

Summary
L'aggregazione della proteina induce stress ossidativo cellulare. Questo protocollo descrive un metodo per il monitoraggio degli Stati intracellulari di proteine amiloidogeniche e lo sforzo ossidativo connesso con loro, mediante citometria a flusso. L'approccio viene utilizzato per studiare il comportamento delle varianti solubile e soggetto a aggregazione del peptide amiloide-β.
Abstract
Misfolding e aggregazione amiloide conformazioni hanno riguardati l'insorgenza e la progressione di parecchie malattie neurodegenerative. Tuttavia, ci sono ancora poche informazioni sulla proteina insolubile come aggregati esercitano loro effetti tossici in vivo. Organismi di modello semplice di procarioti ed eucarioti, come batteri e lieviti, hanno contribuito notevolmente alla nostra comprensione attuale dei meccanismi dietro la formazione intracellulare dell'amiloide, propagazione di aggregati e tossicità. In questo protocollo, l'uso del lievito è descritto come un modello da dissezionare il rapporto tra la formazione di aggregati proteici e del loro impatto su stress ossidativo cellulare. Il metodo combina la rilevazione dello stato solubile/aggregati intracellulare di proteina amiloidogenica con la quantificazione del danno ossidativo cellulare derivanti dalla sua espressione mediante citometria a flusso (FC). Questo approccio è semplice, veloce e quantitativa. Lo studio illustra la tecnica correlando lo stress ossidativo cellulare causato da una vasta serie di varianti del peptide amiloide-β con loro propensioni di aggregazione intrinseco rispettivi.
Introduction
Proteostasis è un determinante fondamentale dei processi di fitness e invecchiamento delle cellule. Nelle cellule, omeostasi della proteina è mantenuto da controllo di qualità di proteina sofisticate reti intendevano ad assicurare il corretto ripiegamento di conformeri proteine misfolded da accompagnatori e/o loro proteolisi mirata con diversi meccanismi ben conservati1 ,2,3,4,5. Un gran numero di studi fornisce il supporto per il collegamento tra l'inizio e la progressione di una vasta gamma di malattie umane e il fallimento di proteostasis, che conduce al misfolding e aggregazione. Per esempio, la presenza di depositi proteici è considerata un marchio di garanzia patologico di molti disordini neurodegenerative, quali Alzheimer, Parkinson e malattie6,7,8, di Huntington prionogenic malattie e non degenerativa amiloidosi9. È stato suggerito che presto assembly oligomerici e protofibrillar nella reazione di aggregazione sono i principale elicitori di citotossicità, stabilendo aberrante interazioni con altre proteine nell' affollato ambiente cellulare10. Inoltre, inclusioni di proteina (PI) possono essere trasmessi tra le cellule, propagando il loro effetto tossico11,12. Di conseguenza, potrebbe essere che la formazione di PI potrebbe infatti costituire un meccanismo di disintossicazione che limita la presenza di specie pericolose aggregati a posizioni specifiche in cella, dove possono essere elaborati o accumulati senza effetti secondari importanti 13 , 14.
Standard in vitro approcci biochimici hanno fornito importanti intuizioni le diverse specie che popolano le reazioni di aggregazione e loro proprietà15,16. Tuttavia, le condizioni utilizzate in queste analisi sono chiaramente diverse da quelli che si verificano all'interno della cellula e, pertanto, chiedo loro rilevanza fisiologica. A causa della notevole conservazione delle vie cellulari come controllo di qualità di proteine, l'autofagia o il regolamento del17,stato redox cellulare18 tra eucarioti19,20,21 ,22,23, il lievito Saccharomyces cerevisiae (S. cerevisiae) è emerso come un modello di cellulare semplice privilegiato per studiare i determinanti molecolari dell'aggregazione proteica e il suo impatto citotossico associato in ambienti biologicamente rilevanti24,25,26.
Propensione di aggregazione della proteina è una caratteristica intrinsecamente codificata nella sequenza primaria. Così, la formazione di amiloide-come le strutture può essere preveduta basato sull'identificazione e la valutazione della potenza di aggregazione-promuovere le regioni polipeptidi27. Tuttavia, nonostante il successo di algoritmi bioinformatici per predire le proprietà di aggregazione in vitro di sequenze proteiche, sono ancora lontano da come queste propensioni si traducono in vivo effetto citotossico di previsione. Gli studi che affrontano il legame tra lo stato aggregato di una data proteina e suoi associato danno cellulare in maniera sistematica possono contribuire ad per aggirare questa limitazione computazionale. Questa connessione è indirizzata nello studio presente, approfittando di un grande insieme di varianti del peptide amiloide-β Aβ42 differiscono solo per un singolo residuo, ma la visualizzazione di un intervallo continuo di aggregazione propensioni in vivo28. In particolare, è descritto un approccio basato su FC per identificare la specie conformazionale contabilità per il danno ossidativo suscitato da aggregazione-incline proteine in cellule di lievito. La metodologia fornisce molti vantaggi come la semplicità, funzionalità di alto-rendimento e accurata misurazione quantitativa. Questo approccio ha permesso di confermare che svolgono un ruolo protettivo contro lo sforzo ossidativo di PI.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. S. cerevisiae culture e l'espressione della proteina
Nota: Varianti Aβ presentano propensioni di aggregazione relativo diversi a causa di una mutazione in un singolo residuo alla posizione 19 (Phe19) del peptide Aβ42 (Figura 1A). Queste varianti del peptide sono contrassegnate con la proteina fluorescente verde (GFP), che agisce come un'aggregazione reporter (Figura 1)29.
- Trasformare i plasmidi codifica per 20 varianti di Aβ42-GFP in cellule di lievito con un BY4741 sfondo parentale (MATa sua3Δ1 leu2Δ0 met15Δ0 ura3Δ0)30. Ogni plasmide codifica per un mutante di Aβ42 fuso alla GFP da un linker GSSGSSG. I plasmidi sono pESC(-URA) e comprendono il marcatore selezionabile URA329.
- Preparare il supporto completo sintetico senza uracile (SC-URA) con i seguenti componenti: 1,9 g/L azoto lievito base senza uracile, 5 g/L di solfato di ammonio e 900 mL di ddH2O combinato con 100 mL di glucosio 20% (p/v).
- Mettere una colonia delle cellule del lievito trasformato in 20 mL di terreno SC-URA contenente 2% di glucosio e far crescere le colture durante la notte a 30 ° C sotto agitazione a 210 giri/min.
- Il giorno successivo, crescere nuove culture di cellule di lievito inoculando 100 µ l della coltura durante la notte in 5 mL di terreno nuovo SC-URA.
- A un OD590 di 0,5, centrifugare le culture a 3.000 x g per 4 minuti, eliminare il supernatante e risospendere le cellule nello stesso volume di terreno nuovo SC-URA contenente 2% raffinosio.
- Incubare a 30 ° C sotto agitazione a 210 giri/min per 30 min. Centrifugare le cellule a 3.000 x g per 4 minuti, quindi eliminare il supernatante. Risospendere le cellule in mezzo SC-URA fresco contenente 2% galattosio per indurre l'espressione di proteine ricombinanti.
- Dopo 16h di indurre l'espressione della proteina in mezzo SC-URA contenente 2% galattosio, vendemmia 1 mL di cellule indotte in microcentrifuga sterile per centrifugazione a 3.000 x g per 4 min.
Nota: Le differenze più rilevanti si verificano dopo un periodo di induzione di cellule di lievito indotto 16 h. esprimendo la varianti di Aβ42-GFP possa essere visualizzato da microscopia di fluorescenza per determinare la distribuzione della proteina ricombinante all'interno delle cellule (Figura 2B).
2. cellula di colorazione
Nota: Cellule fresche indotto non sono necessari come controllo negativo per stabilire una soglia fluorescente durante un'analisi FC.
- Prendere le cellule di lievito indotta da h 16 e determinare la densità ottica a 590 nm (OD590). Diluirli in una soluzione salina tamponato fosfato sterile (PBS) a un OD590 di 0.1.
- Preparare 1 x tamponato fosfato salino come segue: aggiungere 8 g di NaCl, 0,2 g di KCl, 1,44 g di Na2HPO4, e 0,24 g di KH2PO4 a 800 mL di ddH2O. riempire la soluzione fino a 1 L di ddH2O dopo aver regolato il pH 7.4 con HCl. filtrare il buffer su un filtro di 0,2 µm.
- Trasferire le sospensioni delle cellule che esprimono adeguatamente etichettate provette (polistirolo mm turno-fondo di 12 × 75) e proteggerli dalla luce. Assicurarsi che sono pronti per essere caricati sul citometro a flusso per l'analisi, insieme alle cellule indotto non e non tinto.
- Aggiungere la sonda di stress ossidativo (ad es., CellROX profondo rosso) per ogni campione a una concentrazione finale di 5 µM e incubare le cellule a 30 ° C per 30 min al buio.
- Dopo l'incubazione, lavare le cellule 3 volte con 1x PBS e risospendere li nello stesso volume del buffer prima di loro analisi di FC.
3. analisi di citometria a flusso di
Nota: Utilizzare un'installazione FC con laser appropriato e filtri per rilevare GFP e segnale di fluorescenza sonda di stress ossidativo. Può essere utilizzato un citometro a flusso dotato di un laser di 488 nm blu per la rilevazione di GFP e un laser 635 nm rosso per la rilevazione della fluorescenza di sonda di stress ossidativo. Acquisizione di fluorescenza emissione di GFP e stress ossidativo sonda viene eseguita con un 530/30 nm BP e un filtro di BP di 660/20, rispettivamente (tabella 1).
- Fare clic su Nuovo foglio di lavoro aperto Pannello e creare le seguenti trame di puntino di acquisizione.
- Selezionare lo Scatter Plot strumento dalla barra degli strumenti e creare una trama con le variabili zona Side Scatter (SSC-A) sull'asse y vs Forward Scatter-Area (FSC-A) in una scala lineare sull'asse x.
- Fare clic sullo Scatter Plot strumento dalla barra degli strumenti e creare una trama con le variabili FL1-Area (FITC) sul asse x vs. FL3-Area (APC) in una scala logaritmica sull'asse y.
- Fare clic sull'icona Strumenti impostazioni . Fare clic sulla scheda compensazione e impostare tutti i livelli di compensazione. Fare clic sulla scheda di acquisizione e selezionare un numero totale di 20.000 eventi da registrare.
- Fare clic sull'icona di Portata per passare la portata a bassa.
- Fare clic sulla scheda Acquisisci per avviare l'esecuzione le cellule non-indotta, non macchiate e regolare la tensione nell' Impostazioni dello strumento di avanti e dispersione di lato fino a quando la popolazione è distribuita nel quadrante centrale a sinistra.
- Fare clic sull'icona del poligono per impostare una regione R1 intorno la popolazione delle cellule escluse detriti cellulari e utilizzare questa popolazione gated P1 = R1 per tutte le trame puntino fluorescente e rappresentanze di istogramma (Figura 3).
- Per regolare la tensione PMT del segnale di fluorescenza, eseguire le cellule senza macchiate nella trama FL1-FL3 dot, tuning il guadagno nella scheda di Impostazione dello strumento , fino a quando le cellule sono distribuite nel quadrante inferiore sinistro.
- Cambiare il campione per le cellule indotte per misurare la fluorescenza di GFP. Nella scheda Impostazione dello strumento , è necessario impostare il guadagno nella FSC-A vs FL1 (FITC) quando la popolazione è distribuita nel quadrante inferiore destro. Definire la popolazione di cellule positive con un cancello (cancello P2).
- Cambiamento del campione indotto non macchiato celle per visualizzare lo sforzo ossidativo dalla fluorescenza su un terreno di dot FSC-A vs FL3 (APC). Sintonizzare il guadagno fino a che la popolazione delle cellule è distribuita nel quadrante superiore sinistro. Porta la popolazione cellulare positiva nel P3.
- Fare due appezzamenti di istogramma con l'icona di istogramma per rappresentare la fluorescenza delle cellule. Per rappresentare l'intensità di fluorescenza, assicurarsi FL1 (FITC) che rappresenta la fluorescenza di GFP è nell'asse x e FL3 (APC) che rappresenta la fluorescenza è nell'asse y, l'applicazione di popolazioni P2 e P3 su una scala logaritmica, rispettivamente.
Nota: Istogramma sovrapposizioni sono un buon modo per illustrare le differenze tra i profili di fluorescenza delle cellule che esprimono diverse varianti Aβ.
4. Immunodetection recombinant della proteina
- Per determinare i livelli di espressione della proteina, raccogliere le colture di lievito indotte per 16 h mediante centrifugazione a 4.000 x g per 6 min e conservare il pellet di cellule a-80 ° C.
- Risospendere le cellule raccolte che esprimono la proteina ricombinante per 16 h in PBS. Preparare sospensioni cellulari di 200 µ l di ogni mutante di Aβ42-GFP a un OD590 di 20.
- Per l'analisi di frazione della proteina totale, 100 µ l di ogni mutante della raccolta mediante centrifugazione a 14.000 x g per 20 min e risospendere il pellet cellulare nello stesso volume di tampone di lisi del lievito Y-a (50 mM Tris-HCl pH 8.0, 1% DMSO, 200 mM NaCl, 1 mM EDTA contenente 10 mg/mL SB3-14 (myr istyl solfobetaina) completati con 1 mM phenylmethylsulfonyl fluoruro (PMSF)).
- Incubare i campioni per 20 min a temperatura ambiente sotto lieve agitazione.
- Determinare la concentrazione di Estratto di proteine usando l'analisi di Bradford. Caricare fino a 5 µ g di estratto proteico di ogni campione su un gel di elettroforesi SDS-PAGE 15% di acrilammide e macchia su una membrana di polivinilidene difluoruro (PVDF) a 100 V per 60 min.
- Preparare un Bradford reagente con 100 mg di Coomassie Brilliant Blue G-250 disciolto in 50 mL di etanolo al 95% e aggiungere 100 mL di acido fosforico 85% (p/v). Poi, quando il colorante è completamente sciolto, stemperate il composto con ddH2O a 1 L e filtrare attraverso carta da filtro cellulosa appena prima dell'uso.
Nota: Il reagente di Bradford dovrebbe essere marrone chiaro. - Preparare un standard di albumina di siero bovino (BSA) che vanno da 5-100 µ g di proteina in 1.000 µ l del reagente Bradford.
- Aggiungere 1-10 µ l di proteina estrarre a 1 mL di reattivo di Bradford e incubare la miscela a temperatura ambiente per 5 min.
- Misurare l'assorbanza dei campioni e la proteina estrarre campioni OD595.
- Per l'analisi, fare una trama con i valori di assorbanza ottenuti per il standard vs. µ g di proteina. Sulla curva standard, determinare le concentrazioni dei campioni originali dalla quantità di proteine, considerando il volume e la diluizione, se presente.
- Preparare un Bradford reagente con 100 mg di Coomassie Brilliant Blue G-250 disciolto in 50 mL di etanolo al 95% e aggiungere 100 mL di acido fosforico 85% (p/v). Poi, quando il colorante è completamente sciolto, stemperate il composto con ddH2O a 1 L e filtrare attraverso carta da filtro cellulosa appena prima dell'uso.
- Risciacquare la membrana con 1x Tris-buffered saline (TBS), 0.1% Tween 20 (Immunotyping) e bloccarlo utilizzando latte in polvere senza grassi 5% (p/v) in 1 x TTBS.
- Per preparare 1 L di 10 x TBS, sciogliere 24 g di Tris base e 88 g di NaCl in 900 mL di H2Odd e regolare il pH a 7,6. Aggiungere ddH2O ad un volume finale di 1 L. Per una soluzione 1x, miscelare 1 parte di stock x 10 con 9 parti di ddH2O.
- Incubare la membrana per 1 h con un anticorpo primario di β-amiloide 6E10 diluito 1:1,000 e per 1 h a temperatura ambiente con una capra di anticorpo secondario coniugato IgG-HRP anti-topo diluito 01:10, 000.
- Sviluppare le membrane con 1 mL di reagente fluorescente Luminata e quantificare le bande di analisi densitometrica come descritto al punto 5.2.
5. analisi dei dati
- Per analizzare i dati ottenuti da FC, creare una tabella che mostra l'intensità di fluorescenza media (MFI) e mediana fluorescenza con relativo errore standard corrispondenti e/o il coefficiente di variazione (CV) per fluorescenza GFP e livelli di stress ossidativo.
- Fare clic sulla scheda "Impostazioni" e l'icona di statistiche per personalizzare i dati statistici necessari per ogni canale.
Nota: Quando si confrontano le varianti della proteina, convertire dati dei valori di fluorescenza in uno sfondo di piega o metrica ad alta risoluzione (RD):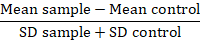
In questa metrica, il campione medio è la media di fluorescenza di campioni di problema, il controllo medio è la media di fluorescenza delle cellule usate come controllo e la SD è la deviazione standard della media di fluorescenza. Fattore di RD è un indice più accurato rispetto valore MFI non solo calcola la differenza tra i campioni, ma anche gli account per la diffusione dei dati. Questo calcolo è specificamente rilevante quando il confronto dei dati provenienti da diversi esperimenti effettuati nel corso del tempo. Una volta sono stati calcolati il RDs, l'uso di un test statistico è consigliabile confermare il significato delle differenze osservate tra varianti della proteina.
- Fare clic sulla scheda "Impostazioni" e l'icona di statistiche per personalizzare i dati statistici necessari per ogni canale.
- Per proteina immunodetection, quantificare i livelli di proteina ricombinante mediante Western blot utilizzando software ImageJ.
- Prima dell'analisi densitometria, convertire le immagini raw originale della membrana sviluppata in modalità di scala di grigi e il formato di file JPEG.
- Regolare la sezione Impostare misure di menu analizza a Dire valore di grigio.
- Fare clic sullo strumento rettangolo da ImageJ e definire un'area di interesse (ROI) con una dimensione coerente per l'analisi di densitometria di banda. Centro di ciascuna banda sull'immagine della membrana all'interno della cornice rettangolare creata e registrare la misurazione uno utilizzando Ctrl + M (il comando di misurazione).
- Misurare lo sfondo locale adiacente per ogni banda, applicando la stessa area ROI. Dopo la misurazione, sottrarre lo sfondo locale corrispondente da ogni banda individuo.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Questo protocollo descrive come impiegare una collezione di 20 varianti del peptide Aβ42 dove Phe19 è stato mutato a tutti naturale proteinogenici aminoacidi28. Le propensioni di aggregazione teorica di queste proteine possono essere analizzati usando due algoritmi bioinformatici diversi (AGGRESCAN e TANGO31,32). In entrambi i casi, questa analisi esegue il rendering di una gradazione progressiva delle tendenze di aggregazione, attribuendo, come regola generale, i valori più elevati di residui idrofobici e il più basso a quelle cariche e polare (Figura 1B, 1C).
Per tenere traccia dello stato di aggregazione intracellulare di diverse varianti di Aβ42 di sperimentalmente, l'esperimento descritto richiede una trasformazione di un plasmide, codifica per Aβ42 e fuso a GFP (Figura 1A) sotto il controllo di GAL1 galattosio-inducibile, in S. cerevisiae. Le cellule di lievito che esprimono Aβ42 varianti dopo un periodo di induzione di 16 h possono essere visualizzate con un microscopio a fluorescenza. Questa visualizzazione permette di confermare la formazione di PI in 10 su 20 varianti della collezione analizzati. Il numero e le dimensioni degli aggregati fluorescenti differiscono da variant a variant (Figura 2). La Figura 2A Mostra la quantificazione di PI da un totale di 500 celle in ognuna delle due culture indipendenti. Qui, possiamo osservare un eccellente accordo tra predetto e in vivo proprietà di aggregazione.
Per risolvere se la formazione di aggregati proteici può innescare lo sforzo ossidativo in questo modello cellulare, questo protocollo utilizza FC per monitorare la fluorescenza di GFP cellulare insieme con la fluorescenza della sonda fluorogenici come indicatore di ossigeno reattiva produzione di specie (ROS). Figura 3 illustra la procedura e ha ottenuto risultati per mutanti Gln e Ile, che corrispondono alle varianti di Aβ42 omogeneamente distribuite e PI-forming, rispettivamente.
In primo luogo, la popolazione delle cellule analizzate è recintato (P1) in FSC-A vs SSC-un punto complotto per rimuovere i detriti delle cellule dall'analisi (Figura 3A). Il segnale di fluorescenza della GFP contro lo sforzo ossidativo sonda della popolazione P1 è rappresentato nei grafici di trama puntino dove le cellule fluorescenti verdi sono gated in P3 (Figura 3B). Figura 3 e 3D Visualizza gli istogrammi creati per ottenere dati statistici per ogni variante espresse, tra cui il CV (tabella 2), al fine di quantificare la media e la mediana fluorescenza di ogni marcatore fluorescente.
Tutti i mutanti dovrebbero essere espressi allo stesso livello, in quanto differiscono per un singolo amminoacido (0,02% della sequenza di Aβ42-GFP). Tuttavia, i macchinari di proteostasis possono rispondere in maniera differenziale a loro propensioni di aggregazione particolare. Di conseguenza, i livelli di espressione della proteina negli estratti cellulari vengono quantificati utilizzando un anticorpo specifico Aβ (Figura 4). Vediamo che, come una tendenza generale, PI-formando Aβ42 varianti sono presenti a livelli inferiori a quelli che rimangono omogeneamente distribuite nel citosol.
Importanti differenze tra le varianti di Aβ42 possono essere osservate quando loro fluorescenza sonda stress ossidativo, i livelli della proteina e le proprietà di fluorescenza GFP sono rappresentati rispetto la loro capacità di formare PI e loro propensioni di aggregazione intrinseco ( Figura 5). Più aggregazione-incline varianti PI-formare suscitano molto stress ossidativo inferiore rispetto alle loro controparti più solubile (Figura 5A, 5B). Con questi risultati, non esiste alcuna correlazione evidente tra la dimensione e il numero di aggregazioni a singola cella (Figura 2) e livelli di stress ossidativo. PI-formare varianti, soprattutto mutanti recante Trp e Phe in posizione 19, sono presenti nella cella a livelli inferiori a quelli distribuiti omogeneamente nel citosol (Figura 5, 5D). L'eccezione a questa è il mutante di Thr, per cui meno del 10% delle cellule formano PI (Figura 1B, 1C). Ciò corrisponde con il fatto che le varianti più incline a aggregazione selettivamente vengono ripulite con il lievito controllo qualità degradazione macchine28. Fluorescenza GFP e livelli della proteina correlano bene per le forme di Aβ42 solubile più, ma non per quelle che formano i PI (Figura 5E, 5F). Questo è probabilmente perché in queste popolazioni cellulari, la fluorescenza proviene da due diversi compartimenti, il citosol e le inclusioni, e loro contributi relativi alla fluorescenza totale differiscono tra mutanti34.

Figura 1 . Analisi di tendenza di aggregazione per la raccolta dei costrutti di fusione di GFP Aβ42 derivato dalla mutazione di residuo alla posizione 19 nel peptide Aβ42 di tutti gli aminoacidi naturali. A. questa immagine mostra un modello 3D di wt Aβ42 fusa alla GFP da un linker (Aβ42, grigio; GFP, verde), basato sul 1EMA PDB per proteina verde fluorescente da Aequorea victoria e il 2OTK per il peptide Aβ Alzheimer, in cui la catena laterale di Phe19 di wt Aβ42 è mostrata in rosso. Il modello è creato utilizzando software Pymol. 19 di residui nella sequenza di Aβ42 occupa una posizione centrale nel cluster centrale idrofobo. I grafici a barre vengono creati con due differenti bioinformatic preannunciatori: B. AGGRESCAN, C. TANGO. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2 . Varianti di Aβ42-GFP PI-formante in S. cerevisiae culture indotte per 16 h di espressione. A. il grafico a barre indica la percentuale di cellule che contengono un numero diverso di PI calcolato da un totale di 500 cellule fluorescenti per ogni variante in due replicati biologici. B. queste immagini sono immagini di microscopia fluorescente rappresentativo delle varianti di Aβ42-GFP selezionate (Phe, Ile, Thr, Trp). Sono state acquisite sotto luce UV utilizzando un filtro di eccitazione per GFP (450-500 nm) e un intervallo di emissione (515-560 nm). La barra della scala rappresenta 10 µm. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3 . Schema per l'analisi del flusso cytometry (FC) of S. cerevisiae cellule esprimenti selezionato Aβ42-GFP varianti. A. il grafico mostra le cellule di lievito con cancello (P1) in appezzamenti dot (FSC-A vs SSC-A) cui detriti cellulari viene rimosso dalla popolazione delle cellule. Le immagini microscopiche di Gln (omogeneamente distribuito) e varianti di Ile (PI-formando) vengono visualizzate accanto a che trame il puntino FC. La barra della scala rappresenta 10 µm. B. Queste immagini di trama dot scatter rappresentano GFP-A vs sonda stress ossidativo, in cui la popolazione gated (P3) comprende cellule solo fluorescenti, escludendo il segnale di fondo. Gli istogrammi di frequenza delle cellule sono di C. GFP ampiezza del segnale (FITC) gestita da P1 e D. CellROX (ampiezza APC) gestita da P3. L'acquisizione di cella è stato effettuato con un citometro a flusso. Ogni grafico rappresenta 20.000 eventi. Q e corrispondono a Gln e Ile Aβ42 mutanti, rispettivamente. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 4 . Quantificazione dei livelli della proteina cellulare. Questa figura mostra macchie occidentali delle frazioni della proteina totale di mutanti di Aβ42-GFP dopo 16h di espressione in S. cerevisiae. Le varianti PI-formanti sono colorate in verde e quelli distribuiti diffusamente nel citosol sono colorati in rosso chiaro. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 5 . Stress ossidativo cellulare, fluorescenza GFP intracellulare e livelli di proteina cellulare per mutanti di Aβ42-GFP determinati dopo 16h di espressione in S. cerevisiae. Le varianti rappresentate sull'asse x sono state ordinate secondo loro propensioni di aggregazione previsto da TANGO (pannello di destra) o AGGRESCAN (pannello di sinistra). Le varianti PI-formanti sono colorate in verde e le varianti non-PI-forming sono colorate in rosso chiaro. A. B. questi grafici a barre Visualizza i valori di fluorescenza di sonda stress ossidativo ottenuti da un'analisi FC dei mutanti Aβ42-GFP. C. D. questi grafici a barre rappresentano i livelli di proteina come quantificato mediante una densitometria di Western blot utilizzando software ImageJ. E. F. questi grafici a barre Visualizza i valori di fluorescenza di GFP dopo un'analisi FC. Le barre di errore per lo stress ossidativo della sonda e valori di fluorescenza di GFP rappresentano il coefficiente di variazione (CV) delle cellule FC-gated. Le barre di errore per i livelli di espressione di proteine rappresentano ± SE (n = 3). Clicca qui per visualizzare una versione più grande di questa figura.
| Canale | Fluoroforo | Lunghezza d'onda di eccitazione (nm) | Filtro passa banda |
| FITC | GFP | 488 | 530/30 |
| APC | CellRox | 635 | 660/20 |
| PerCP | IP | 488 | 585/42 |
Tabella 1. Sorgente laser, filtri passa-banda e fluorofori utilizzati nel citometro a flusso.
| Fluorescenza GFP | CURRICULUM VITAE | CellROX fluorescenza | CURRICULUM VITAE | |
| A | 9316 | 96,4 | 1964 | 127.5 |
| C | 9709 | 91,9 | 1275 | 172,2 |
| D | 11213 | 101.7 | 3443 | 155,8 |
| E | 12256 | 101.1 | 3220 | 152,2 |
| F | 3010 | 96,1 | 1245 | 146.4 |
| G | 11541 | 97,2 | 2947 | 158 |
| H | 7895 | 98,2 | 3582 | 120,8 |
| Ho | 7365 | 97,2 | 1416 | 141,3 |
| K | 10839 | 100.4 | 3102 | 122.1 |
| L | 7605 | 96,9 | 1401 | 161,8 |
| M | 8149 | 96 | 1308 | 170,4 |
| N | 12741 | 97,5 | 3403 | 134,9 |
| P | 9768 | 102,8 | 2629 | 143,6 |
| Q | 13066 | 91,3 | 3354 | 169,9 |
| R | 8537 | 101.3 | 2839 | 127.5 |
| S | 12053 | 99,1 | 3313 | 174,3 |
| T | 10615 | 97,7 | 2213 | 107.7 |
| V | 9169 | 96,1 | 1878 | 121.7 |
| W | 1715 | 94.7 | 1531 | 100 |
| Y | 7574 | 94,5 | 1234 | 138 |
Tabella 2. Elenco dei valori per fluorescenza GFP e stress ossidativo sonda fluorescenza ottenuti dall'analisi FC delle cellule di lievito che esprimono mutanti di Aβ42-GFP per 16 h. Questa tavola mostra l'intensità media di fluorescenza (MFI) e il CV per ogni mutante.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Una vasta gamma di malattie è legata all'accumulo di proteine misfolded in depositi cellulari6,7,8,33. Molti sforzi sono stati fatti per svelare i meccanismi molecolari che determinano l'insorgenza di queste malattie utilizzando approcci computazionali, che non tengono in concentrazioni di proteina di conto, o in vitro si avvicina, in cui la concentrazione nella proteina rimane costante durante la reazione. Tuttavia, all'interno della cellula, proteine sono costantemente sintetizzate e degradate in un ambiente affollato e non omogeneo. Questo spiega le frequenti discrepanze tra le proprietà di aggregazione in silico, in vitro e in vivo di proteine amiloidogeniche.
Ci sono diverse ragioni perché semplici modelli cellulari, come il lievito, sono una scelta ragionevole per studiare l'aggregazione e associati tossicità delle proteine legate a malattie neurodegenerative in un contesto più biologico. Per esempio, ci danno la possibilità di analizzare l'impatto di aggregazione misfolding di proteine su una popolazione cellulare intatta utilizzando analisi veloce come quello implementato qui. Tuttavia, dovremmo considerare che livelli ossidativi possono differire tra ceppi di lievito. Così, per confrontare tra ceppi, o anche tra diverse proteine, ossidanti e riducenti, quali DTT e diammina, devono essere utilizzati per stabilire una scala di riduzione/ossidazione.
Nell'esempio illustrato in questo articolo, l'analisi bioinformatica, quantificazione della proteina cellulare, formazione immagine di localizzazione della proteina e l'analisi simultanea di FC di GFP attività ed i livelli di stress ossidativo sono stati integrati per identificare il molecolare specie responsabile del danno ossidativo suscitato da reazioni di aggregazione di proteine. I risultati dimostrano che le varianti di Aβ42 solubile più, piuttosto che più aggregazione-incline, promuovere il più alto sforzo ossidativo. Ciò indica la specie diffusibile, essendo il più pericoloso di specie di Aβ42 in vivo. La bassa tossicità di aggregare varianti sembra rispondere sia per il fatto che queste conformazioni sono sequestrate in PI loro degradazione preferenziale di macchine di qualità di proteine, con conseguente più bassi livelli di proteina che quella del loro solubile controparti.
Il metodo descritto non è limitato all'analisi dello stress ossidativo prodotto da Aβ42 solubile/aggregati specie e può essere applicato anche per studiare una varietà di disordini di aggregazione delle proteine. Inoltre, la tecnica potrebbe essere utile per monitorare l'effetto di inibitori dell'aggregazione su stress ossidativo cellulare, che permette di scartare per ulteriori applicazioni cliniche quelle molecole che promuovono l'accumulazione delle specie di proteina attiva ossidativo. Infine, come una sonda fluorogenic è disponibile, l'approccio offre notevoli opportunità per identificare le specie molecolari responsabili di altri effetti tossici legati all'aggregazione di proteine in modo rapido in modo semplice, fornendo dati quantitativi.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Materials
| Name | Company | Catalog Number | Comments |
| Yeast cells BY4741 | ATCC | 201388 | Genotype: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| pESC(-Ura) plasmid | Agilent Genomics | 217454 | Yeast expression plasmid with a Gal promotor. Selectable marker URA3 |
| Yeast Synthetic Drop-out Medium Supplements | Sigma | Y1501 | Powder |
| Yeast Nitrogen Base Without Amino Acids | Sigma | Y0626 | Powder |
| Raffinose | Sigma | R7630 | Powder |
| Glucose | Sigma | G7021 | Powder |
| Galactose | Sigma | G0750 | Powder |
| Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | Solution 10X |
| CellROX Deep Red Reagent | Life Technologies | C10422 | Free radical cell-permeant fluorescent sensor, non-fluorescent while in a reduced state, and exhibits bright fluorescence upon oxidation by reactive oxygen species (ROS), with absorption/emission maxima at 644/665 nm. |
| Y-PER protein extraction reagent | Thermo Scientific | 78990 | Liquid cell lysis buffer |
| Acrylamide/Bis-acrylamide | Sigma | A6050 | Solution |
| Bradford dye reagent | Bio-Rad | 5000205 | Dye reagent for one-step determination of protein concentration |
| β-amyloid antibody 6E10 | BioLegend | 803001 | Mouse IgG1. The epitope lies within amino acids 3-8 of beta amyloid (EFRHDS). |
| Goat anti-mouse IgG-HRP conjugate | Bio-Rad | 1721011 | |
| Membrane Immobilon-P, PVDF | Millipore | IPVH00010 | |
| Luminata forte | Merk | WBLUF0100 | Premixed, ready to use chemiluminescent HRP detection reagent |
| Phenylmethanesulfonyl fluoride solution (PMSF) | Sigma | 93482 | Protease inhibitor. Dissolved at 0.1 M in ethanol |
| FACSCanto flow cytometer | BD Biosciences | 657338 | Equipped with a 488 nm blue laser for the detection of GFP, and 635 nm red laser / 530/30 nm BP filter and 660/20 BP filter |
| Mini Trans-Blot Electrophoresis Transfer cell | Bio-Rad | 1703930 | Protein transference system |
| Mini-PROTEAN Tetra Handcast Systems | Bio-Rad | 1658000FC | Electrophoresis system |
References
- Frydman, J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annual Review of Biochemistry. 70, 603-647 (2001).
- Hartl, F. U., Bracher, A., Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature. 475, 324-332 (2011).
- Wong, E., et al. Molecular determinants of selective clearance of protein inclusions by autophagy. Nature Communications. 3, (2012).
- Winkler, J., Tyedmers, J., Bukau, B., Mogk, A. Chaperone networks in protein disaggregation and prion propagation. Journal of Structural Biology. 179, 152-160 (2012).
- Glickman, M. H., Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiological Reviews. 82, 373-428 (2002).
- Dobson, C. M. Getting out of shape. Nature. 418, 729-730 (2002).
- Chiti, F., Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annual Review of Biochemistry. 75, 333-366 (2006).
- Aguzzi, A., O'Connor, T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery. 9, 237-248 (2010).
- Rapezzi, C., et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nature Reviews Cardiology. 7, 398-408 (2010).
- Deas, E., et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson's disease. Antioxidants & Redox Signaling. 24, 376-391 (2016).
- Brundin, P., Melki, R., Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature Reviews Molecular Cell Biology. 11, 301-307 (2010).
- Frost, B., Diamond, M. I. Prion-like mechanisms in neurodegenerative diseases. Nature Reviews Neuroscience. 11, 155-159 (2009).
- Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431, 805-810 (2004).
- Ross, C. A., Poirier, M. A. Opinion: what is the role of protein aggregation in neurodegeneration? Nature Reviews Molecular Cell Biology. 6, 891-898 (2005).
- Mishra, R., Sjölander, D., Hammarström, P. Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Molecular BioSystems. 7, 1232-1240 (2011).
- Klunk, W. E., Jacob, R. F., Mason, R. P. Quantifying amyloid by congo red spectral shift assay. Methods in Enzymology. 309, 285-305 (1999).
- Khurana, V., Lindquist, S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nature Reviews Neuroscience. 11, 436-449 (2010).
- Tenreiro, S., Outeiro, T. F. Simple is good: yeast models of neurodegeneration. FEMS Yeast Research. 10, 970-979 (2010).
- Moosavi, B., Mousavi, B., Macreadie, I. G. Yeast model of amyloid-β and Tau aggregation in Alzheimer's disease. Journal of Alzheimer's Disease. 47, 9-16 (2015).
- Tenreiro, S., Munder, M. C., Alberti, S., Outeiro, T. F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. Journal of Neurochemistry. 127, 438-452 (2013).
- Yang, J., Hao, X., Cao, X., Liu, B., Nyström, T. Spatial sequestration and detoxification of huntingtin by the ribosome quality control complex. eLife. 5, (2016).
- Braun, R. J., Büttner, S., Ring, J., Kroemer, G., Madeo, F. Nervous yeast: modeling neurotoxic cell death. Trends in Biochemical Sciences. 35, 135-144 (2010).
- Figley, M. D., Gitler, A. D. Yeast genetic screen reveals novel therapeutic strategy for ALS. Rare Diseases. 1, e24420 (2013).
- Cooper, A. A., et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 313, 324-328 (2006).
- Johnson, B. S., McCaffery, J. M., Lindquist, S., Gitler, A. D. A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 105, 6439-6444 (2008).
- Bharathi, V., et al. Use of ade1 and ade2 mutations for development of a versatile red/white color assay of amyloid-induced oxidative stress in saccharomyces cerevisiae. Yeast. 33, 607-620 (2016).
- Pallarès, I., Ventura, S. Advances in the prediction of protein aggregation propensity. Current Medicinal Chemistry. , (2017).
- Villar-Piqué, A., Ventura, S. Protein aggregation propensity is a crucial determinant of intracellular inclusion formation and quality control degradation. Biochimica et Biophysica Acta. 1833, 2714-2724 (2013).
- Navarro, S., Villar-Piqué, A., Ventura, S. Selection against toxic aggregation-prone protein sequences in bacteria. Biochimica et Biophysica Acta. 1843, 866-874 (2014).
- Morell, M., de Groot, N. S., Vendrell, J., Avilés, F. X., Ventura, S. Linking amyloid protein aggregation and yeast survival. Molecular BioSystems. 7, 1121-1128 (2011).
- Conchillo-Solé, O., et al. AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinformatics. 8, 65 (2007).
- Fernandez-Escamilla, A. M., Rousseau, F., Schymkowitz, J., Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology. 22, 1302-1306 (2004).
- Renner, M., Melki, R. Protein aggregation and prionopathies. Pathologie Biologie.(Paris). 62, 162-168 (2014).
- Carija, A., Navarro, S., de Groot, N. S., Ventura, S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biology. 12, 699-711 (2017).
