP para la edición de p. hawaiensis : preparar alícuotas de un solo uso Cas9 diluyéndolas con agua libre de nucleasas y rojo de fenol (para la visualización de las inyecciones) para una concentración final de 6,25 μm de Cas9 y 0.15% de rojo de fenol.
- Montar el complejo RNP por un 2-5 x exceso molar de gRNA a la proteína Cas9 en un volumen total de 6 μl. Añadir 12 pmol de Cas9 a gRNA, elevando la concentración final de Cas9 a 2 μm, gRNA concentración a 4-8 μm y la concentración de rojo de fenol de 0.05% de la mezcla.
- Incubar la mezcla a temperatura ambiente durante 10 min al complejo de la RNP.
- Saltar al paso 6 para las siguientes instrucciones en la edición de p. hawaiensis Protocolo.
2. cultura y preparación de la célula
Nota: Realice pasos 2.1.1 a 3.3.3 en un gabinete de seguridad biológica.
-
Humanos criopreservados compra movilizaron a sangre periférica CD34+ HSPCs de un vendedor.
- Descongelar ~ 1 x106 HSPCs en un agua de 37 ° C baño de 3 minutos y transferir a un tubo cónico de 15 mL. Añadir 10 mL de un medio libre de suero de la expansión de una fuente comercial y girar la mezcla a 100 x g por 10 minutos quite el sobrenadante y resuspender las células en 2 mL de SFEM suplementado. Las células en placas de 6 pocillos de la placa y el cultivo en un incubador de 37 º C durante 24-48 h antes de la electroporación de la RNP.
- Contar las células con un hemocitómetro y transferir el número de HSPCs necesitada (150.000-200.000 HSPCs por cubeta a electroporated) a un tubo de centrífuga.
- Girar el tubo a 100 x g por 10 min para que sedimenten las células.
-
Compra humana primaria CD4+ T de las células de un vendedor o aislarlos de sangre humana por el de centrifugación del gradiente de densidad29.
- Antes de la activación de células T, la capa bien 48 placas con αCD3 (UCHT1) y αCD28 (CD28.2). Cubrir las placas con 500 μl de 10 μg/mL αCD3 y αCD28 de 10 μg/mL en PBS durante al menos 2 h a 37 ° C.
Nota: Para algunos loci, NHEJ puede lograrse sin estimulación previa, pero como este paso maximiza su eficiencia. - Células en cultivo la T durante 48 h a 37 ° C en placas de anticuerpo αCD3/αCD28 en Medio RPMI completo [RPMI-1640 suplementado con 5 mM de HEPES, 2 mM de alternativa comercial a la L-glutamina, 50 μg/mL de penicilina/estreptomicina, 50 μm de 2-Mercaptoetanol, 5 mM de no esenciales aminoácidos, 5 mM de piruvato de sodio y 10% FBS (vol/vol)]. Células en cultivo la T con una densidad de células de T 2.000.000 en 500 μl de medio por pozo de una placa de 48 pozos.
- Cuenta el T las células usando un hemocitómetro y transferencia el número total de células T para la electroporación experimento (células de T de 100.000-1.000.000 por cubeta a electroporated) a un tubo de centrífuga.
- Girar el tubo a 90 x g por 8 min para que sedimenten las células. Si las células han sido densidad gradiente separados dentro de 2 días, los hace girar a 200 x g por 8 min.
- Antes de la activación de células T, la capa bien 48 placas con αCD3 (UCHT1) y αCD28 (CD28.2). Cubrir las placas con 500 μl de 10 μg/mL αCD3 y αCD28 de 10 μg/mL en PBS durante al menos 2 h a 37 ° C.
-
Para ambos tipos de células, aspirar el sobrenadante con un pipeta/vacío, eliminando cualquier burbuja.
- Resuspender suavemente las células con 20 μl de buffer de electroporación por cubeta.
- Añadir 20 μl de las células (150.000-200.000 HSPCs o células de T de 100.000-1.000.000) a cada cubeta, que ya contiene 10 μl de la RNP y mezclar bien mediante pipeteo arriba y abajo sin crear burbujas.
3. RNP electroporación
- Electroporate las cubetas después de poner en un nucleofector. Para HSPCs, utilice el código de pulso ER100. Para las células T, utilice el código de pulso EH-115.
-
HSPCs solamente: Añadir 100 μl de un medio SFEM suplementado (calentado a 37 ° C) para cada cubeta inmediatamente después de la electroporación y dejar que las células recuperan para 10-15 min.
- Transferir las células a cultura en un fondo redondo de 96 pocillos de la placa y agregar un adicional 100 μl del medio suplementado SFEM durante 24 h.
- Cambiar a un medio fresco de SFEM suplido e incúbelos por un adicional 24-72 h.
- Eliminar las células para genotipificación ellos 48-96 h post-electroporación. Girar las células a 300 x g durante 5 minutos y retirar el sobrenadante antes de comenzar la extracción de ADN (paso 8.2).
-
Las células T sólo: Añadir 80 μl de RPMI completo medios de cultivo previamente calentado a 37 ° C desde el depósito a cada cubeta o bien, utilizando una pipeta multicanal (si es necesario).
- Incúbelos a 37 º C durante 15 minutos.
- Añadir el medio apropiado, anticuerpos, citoquinas, etc. a la placa de destino y calentarlos previamente en una incubadora de 37 ° C.
- Transferencia 107 μl de las células electroporated de los pozos de una placa de 96 pocillos de fondo redondo con una pipeta multicanal (si es necesario).
- Para obtener información sobre la evaluación de los resultados de la edición, vaya al paso 8.
4. preparación de C. elegans
-
1 día antes de la microinyección: preparar los cojines de la agarosa para la microinyección.
- Hacer una solución de agarosa al 3% (p/v) en agua agregar agarosa al agua y llevando la solución a ebullición sobre una placa caliente o en el microondas.
- Organizar 24 mm x 50 mm x 1,5 mm tapa portaobjetos sobre una mesa y utilizar un pipeta Pasteur de vidrio colocar una gota pequeña (~ 15 μl) de solución de agarosa en la diapositiva. Aplanar rápidamente la caída de agarosa colocando otro cubreobjetos en la parte superior. Permita que la agarosa solidificar y después quitar uno de los cubreobjetos.
- Deje el cubreobjetos recubierto de agarosa boca arriba sobre una mesa durante la noche para secarse. Después de 24 h, guarde las pastillas de agarosa en un recipiente limpio y seco.
Nota: Éstos se pueden utilizar indefinidamente.
- Tire las agujas de microinyección: con Capillares de vidrio de borosilicato de filamentos (diámetro externo diámetro de 1,0 mm e interior 0,58 mm), tire las agujas basadas en Mello y Fire57 y otros recursos58. Las agujas se pueden utilizar inmediatamente o se pueden almacenar en un recipiente limpio y seco, apoyado por soportes de arcilla.
- Para el mantenimiento de los gusanos, preparar un agar de medios de crecimiento de nematodos (NGM) vierte en placas Petri y manchado con bacterias OP50 (para protocolos estándar C. elegans mantenimiento y recetas para los medios de crecimiento, véase Stiernagle59).
- Los gusanos para la microinyección de la etapa: 12-24 h antes de la microinyección, escoger hermafroditas L4-puesta en escena a una nueva placa NG-agar con bacterias OP50 e incúbelos durante la noche a 20 ° C. Para cada mezcla de blanco/inyección Cas9, recoger gusanos de ~ 30 a la placa.
-
Día de microinyección: Carga la aguja microinyección tirado con la solución de la RNP sobrenadante preparada en el paso 1.5.
- Pipetear el sobrenadante del paso 1.5.4 en una pipeta capilar tirado y relleno de zanjas en la solución de la pipeta capilar en la aguja de microinyección preparado (generalmente menos de 0.1 μl de carga).
- Monte la aguja cargada sobre el aparato de microinyección unido a un micromanipulador. Ajustar la presión del aparato de inyección 250 kPa y la presión de equilibrio a 25 kPa.
-
Romper nuevamente la punta de la aguja cargada para generar un borde afilado de la aguja. Lugar un 15 x 15 mm x 1,5 mm cubreobjetos cuadrados en la parte superior de un 24 mm x 50 mm x 1,5 mm cubreobjetos.
- Superposición de un borde del cubreobjetos cuadrado con aceite de halocarburos 700.
- Coloque la aguja en el aceite, en el borde del cubreobjetos cuadrados de 15 mm.
- Utilizando una mano para guía de la platina del microscopio y el cubreobjetos, la diapositiva del cepillo hacia arriba y el borde de la aguja mientras presiona el pedal/botón de inyección. Romper la punta de la aguja hacia atrás, aumentando el flujo del líquido de la aguja. Lograr un flujo óptimo haciendo la inyección mezcla flujo a lo largo del borde de la aguja, formando burbujas/s ~ 1.
- Confirman que los gusanos L4 escogidos 12-24 h antes de la microinyección jóvenes al desarrollo efectuados en el día de la inyección. Recoger los gusanos adultos jóvenes a una placa de agar NG que carece de bacterias OP50 y que puedan arrastrarse alrededor de 5 minutos. Esto reduce la cantidad de bacterias a la almohadilla de la inyección, minimizando las obstrucciones de la aguja.
- Coloque una inyección pad/cubreobjetos de agarosa en un ámbito de disección. Usando un punzón de gusano, poner una pequeña pista de aceite halocarburos a lo largo de un borde de la almohadilla.
-
Mediante la selección de gusano cubierta en aceite, levante varios gusanos la placa NG-agar y en la pista de aceite. Con un pelo fino atado a una pipeta, como un bigote de gato o pestañas, colocar las lombrices en paralelo, suavemente empujando los gusanos en el cojín de agarosa. Hasta cómodo con el procedimiento de microinyección, solo Monte e inyectar un gusano en un momento.
Nota: La agarosa seco absorberá la humedad de los gusanos, causando que se adhieren a la plataforma. En consecuencia, se debe trabajar rápidamente como pueden desecar los gusanos.- Una vez en posición y unido a la placa, superponer los gusanos con el otro escoger algunas gotas de aceite de halocarburos (~ 20 μl) de la punta del gusano.
5. C. elegans gónada microinyección con RNPs y cuidado después de la inyección
Nota: El protocolo de microinyección es adaptado de Mello y Fire57y descrito en detalle en otra parte60,61.
-
Colocar el cubreobjetos con los gusanos montados en el microscopio de la inyección. Con bajos aumentos (objetivo de 5 X, 10 X ocular), coloque los gusanos perpendiculares a la aguja de inyección.
- Interruptor a un gran aumento (objetivo 40 X, 10 X ocular), vuelva a colocar la aguja adyacente en el brazo de la gónada correspondiente a la región cerca de los núcleos en mediados de-a tarde-paquiteno.
- Mediante el micromanipulador, mueva la aguja contra el gusano, presionando ligeramente la cutícula. Luego, con una mano, golpee el lado de la platina del microscopio para sacudir a la aguja a través de la cutícula. Presione el pedal/botón de inyección y lentamente el brazo de la gónada se llenan de la mezcla de inyección y retire la aguja.
- Repita este paso con el otro brazo de la gónada.
-
Una vez que los gusanos se inyectan, retire la almohadilla de agarosa/cubreobjetos y colocarlo bajo un microscopio de disección.
- Con una pipeta capilar tirada, desplazar el aceite de los gusanos mediante pipeteo un buffer M9 sobre ellos. Realizar este tratamiento para los gusanos del agar.
- Después de 10 minutos, cuando los gusanos están golpeando alrededor en el búfer, moverlos a una placa de agar NG con OP50 bacterias usando la pipeta capilar tirada. Coloque la placa a 20 ° C durante 2-3 horas hasta que los gusanos se han recuperado y se están moviendo alrededor.
- Una vez recuperado, individualmente transferir los gusanos a las placas de agar NG con OP50 y transferir las placas a una incubadora de 25 ° C.
-
Permiten la P0-inyectado gusanos crecen y progenie durante 3 días. Pantalla de la descendencia de1 F.
- Si usa co-CRISPR o conversión Co62,63,64,65, entonces seleccione los gusanos de candidato para la detección basada en si tienen el fenotipo mutante del gen de referencia. Transferir estos gusanos marcados a NG nuevo-placas con OP50 individualmente y les permiten poner progenie de F2 a 20 ° C.
Nota: El fenotipo usado para detección de co-CRISPR o selección debería proporcionar una estimación temprana para el éxito de Cas9 edición. - Si no está presente el fenotipo CRISPR co, microinject un plásmido de control positivo para ayudar a mejorar la eficiencia de la microinyección.
Nota: por ejemplo, como un plásmido en la mezcla de inyección que codifica tagged mCherry MYO-2 le ayudará a evaluar la eficiencia de la inyección. Gusanos con éxito inyectados con pCFJ90 tendrá algunas crías con pharynxes fluorescentes.
- Si usa co-CRISPR o conversión Co62,63,64,65, entonces seleccione los gusanos de candidato para la detección basada en si tienen el fenotipo mutante del gen de referencia. Transferir estos gusanos marcados a NG nuevo-placas con OP50 individualmente y les permiten poner progenie de F2 a 20 ° C.
- Examinar los gusanos de1 F la presencia de la edición deseada. Escoger a la madre de1 F a un pozo individual de una placa de 96 pocillos, lyse le y examinar su ADN por amplificación de la polimerización en cadena de inserción específicos, análisis de la secuencia de ADN o topógrafo nucleasa ensayo (CEL-1)66.
Nota: Estos ensayos se pueden realizar cuando se utiliza una proyección co-CRISPR/co-conversion u otros o selección regímenes65,66,67,68. - Para obtener información sobre la evaluación de los resultados de la edición, vaya al paso 8.
6. preparación de hawaiensis p.
- 1 día antes de la microinyección, enriquecer los embriones tempranos estableciendo un 'tanque par' la noche anterior; recién separados de las hembras contienen embriones recién fecundados. Ver Rehm et al. 69 para más detalles.
- En el día de microinyección, recoger los embriones de Parhyale unicelulares (0-4 h post-fecundación) por anestesiar las hembras grávidas con aceite de clavo de 0.02% en agua de mar y raspar suavemente los embriones en su bolsa incubadora ventral usando una llama-tirado y pipeta de cristal redondeado y un aburrido par de fórceps #3.
7. p. hawaiensis embrión microinyección con RNPs y cuidado después de la inyección
- Rellenar un tubo capilar tirado con aproximadamente 1 μl de la mezcla de inyección RNP descrito anteriormente.
-
Utilizar nitrógeno comprimido para microinject cada embrión según Rehm et al. 69.
- Inyectar los embriones Parhyale bajo un microscopio de disección utilizando un microinyector y un micromanipulador. Cargar 1,5 μl de la mezcla de inyección en la parte posterior de un tubo capilar tirado (4 pulgadas - 1,0 mm con filamentos, tirado utilizando una micropipeta tirando aparato) utilizando una pipeta de microloader.
- Configurar la aguja sobre el aparato de inyección y romper la punta de la aguja (una cantidad muy pequeña) con un par de pinzas en el ámbito de la disección. Calibrar el volumen entregado por inyectar en aceite 700 de halocarburos y medir el diámetro de la burbuja.
- Corte un 'canal' fuera del agente curado mediante una hoja de afeitar. Llenar hasta la mitad con agua de mar esterilizada por el filtro y se alinean los embriones Parhyale en el canal para estabilizar.
- Inyectar los embriones utilizando la configuración de la microinyección, estabilización de cada embrión con un par de fórceps durante la inyección. Después de la inyección, use una pipeta de vidrio para transferir los embriones sobre a una placa de cultivo fresco 60 mm llenada hasta la mitad con agua de mar esterilizada por el filtro.
-
Si la primera división ya ha ocurrido para formar un embrión de 2 células (4-6 h tras la fertilización), generar completamente mutantes animales mediante la inyección de dos blastómeros. Para asegurar una total ruptura de la etapa 2-células, conjuntamente inyectar los blastómeros con dextrano FITC o TRITC y observar que la señal se limita a una sola blastómera bajo un fluorescente alcance de disección después de la inyección.
- Por otra parte, generar 'mitad mutante' animales mediante la inyección de sólo uno de los dos blastómeros en la fase de 2 células (aproximadamente dividida izquierda dependiendo del tejido y la posición del eje A-p).
- Inyectar una célula de un embrión de 8 células (post-fecundación 7.5-9 h) para restringir la edición a una sola capa de germen. Ver Gerberding et al. 70 para ver un mapa de los primeros linajes de blastómero.
-
Incubar los embriones en platos de cultivo de 60 mm (no más de 25 por plato), llenados hasta la mitad con agua de mar esterilizada por el filtro, 'previamente oxigenada' usando un borboteador de acuario o agitando con fuerza.
- Coloque los platos de los embriones en un recipientes de plástico sellados libremente forrado con toallas de papel húmedo para mantener la humedad y colocarlas en un incubador de 26 ° C con un ciclo de luz-oscuridad de 12 h.
- Transferencia de los embriones sobreviven para limpiar platos de agua de mar cada pocos días.
Nota: Los embriones pueden cultivarse a temperatura ambiente, aunque se desarrollan mucho más lentamente.
-
Disecar y arreglar los embriones en diferentes etapas de un análisis de la expresión en situ hibridación o anticuerpo tinción (véase Browne et al. 71 para una guía de puesta en escena y referencias adicionales para la disección y fijación72 en situ hibridación73y74la coloración del anticuerpo).
- Hacer agujas de disección ensartando un pedazo doblado de alambre de tungsteno aproximadamente 0.5 en longitud en el extremo de una aguja de insulina. Afilar la aguja en el hidróxido de sodio bajo una corriente. Utilizar una jeringa de 1 mL como el mango de la aguja de disección.
- Un pozo de un plato de cristal 3-bien a mitad de camino se llenan de una solución recién hechos de 9 partes PEM tampón (0,1 M de pH tubos 6.95, 2 mM de EGTA, 1 mM de MgSO4), PBS 10 x parte 1 y 1 parte 32% PFA. Coloque los embriones de 3-5 en el plato y hágale un agujero pequeño en cada embrión, utilizando una aguja de tungsteno afilada para poke uno ligeramente rasgunõs para estabilizar, permitiendo que la yema salga y el fijador se ejecute en.
- Usando un par de agujas de tungsteno afilada, suavemente a fastidiar el exteriores dos membranas que rodean el embrión Parhyale . Disecar en fijador para hacer más robustos los embriones pero trabajo rápidamente para evitar que la membrana ser fijo al embrión, que hace más difícil la eliminación de la membrana. Permitir que los embriones destinados a fijar para un total de 15-20 minutos para la tinción de anticuerpo o 40-50 min para hibridación en situ .
- Imagen energizados neonatos y analizarlas para fenotipos morfológicos y conductuales o fijar y mancha para análisis más detallados. Criar las crías a la madurez sexual en 2 a 3 meses para establecer nocaut y líneas transgénicas (ver Kontarakis y Pavlopoulos75 para el cuidado de pichones y otros datos útiles).
8. evaluar los resultados de la edición
- En su caso, busque un fenotipo visual o funcional en las células editadas u organismos.
Nota: Este proceso varía ampliamente por el uso, y algunos ejemplos se describen al final de sus pasos pertinentes del protocolo anteriores. Después de corregir la mutación de células falciformes en HSPCs, análisis de la producción de hemoglobina por eritroblastos diferenciados mediante HPLC (figura 1A). Un golpe de gracia del gene del receptor de IL-2 en las células T puede confirmarse por la coloración superficial y flujo cytometry (figura 1B). Para evaluar fenotipos C. elegans y p. hawaiensis , observar la morfología animal y comportamiento bajo un microscopio fluorescente o luz (figuras 1C y 1D). - Para determinar la eficiencia y el tipo de la edición genómica generada, lyse las piscinas de células editadas y extraer su ADN genómico utilizando un kit de extracción comercial21.
-
Para una estimación rápida de la formación de indel, amplificación de PCR por lo menos 200 pares de bases en el corte del sitio y realizar un endonuclease1 T7 (T7E1)76 o topógrafo (1 CEL nucleasa) ensayo77.
- Si una formación de indel en el Cas9 de corte sitio o HDR exitosa será crear o eliminar un sitio de restricción conocido, considere el uso de una enzima de la restricción digestión para estimar la eficiencia edición6. El ensayo de polimorfismo (RFLP) de longitud de fragmento de restricción puede ser una forma conveniente para comprobar la eficacia pasa a ser disponible.
- Para una cuantificación precisa de la eficacia de la edición y la determinación de resultados edición predominantes, envíe el amplicon PCR para un estándar Sanger secuenciación con cebadores adelante y atrás.
Nota: Si al analizar una sola copia o un organismo, el análisis de resultados de Sanger es simple, como se muestra en la figura 2A. Si al analizar un grupo de células, entonces analizar los cromatogramas con la herramienta en línea78, como se muestra en la figura 2B. - Para una cuantificación completa y secuencias de edición los resultados, realizar secuenciación profunda27,54, como se muestra en la figura 2C.
- Para evaluar un conjunto particular de cambios fuera del objetivo, los sitios de destino previstos de amplificación PCR y enviar para NGS. Para habilitar la detección de translocaciones cromosómicas, realizar guía seq79 o alto rendimiento, desplazamiento de genoma secuencia (HTGTS)80. Para un cuadro completo de las ediciones fuera de objetivo en una población clonal, realizar secuenciación del genoma completo (WGS)81,82,83.
Nota: Hay una variedad de métodos para cuantificar ediciones del genoma en y fuera del objetivo, explicó además en revisión varios artículos84,85,86.
Representative Results
Estos experimentos ver cómo premontado Cas9 RNP puede utilizarse para manipular el genoma de células primarias y organismos enteros. Investigadores purificarán o comprar sgRNA y proteína Cas9, combinan los dos componentes para formar el complejo e introducen el RNP en sus células o un organismo de interés. Después de permitir suficiente tiempo para la edición para ocurrir y de descendencia de la generación siguiente de nacer (si corresponde), comprobar los fenotipos o recolectar células para genotipificación. Se pueden observar fenotipos mediante ensayos funcionales, ensayos de expresión, visualización (ojo o con microscopia) u otros métodos, según el experimento.
Por ejemplo, HSPCs que se han editado para corregir la mutación β-globina que causa la enfermedad de células falciformes pueden diferenciados en eritrocitos y repetir el ensayo de producción de sano o de hoz hemoglobina27,87 (figura 1 A). Las células T editado para noquear el gen de receptor de alta afinidad IL-2, CD25 (IL2RA), puede ser analizado por la coloración superficial y flujo cytometry88y funcionalmente analizados para detectar una señalización respuesta a la estimulación de la IL-2 (figura 1B ). Las células T también pueden ser reprogramadas en muchos aspectos clínicamente importantes que requieren evaluación de fenotipos diferentes, incluyendo la eficacia de VIH infección89 y células de la eficacia antitumoral en vivo del coche-T11.
Usando un enfoque de proyección co-CRISPR/co-conversion, gusanos C. elegans se editan simultáneamente en dos loci62. HDR en el gene de referencia de dpy 10 usando un ssODN reparación resultados de plantilla en una mutación de la ganar-de-función de dominante anotó fácilmente dpy 10 . Heterozigótico F1dpy-10(gof) animales son rodillos (Rol) y animales homocigóticos dpy-10(gof) rechoncho (Dpy). La presencia del fenotipo indica Cas9 edición se produjo en estos animales y mejora las probabilidades de identificar un evento de edición en el segundo lugar en el Rol o Dpy F1 animales. Un exitoso experimento de edición debe resultar en 33-50% de los inyectados P0 gusanos que rinde 20 o más crías de1 de F que son de Rol o Dpy90. Es entonces posible elegir animales de Rol no volver dpy 10 a tipo salvaje y seleccione Editar homocigótica de interés. Como regla general, la concentración de la crRNA del gen de referencia co-CRISPR debe ser la mitad que de la crRNA dirigidos al gen de interés. Si no se recupera una edición en el gen de interés, se pueden ajustar las proporciones de los dos RNAs CRISPR para aumentar la probabilidad de recuperar la mutación deseada. Por ejemplo, aumentando la cantidad de crRNA para el gen de interés en relación con el crRNA del gen de referencia aumentará el porcentaje de gusanos que poseen modificaciones en el gen de interés dentro de la población de gusanos que poseen también ediciones en el locus del gen de referencia. Las frecuencias de conversión Co varían, pero las tarifas son típicamente 20-60%, cediendo a menudo ediciones homocigóticas en la generación de1 F (figura 1C).
P. hawaiensis crías que han sido editados para noquear el gen Abdominal-B (Abd-B) Mostrar anormalidades morfológicas claras3 (figura 1D). Este gen es necesario para modelar abdominal correcto, y sus resultados de interrupción en torácica tipo saltar y caminar piernas reemplazando las piernas natación y anclaje que suelen presentan en el abdomen.
Determinación de edición los resultados a nivel genotípico genómica requiere secuenciación o un ensayo en vitro que detecta cambios de la secuencia. Aquí, mostramos datos representativos de la secuencia de nuestro modelo de la célula y organismos, destacando diferentes enfoques para la cuantificación de la edición. Tenga en cuenta que las etiquetas de figura están generalizadas porque todos los métodos mostrados aquí pueden aplicarse a cualquier sistema biológico.
Enfoques basados en la secuencia varían en complejidad técnica y la profundidad de los resultados. Para poblaciones clónicas editadas u organismos individuales fácilmente separable, individuos editados pueden ordenados después de la extracción de ADN genómica. Resultados de secuenciación de Sanger estándar revelará el cambio de secuencia en el sitio de corte Cas9 en un individuo dado, con la hipotética mutágeno ' frameshift ' que le alteran su función (figura 2A). La herramienta en línea para la secuencia es otro enfoque basado en la secuenciación de Sanger que puede aplicarse a poblaciones mixtas en lugar de mutantes individuales78. Secuencias son analizadas con una herramienta en línea que puede aproximar la eficacia general de edición así como los resultados de la secuencia predominante. Los datos representativos se muestran en la figura 2B.
El más completo método de secuenciación descrito aquí es profunda secuencia (a veces se denomina secuenciación de alto rendimiento o de próxima generación). Este método proporciona las secuencias de ADN de genomas individuales en una población mixta. Estos datos pueden ser ilustrados en una variedad de maneras. Aquí, hemos clasificado lecturas individuales de la secuencia de células editadas basadas en los resultados de edición (figura 2C). Mayoría de las células se edita a través de la vía NHEJ, que típicamente resulta en la interrupción del gene. En otros, el gene de la blanco ha ha cambiado hacia fuera para una versión alternativa via HDR27.
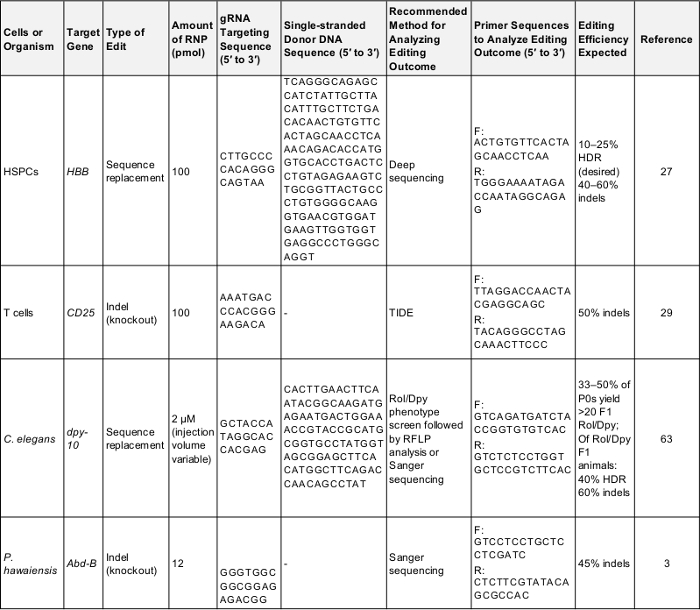
Tabla 1: controles positivo genoma preliminar edición experimentos. Esta tabla muestra la información clave necesitada para realizar un genoma de la primera edición de experimento en cada una de las células y los organismos descritos en el presente Protocolo. Siguiendo estos parámetros pueda producir un resultado exitoso que puede utilizarse para probar el protocolo o como base para la comparación una vez que el experimentador se dirige a un gen de su propio interés. F: adelante, R: revés, HDR: reparación dirigida por homología. Haga clic aquí para descargar esta tabla.

Figura 1 : Representante fenotípica resulta de RNP Cas9 edición de las células humanas primarias y organismos. (A) es un rastro HPLC, mostrando que después de genoma exitosa edición, HSPCs que se diferencian en la última etapa eritroblastos producirá hemoglobina más funcional de la hemoglobina de la hoz. Eritrocitos mutantes producen hemoglobina drepanocítica (HbS), mientras que las células editado con éxito producirá hemoglobina saludable (HbA y HbA2) así como hemoglobina fetal (HbF). Se graficó la absorbancia en unidades arbitrarias (UA). Este panel fue publicado por primera vez en DeWitt et otros. 27. se reimprime con el permiso de la Asociación Americana para el avance de la ciencia. (B) a la izquierda, para cada condición, este panel muestra datos de citometría de flujo mostrando que teñido de superficie de las células de T no expresan CD25, después de que el gen de CD25 ha sido noqueado con la RNP. La abundancia de CD25 se traza en el eje x con el tamaño de celda en el eje y. A la derecha, para cada condición, este panel muestra la cuantificación de la fosfo-Stat5 (pStat5) después de una inducción con IL-2. La señalización se reduce cuando el receptor de IL-2 está ausente (CD25 KO). La abundancia de pStat5 se traza en el eje x y los datos resultantes de tres diferentes niveles de entrada de IL-2 se comparan verticalmente. (C) este panel muestra una elegans de Caenorhabditis co-CRISPR/co-conversion pantalla a dpy-10 como el marcador de la conversión. Dos guía RNAs Diana dos loci, dpy-10 y su favorito gene (yfg), en el mismo P0-animal inyectado. HDR en dpy 10 da como resultado un fenotipo Rol o Dpy. La selección de Rol o Dpy F1 animales aumenta las posibilidades de identificar cambios en el segundo lugar. (D) este panel muestra que wildtype Parhyale hawaiensis crías tienen abdómenes normales con piernas natación y anclaje. Las crías de knock-out de Abd-B (personas de0 F) desarrollan un abdomen transformado hacia el tórax. Así, la natación y las piernas de anclajes son pasadas y sustituidas por las piernas Salta y camina asociadas con un tórax normal. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2 : Resultados típicos de edición de métodos de análisis de resultados. (A) este panel muestra ejemplos de la Sanger secuenciación de F1 p. hawaiensis organismos individuales, incluyendo la secuencia de tipo salvaje y tres indels diferentes que alteran la función del gen cambiando el marco de lectura abierto. (B) marea estos resultados muestran la variedad de inserciones y borrado eventos que ocurrieron en un sitio de destino Cas9 en un pool de células T secuenciados. El eje x indica la longitud de una determinada inserción o deleción de nucleótidos. (C) los resultados de secuenciación profunda no demuestran ninguna edición del genoma sin nucleofection o gRNA y exitosa edición con RNP Cas9 intacto, agrupadas por resultados de reparación de DNA en HSPCs. haga clic aquí para ver una versión más grande de esta figura.
Discussion
Establecimiento de un genoma robusto protocolo de edición en una celda de línea u organismo de interés requiere la optimización y empírica prueba de varios parámetros clave, discutidos en esta sección. Algunas variaciones de los enfoques generales presentados aquí es muy animado. La limitación fundamental de este protocolo es la aplicación de estos métodos a otras células u organismos pueden conducir a un resultado diferente dependiendo de las especies estudiadas, y un diseño experimental que lleva a un golpe de gracia del gene de alta eficiencia no puede promover la inserción de ADN. Por lo tanto, le recomendamos comenzar con los métodos presentados aquí y solución de problemas como se describe a continuación.
Solución de problemas de calidad de reactivo de edición genómica:
Generación o la compra de reactivos de alta calidad es un paso crítico en cualquier genoma edición protocolo. Cas9 proteína puede ser purificada en el laboratorio o adquirida comercialmente. Muchos protocolos nota una concentración final de Cas9 en recetas de RNP, pero el gen óptima edición de actividad dependerá de la actividad específica de un preparado de proteínas individual Cas9, que varía según la fuente. Una vez en marcha el protocolo presentado aquí es considerar optimizando la cantidad de RNP utilizada por valorar niveles de Cas9 para establecer una concentración óptima: uno que proporciona clivaje de ADN muy específicos sin escote de objetivo innecesario causado por excesiva Cas940.
Guía RNA pureza y homogeneidad pueden ser también determinantes del genoma edición éxito22. SgRNAs adquirido o componentes separados de crRNA y tracrRNA son reactivos generalmente de alta calidad y una variedad de modificaciones químicas están disponibles para combatir los problemas de degradación del RNA o a dotar de características adicionales para la RNP91. Mientras gRNAs modificado químicamente puede no ser necesario para el genoma estándar edición de experimentos, algunos grupos han observado mayores edición de eficiencias con tales reactivos, por lo que pueden ser vale la pena intentarlo después de dominar el proceso y cuando gRNA degradación parece ser un tema22,91. Transcripción in vitro y gel posterior purificación es una alternativa económica, que puede ser suficiente para la rutina genoma edición experimentos17,21,49,50. Además, varios enfoques que se aplican comúnmente para producir gRNA homogéneo poblaciones en vivo, incluyendo ribozyme y tRNA-basado en la supresión de guías individuales, puede ampliarse a in vitro preparación de RNA para generar limpiador productos92.
RNA de la guía y de los donantes de ADN diseñan consejos:
Guía selección de RNA es un factor crítico en el logro de objetivos altamente eficiente edición minimizando las posibilidades de escote fuera de objetivo. Para ayudar en la selección de la guía, varios estudios han utilizado pantallas de alto rendimiento juntados con la secuenciación de próxima generación para compilar las características de la secuencia de guías éxito47,79,93,94, 95,96. Estas características se han utilizado para desarrollar algoritmos de predicción y herramientas en línea para ayudar en la guía selección44,45,46,47,48. Estos algoritmos están basados en pantallas con sistemas basados en el ADN para expresión de RNA de la guía. Guías se expresan usando un promotor Pol III, y su expresión es por lo tanto propensa a las limitaciones asociadas con la transcripción de Pol III, como terminación prematura al encontrar pistas del uracil97,98, 99. Sin embargo, hizo uso de RNPs con in vitro-sintetizada guía RNAs omite esas preocupaciones y simplifica las limitaciones en el diseño de la guía. Una característica común que surgió de estos algoritmos y se ha confirmado en numerosos estudios con la edición del genoma altamente eficaz, es la presencia de una purina, particularmente una guanina, en el extremo 3′ de secuencia de objetivos específicos de la guía. Esta función de guía ha tenido mucho éxito entre los organismos que van desde mamíferos C. elegans, moscas de la fruta y pez cebra65,100,101. Además, para C. elegans, diseñando guías con un dinucleótido GG en el extremo 3′ de región dirigida a de la guía es una estrategia efectiva para la predicción altamente eficaz guía RNAs65. Idealmente, la prueba a múltiples guías en paralelo para determinar que es más acertado para una aplicación determinada.
Al intentar introducir una secuencia de ADN en el genoma, el diseño de la donante o la plantilla de ADN también es crucial. Los donantes del oligonucleótido de cadena simple (ssODNs) se insertan más fiable que otras plantillas de reparación típica, lineal de doble hebra y plásmido ADN54,55,102. En algunos lugares geométricos, eficiencia HDR puede mejorarse con ssODNs que son complementarios a los ajenos o desplazado el filamento de la DNA y poseen armas de homología que son asimétricos en longitud27,55. Puesto que la plantilla de reparación se inserta en el sitio de corte e incluye la secuencia objetivo, deben adoptarse medidas para evitar que Cas9 hendiendo el ADN del donante antes o después de la inserción genómica. Esto se logra haciendo mutaciones silenciosas a la secuencia de PAM o región de la semilla, evitando el reconocimiento por Cas9 conservando la función de los genes insertados21,103. Aunque los cambios de nucleótidos incluso al PAM es probable que suprimir enlace104, trate de cambiar por lo menos cuatro nucleótidos para estar seguro.
Importancia y usos futuros:
Edición con CRISPR Cas9 genómica se ha convertido en un método de gran alcance que permite la fácil manipulación genética de cualquier organismo. Edición con el RNP Cas9 toma un poco más de esfuerzo al principio pero es fácil de usar una vez que los reactivos y protocolos se establecen en un laboratorio. Edición de celdas con RNP premontada en lugar de DNA plasmídico conduce a una mayor eficiencia general de edición, incluyendo la inserción del gen difícil de lograr mediante HDR, con menos efectos off-target24,25,26 , 27 , 29. Además, experimentadores evitar problemas con la expresión de genes, RNA degradación, plegamiento de la proteína y la asociación entre el gRNA y Cas9 moléculas sintetizadas por separado dentro de la célula22,23. Edición de RNP también evita preocupaciones de seguridad sobre mutagénesis de insertional y expresión sostenida que puede surgir cuando los métodos de distribución viral son utilizados clínicamente14. Debido a estas ventajas, muchos científicos realizando pre-clínicos, experimentos de prueba de concepto favorecen RNP edición para aplicaciones terapéuticas humanas. Tanto en vivo como ex vivo genoma basadas en RNP edición enfoques son en desarrollo para tratar o incluso curar una variedad de condiciones, de enfermedades genéticas como Duchenne distrofia muscular105 y enfermedad de células falciformes27 a 29 y cáncer VIH11. Curiosamente, Cas9 RNP se emplea cada vez más como un método de entrega para la ingeniería agrícola, ya que permite la 'Libre de ADN' edición de plantas33,34,36.
Disclosures
Los autores Alexander Marson y Jacob E. Corn son fundadores del centro de atención terapéutica. Jacob E. Corn es un asesor de misión terapéutica y su laboratorio ha recibido apoyo a la investigación patrocinada de AstraZeneca y Pfizer. Alexander Marson es asesor de Juno terapéutica y terapéutica del Pacto, y su laboratorio ha recibido apoyo a la investigación patrocinada de Juno Therapeutics, Epinomics y Sanofi. Su laboratorio también ha solicitado patentes relacionadas con tecnología Cas9 RNP.
Acknowledgments
Agradecemos a muchos miembros anteriores de nuestros laboratorios y la comunidad edición de genoma del área de la Bahía por sus contribuciones al desarrollo de estos métodos. Agradecemos a Ross Wilson para la lectura crítica de este manuscrito.
Investigación de Alexander Marson es apoyado por un regalo de la Jake Aronov y concesión de la sociedad nacional de esclerosis múltiple (CA 1074-A-21). Alexander Marson tiene un premio de carrera para los científicos médicos de la Burroughs Wellcome Fund y es investigador Chan Zuckerberg Biohub. Investigación de Jacob E. Corn es apoyada por la Li Ka Shing Foundation, la herencia médica Instituto de investigaciones médicas y el Instituto de California para la medicina regenerativa. Behnom Farboud y de Barbara J. Meyer la investigación es financiada en parte por grant NIGMS R01 GM030702 a Barbara J. Meyer, quien es un investigador del Howard Hughes Medical Institute. Investigación Erin Jarvis y Nipam H. Patel es financiada en parte por la subvención de NSF IOS 1257379 y Erin Jarvis reconoce apoyo de un GRFP NSF y una beca de postgrado de Philomathia.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
