

Summary
顕微鏡の有機体のゲノムのシーケンス処理中に汚染大きい問題に残る。ここでは、汚染のリスクを最小限に抑えるため全ゲノム増幅 DNA のわずか 50 pg を単一の標本からクマムシのゲノムをシーケンス処理する方法を示します。
Abstract
クマムシは、乾燥とすることができますに直面するときと呼ばれる乾燥耐性を元の状態に返すときに水が供給されて ametabolic 状態顕微鏡動物です。時々 これらの動物の遺伝子の水平伝達の範囲についてたとえば、誤った解釈につながるクマムシ リスク細菌汚染など顕微鏡動物のゲノム配列を決定。ここでは、我々 は単一の標本からHypsibius dujardiniクマムシのゲノムを配列する超低入力メソッドを提供します。50 の効率的な抽出と厳密な洗浄および汚染物質の除外を用いて 〜 200 pg 単一の個人からのゲノム DNA、DNA シーケンシング装置でシーケンス化されたライブラリを構築しました。これらのライブラリは、高い再現性で、公平をされ他h. dujardiniのゲノム シーケンスされた読み取りのインフォマティクス解析は汚染の最小限の量を示した。このメソッドは、以前の方法を使用していないシーケンスが難のクマムシに適用できます。
Introduction
クマムシは、乾燥に直面するとき、乾燥耐性と呼ばれる ametabolic 状態を入力することができます顕微鏡動物です。彼らは水1,2の吸収による回復します。Ametabolic 状態ではクマムシが極端な温度3高圧4、5ガンマ線、x 線、紫外線光6の高用量を含む様々 な極限環境に耐えることができます。7,8、および宇宙空間9。ゲノムデータは乾燥耐性の分子機構の研究のために不可欠な基礎です。
クマムシのゲノムをシーケンス処理する前の試みは、細菌汚染10、11,12,13,14の兆候を示しています。このような小さな生物からゲノム配列決定多数の動物を必要とし、細菌汚染になりやすいです。したがって、我々 は既に15汚染のリスクを最小限に抑えるため、クマムシの単一の標本から超低入力メソッドを使用してシーケンス プロトコルを確立されています。これらのデータを使用して、さらに実施した高品質の順序とH. dujardini16,17のゲノムの再構築。ここで述べるこの(図 1tardigrade 一個人からゲノム配列決定法)。このシーケンスの結果の検証は本稿の焦点を越えて、私たちの以前のレポート16で既に、徹底的に議論されています。
このメソッドは、2 つの部分で構成されています: 最低汚染可能と DNA のピクトグラム レベルの質の高い抽出単一クマムシの分離。海中でもは飢えたと抗生物質と同様に、水ですすぎ、任意の細菌汚染の除去を確実に 500 倍の倍率での顕微鏡で観察しました。以前の推定値と測定、クマムシの単一の個人にゲノム DNA の16、凍結融解のサイクルによってまたは手動の均質化によって、キチン質の外骨格を割れによって抽出される約 50 200 pg が含まれていることを示しています。この DNA は、図書館の建設に提出し、DNA シーケンシング装置でシーケンス処理します。追加情報分析は、以前の tardigrade シーケンス プロジェクトと比較して汚染の低レベルと同様、高品質のシーケンスを示しています。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. 準備
- 90 mm プラスチック製培養皿で 10 mL、1% ペニシリン/ストレプトマイシン DW のとカクテルの溶媒として蒸留水 (DW) を使用して 2% の agarose のゲルを準備します。ゲルは 18 ° C で設定インキュベーターで 2 〜 3 週間に保存できます。
注: は、低温は agarose のゲル、ゲルと、クマムシをトラップできる培養皿の壁の非常に小さいギャップの結果を縮小するための 10 の ° C の下のゲルの任意のストレージを避けてください。
2. 試料調製及び汚染物質の排除
- 単一のクマムシを収集、準備された寒天プレート上に配置、それを洗う - 任意の残りの粒子を除去する社殿と 3 x x 2。
- 腸から任意の余分な食品を削除する 24 h 18-22 ° C でクマムシを孵化させなさい。
- ペニシリン/ストレプトマイシンの抗生物質細菌汚染を削除し、浄化した動物 P10 ピペットを使用してきれいなスライド ガラス上に 2-6 h に飢えたクマムシを配置します。
- 500 倍の倍率での顕微鏡下でクマムシを観察し、残存細菌がないことを確認します。
注: 顕微鏡の高倍率は望ましいが、光学顕微鏡はまたなく観察を適用するされることがあります。 - 最大液 5 μ L の P10 のピペットを使用して個々 の収集、低結合ポリメラーゼの連鎖反応 (PCR) チューブに入れます、できるだけ余分な水分を削除します。
注: マイクロ チューブ、PCR チューブ ・ ピペット チップ必要がありますすべて低結合する DNA の損失を最小限に。
注: を行う急速な乾燥、できるだけ均質化または動物の死は、ゲノム DNA を損傷する可能性が大きく。
3. 均質化と DNA の抽出
- 次の方法のいずれかでそのゲノム DNA (gDNA) を取得する動物を均質化します。
注: (列と基づくビーズ) 他のキットはこの非常に低い入力で効果がないため DNA 抽出手順の材料表に示されている指定されたキットを使用するが重要です。ゲノムの換散バッファーを使用する β-メルカプトエタノール前に 0.5% と指定しました。- 凍結融解サイクル18動物をホモジナイズしてください。
- 手順 2.5 の直後、クマムシを含む PCR チューブに換散バッファーの 100 μ L を追加します。
- 37 ° C に加温熱ブロックに移動 10 分、10 分と、液体窒素で PCR チューブを配置2 x この手順を繰り返します。
注: 均質化は不十分であるまたは手動粉砕後に実行できる場合は、この手順を繰り返すことができます。
- 手動で動物をつぶします。
- 直後、顕微鏡下のステップ 2.5、PCR の管の壁に動物を押すことによってピペットの P10 の先端を持つ個人を粉砕し、すぐに換散バッファーの 100 μ L を追加します。
注: 海中でもすることができます簡単にスリップし、非効率的な破砕 gDNA を抽出に失敗になりますので、顕微鏡の下でこのプロシージャを観察するが重要です。換散バッファー有機体に侵入することができますので、tardigrade のキューティクルが壊れていることを確認します。
- 直後、顕微鏡下のステップ 2.5、PCR の管の壁に動物を押すことによってピペットの P10 の先端を持つ個人を粉砕し、すぐに換散バッファーの 100 μ L を追加します。
- 凍結融解サイクル18動物をホモジナイズしてください。
- 管に発生する換散のため室温で 30 分間インキュベートします。
注: 最低 30 分、効率的な換散のため必要ですが、孵化は長くすることができます。 - きれいな 1.5 mL 低結合マイクロ チューブに溶解混合物の全集 (100 μ L) を転送します。
- 均質化のために使用された、空、今低結合 PCR チューブに換散バッファーの 100 μ l 添加し混合物を分注後、ステップ 3.3 で使用される 1.5 mL 低結合マイクロ チューブに転送。2 x この手順を繰り返します。
- ステップ 3.4 のように低結合 PCR チューブに換散バッファーの 300 μ L を追加し、ピペッティングした後低バインディングの 1.5 mL マイクロ チューブに混合物を移動します。1.5 mL 低結合マイクロ チューブは、このステップの後 600 μ L の混合物を含める必要があります。
注: この手順は、サンプルを複数回洗浄することにより PCR チューブ壁にバインドされた gDNA の損失を最小限に抑えることです。 - コレクションの管に置かスピン列に溶解混合物の 600 μ L の合計を追加し、1 分 10,000 × g で遠心分離します。
- 1 分の 10,000 × g で遠心し、列に流れを再適用されます。
注: この手順は確実に重要な gDNA のほとんどが列にバインドされています。 - スピン列に洗浄バッファーの 500 μ l 添加しきれいな 1.5 mL のマイクロ チューブにスピン列 1 分転送 10,000 x g で遠心分離機します。
- 10 mM トリス-HCl、pH 8.5、スピンの列、5 分待って、室温での 20 μ L を適用し、1 分の 10,000 × g で遠心分離機します。
注: ライブラリの準備酵素と干渉するため、溶出バッファーは、EDTA を含んでいないする必要があります。 - スピンの列と常温静置後の 5 分後のフローに再適用、10,000 × g で 1 分間遠心します。
注: この手順は、列にバインドされた gDNA の最大溶出するために重要です。
4. 図書館建設シーケンス
-
DNA の断片化
- DNA 断片化の 15 μ L マイクロ チューブに gDNA 溶離液の 15 μ L を転送し、1 分卓上遠心機を使用して管を遠心分離します。
注: 指定したマイクロ チューブ材料の表に示すように低入力に最適です。低ボリューム超音波と断片化は危険で、これは DNA の非常に低濃度のため酵素断片化で置換することはできません。 - 550 gDNA をフラグメント bp。
注: を用いて 550 bp 設定は次のとおり: 入射ピークパワー 30 W、デューティー比を = = 20%、バーストあたりのサイクル数 = 50、治療時間 = 23 s。 - 徹底的なピペッティングした後クリーン低結合 PCR チューブに断片化された DNA の混合物の 10 μ L を転送します。
注: 実験をここで停止することができます。4 ° C または-20 ° C で DNA を保持します。
- DNA 断片化の 15 μ L マイクロ チューブに gDNA 溶離液の 15 μ L を転送し、1 分卓上遠心機を使用して管を遠心分離します。
-
図書館建設のシーケンス
注: 入力 DNA の低い量のため、次の手順でテーブルの材料で指定されたキットを使用するが絶対に重要です。- テンプレートの準備バッファーの 2 μ L とテンプレート準備酵素の 1 μ L を加えて徹底的にピペットでそれらをミックスします。
- 以下の条件でサーマルサイクラーにテンプレート準備反応を実行: 25 分、20 分、4 ° C ホールド 101-105 ° C で加熱蓋に 55 ° C の 22 ° C反応が完了するは、次の手順に進みます。
- テンプレート作製の反応生成物にライブラリ合成バッファーの 1 μ L とライブラリ合成酵素の 1 μ L を追加し、(4 ° C ホールド) と 40 分の 22 ° C で混合物を孵化させなさい。反応が完了するは、次の手順に進みます。
- ライブラリ増幅マスター ミックス (ライブラリ増幅バッファーの 25 μ L、ライブラリ増幅酵素の 1 μ L、ヌクレアーゼ フリー水の 4 μ L) の 30 μ L とインデックスの試薬の 5 μ L を追加します。
- ピペットを徹底的にミックスのすべてと卓上遠心分離機で簡単に混合物を遠心分離機します。
- 表 1に示した条件で PCR を行います。
| 温度 | 時間 | サイクル |
| 72 ° C | 3 分 | |
| 85 ° C | 2 分 | |
| 98 ° C | 2 分 | |
| 98 ° C | 20 秒 | 4 サイクル |
| 67 ° C | 20 秒 | |
| 72 ° C | 40 秒 | |
| 98 ° C | 20 秒 | 16 サイクル |
| 72 ° C | 50 秒 | |
| 4 ° C | ホールド |
表 1: PCR の条件。
-
PCR の製品の浄化
注: この手順は、他の浄化方法と代替可能します。分光光度計を使用して生成される DNA の純度を確認する別の方法を使用する場合を確認します。260/280 と 260 の吸光度比/230 1.8 上両方する必要があります。- 磁気ビーズを 50 μ l 添加、ピペットのソリューションは、10 x、簡単に卓上遠心機で遠心力場それ。
- 常温で 2 分のためのソリューションを孵化させなさい。
- 5 分、マグネット スタンドにそれを孵化させなさいまたはソリューションが完全になるまでをオフにし、上清を除去します。
- マグネット スタンドに低結合 PCR チューブに作りたての 80% エタノール 200 μ L を追加、30 s と削除上清を待ちます。2 x この手順を繰り返します。ビーズが不可でした。
- 簡単に卓上遠心と低結合 PCR チューブを遠心し、マグネット スタンドに任意の余分なエタノールを削除します。ビーズを風乾、オーバードライを避けてください。
- 10 mm トリス-HCl、pH 8.5 15 μ L でビーズを再懸濁します徹底的に磁気ビーズが均一に分散されるようにソリューションをピペット、簡単なの遠心分離後、室温で 2 分の管を孵化させなさい、2、マグネット スタンドにインキュベートソリューションがクリアされるまでの分。
注: シーケンスの化学と干渉するため、溶出バッファーは、EDTA を含んでいない必要があります。 - 新しい低結合 PCR チューブ、ペレットを乱すことがなく、培養上清を転送します。
注: 実験をここで停止することができます。4 ° c または-20 ° C、長期に渡る保存 DNA を保持します。
5. 品質チェック、定量化、および DNA のシーケンス
注: 品質チェックは DNA の低い量のためにこの工程の前に行わない。
-
DNA ライブラリのサイズ分布の検証
注: サイズ分布のデジタル レポートと他の高感度電気泳動システムを使用できます。- 電気泳動バッファー試薬を戻り、ゲルのカートリッジ (25 1,000 bp の範囲で) 部屋の温度にします。
- 配列ライブラリの 1 μ L を電気泳動バッファー試薬の 3 μ L を追加し、渦と 1 分の徹底的にそれらをミックスし、簡単に卓上遠心分離機、遠心分離機のそれら。
- 電気泳動を行い、関連ソフトウェアのライブラリ サイズ分布を検証します。主なフラグメント ピークは、1,000 に約 300 から広く及ぶべき bp。
-
DNA の定量化
他の蛍光ベースのメソッドまたは量的な PCR 注: を使用することができますが、それはシーケンス ライブラリを定量化するために十分に正確ではないと、吸光光度法を避けてください。- ソリューション バッファーの 796 μ L と蛍光試薬の 4 μ L を追加し、徹底的にそれらをミックスします。2 つの測定管に取り組んで解決策の 190 μ L と 1 つの試験管に 197 μ L を調剤します。
- 作業ソリューションの 190 μ L を含む各試験管に DNA 既知濃度基準の 10 μ L と準備作業ソリューションの 197 μ L を含むアッセイ チューブ ライブラリの 3 μ L を追加します。
- 渦管を簡単に卓上遠心分離の遠心分離機のそれらと。3 μ の設定で、蛍光光度計を用いた DNA を定量化します。
-
Dna ライブラリ
注: シーケンス処理プラットフォームは、指定されたライブラリ構築キットと互換性があります。- 製造元のプロトコルに基づいて配列ライブラリを準備します。
- シーケンス楽器に試薬カセットとフロー セルを設定し、次の製造元のプロトコル情報を実行シーケンスを入力します。
- シーケンスを実行します。
注: 2 つのシーケンスの実行を行った: 4 つのサンプルを 1 つの実行の多重化と同様、1 つのサンプル/実行します。
6. 計算解析
- ベース呼び出しと、必要な場合は、読み取りをデマルチプレックスします。
- FastQC19シーケンス データの品質を検証します。
注: 得られたデータのより詳細な検証、当社の以前のレポート16を参照してください。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
汚染物質の除外:
このプロトコルには、海中でも、汚染を最小限に抑えるために抗生物質治療と殺菌の徹底した洗浄が含まれます。また、これらのプロセスの完全性を確保するため、視覚的なチェック プロセスが含まれます。検証 (ステップ 2.4 プロトコルの) 中に作られた顕微鏡イメージは図 2に示します。500 倍の倍率で観察するとは、個々 のクマムシを動かす小さな粒子として細菌細胞を見ることができます。
DNA ライブラリの品質の検証:
構築された Seq DNA ライブラリの総量は約 109.5 の16ng (7.3 ng/μ x 15 μ L) です。断片化の長さ分布を検証するため、電気泳動パターンは、ようになります図 3.550 に断片化のサイズ設定、bp DNA の切断機と、ライブラリは 550-600 bp、シーケンス アダプターを含むをする必要があります。それは、シーケンス ライブラリの大部分が 200-1000 bp 間に含まれる、複製 (N1 - N4) 間で一貫して観察できます。
シーケンス データの解析:
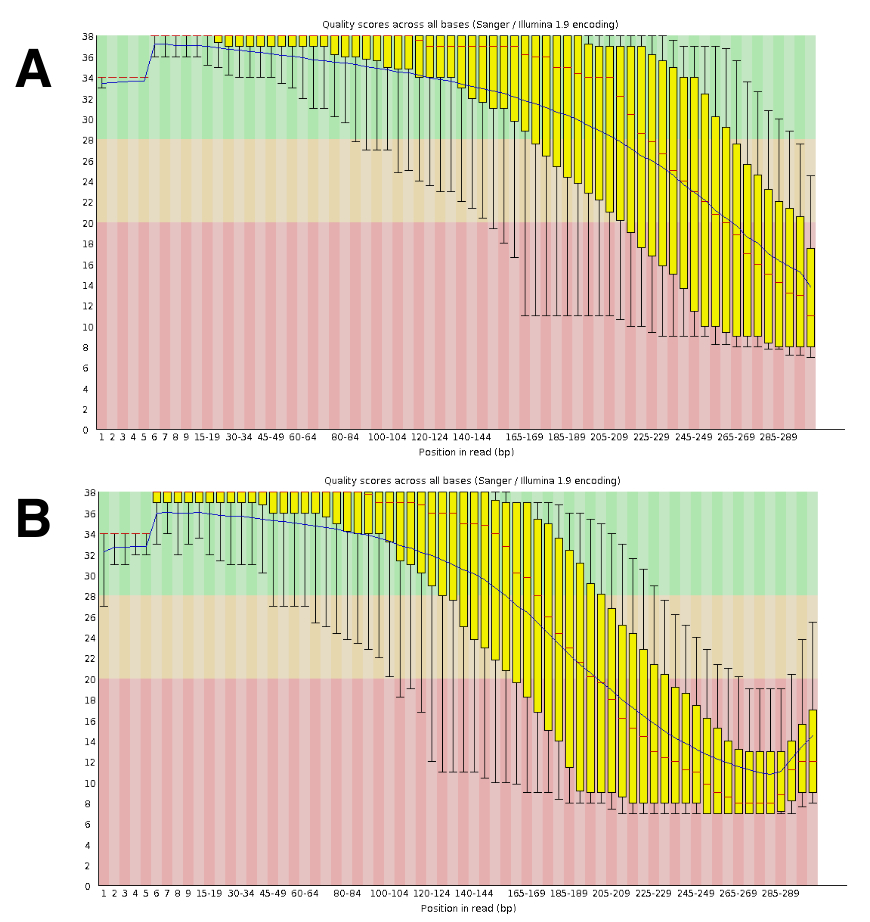
DNA の配列には、約 20-25-メートルにペアになっている読み取りあたりの実行が生成されます。FastQC (図 4) を使用して、品質の検証を行った。シーケンスされた読み取りに沿って品質の分布は、典型的な 300 bp ペア実行です。

図 1: このプロトコルのワークフロー。この図では、このプロトコルの概要を示しています。この図の拡大版を表示するのにはここをクリックしてください。

図 2: 細菌無料緩歩動物の代表的な写真です。この図は、さらに拡大イメージ (下) と共に汚染 (左) 緩歩動物と洗浄 (右) 緩歩動物 (Hypsibius dujardini) の画像を示します。海中でも周りのロッド型細胞は汚染物質を矢印で示されます。スケール バーを示します 100 μ m.この図の拡大版を表示するのにはここをクリックしてください。

図 3: DNA ライブラリ構築のフラグメント長分布の検証します。このパネルには、シーケンス処理ライブラリのサイズの分布が表示されます。紫と緑の線はそれぞれ 1,500、25 bp の上限と下限のマーカーを示します。L = はしご、S = 1 サンプル/実行、N1 - N4 = 4 複製/実行。この図の拡大版を表示するのにはここをクリックしてください。

図 4: FastQC と Seq DNA 品質検証の例です。DNA シーケンス データは、FastQC、シーケンスのパフォーマンスを検証するために送信されました。塩基品質あたり DRR055040 の代表的な結果 (DDBJ シーケンス読み取りアーカイブ DRA00445516) を示します。(A) このパネルは前方の読み取り (R1) を示しています。(B) このパネルは、逆方向の読み取り (R2) を示しています。この図の拡大版を表示するのにはここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
細菌汚染は、微生物のゲノム配列決定に脅威を与えます。Tardigrade ゲノムに関する先行研究を広範な情報学研究の方法12,20を使用して汚染をフィルター処理、一方我々 は汚染のリスクを最小限に抑えるために単一の個人からゲノムをシーケンスです。個々 緩歩動物ゲノム DNA16の約 50-200 pg が含まれているキチン質の外骨格の厚い層で包み込まれているので、汚染物質や高品質の DNA の抽出の排除、このプロトコルでは重要なポイントです。既存のクマムシ文化は、無菌ではありません、それらは、多くの汚染物質の表面だけでなく、自分の腸内に食物残渣野生のキャリーから収集します。以前クマムシのゲノム プロジェクトは、1 つサンプル12,14結果、細菌汚染による影響を受ける可能性が高いことを意味としてまとめて 10,000-100,000 人をシーケンスしました。彼らの報告書、Boothbyらその負の走光性行動14を使用してH. dujardini個人を収集し、グループには抗細菌のすべてのメソッドが採用していなかった。
視覚的に調べるの汚染物質がある場合、我々 は 500 X 顕微鏡下で個人を検討し、抗生物質 (ペニシリン/ストレプトマイシン) でクマムシを培養しました。単一の個々 を分離して慎重に任意の汚染物質の検査で、可能な限り汚染のリスクを最小化。汚染の低水準はよく16としてシーケンス データから確認されました。DNA 抽出に関しては熱均質化18と同様、マニュアルの均質化を採用しました。液体窒素と 37 ° C に個々 の tardigrade を送信すると、亀裂は、キチン質の外骨格で誘導された、換散バッファーは体に浸透し、細胞を溶解することができた。DNA の収量が予想よりも低いとき、は、収率を最大限に熱と手動の均質化を行うことができます。
この資料に記載されている方法には、いくつかの制限があります。まず、凍結融解のサイクルによって均質化は線虫; に関する研究から適用されました。したがって、メソッドは、脱皮動物に対して有効のみです。第二に、DNA シーケンス ライブラリ段階で DNA 断片の増幅、による PCR エラーの可能性は無視できません。したがって、シーケンス データは高精度の読み取りを必要とする分析のため推奨されません (すなわち、 SNP の分析)。さらに、我々 は、プロトコルに記載されている、材料表に示す指定された SNA Seq キットの使用は入力 DNA の低い量のために不可欠。この DNA ライブラリ構築キット郭; 増幅前にイルミナ アダプター シーケンスしたがって、このライブラリは、PacBio やナノ細孔中の技術を使用して長いリード シーケンスを適用できません。最後に、このプロトコルの中に構築された DNA ライブラリの品質チェックはシーケンス ライブラリ構築後 1 回だけ発生します。これは DNA の低入力によりほとんどの DNA の定量化以来、電気泳動方法は、DNA の 50-200 pg を検出できません。そのため、PCR の拡大の後にのみ電気泳動 (図 1) および蛍光ベース数量などの品質チェックを実施しています。
このデータのバイオインフォマティクス解析の詳細については、この記事の範囲を超えて、します。しかし、我々 は簡単にいくつかの分析を行ったを表明しています。FastQC19シーケンス データの品質チェックは、ゲノムのアセンブリにあたりベース資質、シーケンスの重複検証されているなどシーケンス データを送信できる計算します。我々 は MaSuRCA v3.1.321 132 Mb のゲノムを組み立てているし、BWA22と QualiMap23他H. dujardiniゲノムのアセンブリ16この DNA シーケンシング ライブラリの計算されたマッピングの統計情報を比較しています。さらに、また私たちの研究の17、汚染物質の排除のためこの DNA シーケンス データを使用しているし、シーケンスされた読み取り、ゲノム全体に均等に分散される観察しています。
非モデル生物のほとんどのプロジェクトは、クマムシ24症例に十分な試料を培養から開始します。文化技術の技術の進歩は、tardigrade 文化の高い数量を有効にしているが、現在培養法は、まだ、無菌ではなく、ゲノムを実施することは不可能されてほとんどクマムシはまだ labs で培養可能なので、トランスクリプトーム配列。この DNA 配列メソッドは単一の個人からでは、あまり研究されている海洋種を含む希少の tardigrade 種を分析可能です。広い体格エリアで比較ゲノム解析を行い、クマムシの乾燥耐性機構のさらなる理解を達成することがあります。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者が明らかに何もありません。
Acknowledgments
著者はゲノム配列決定でテクニカル サポートのため希阿部、希高井菜穂子石井をありがちましょう。この作業によって支えられた費日本社会のための科学振興会研究員、科研費科研費若手 (No.22681029)、科研費科研費研究 (B)、第 17 H 03620、日本学術振興会からので、基本的な科学研究プロジェクト、住友財団 (No.140340) から、山形県・鶴岡市から研究資金によって部分的に助成。クロレラクマムシをフィードするために使用されたベルフォートは、クロレラ工業 (株)
Materials
| Name | Company | Catalog Number | Comments |
| SZ61 microscope | OLYMPUS | ||
| BactoAgar | Difco Laboratories | 214010 | |
| Penicillin Streptomycin (10,000 U/mL) | Gibco by life technologies | 15140-148 | |
| VHX-5000 System | Keyence | ||
| 0.2mL Silicone coating tube | Bio Medical Science | BC-bmb20200 | |
| Quick-DNA Microprep Kit | ZYMO Research | D3021 | Use of this kit is absolutey critical; see step 3.1 |
| 1.5 mL microtube | greiner bio-one | 616-201 | See 4.1.1 |
| HIgh speed refrigerated micro centrifuge | TOMY | MX-307 | |
| Covaris M220 | Covaris Inc. | 4482277 | |
| ThruPLEX DNA-Seq kit | Rubicon Genomics | CAT. NO. R400406 | Use of this kit is absolutey critical; see step 4.2 |
| Thermal Cycler | Bioer Technology | TC-96GHbC | |
| AMPure XP reagent | BECKMAN COULTER Life Science | A63881 | |
| Ethanol | Wako | 054-027335 | |
| EB buffer | QIAGEN | 19086 | |
| 2200 TapeStation | Agilent | G2965AA | |
| D1000 Reagents | Agilent | 5067-5583 | |
| D1000 ScreenTape | Agilent | 5067-5582 | |
| Qubit dsDNA BR Buffer/Reagent | ThermoFisher Scientific | Q32850 | |
| Cubee Mini-Centrifuge | RecenttecGenereach | R5-AQBD01aqbd | |
| MiSeq 600 cycle v3 | Illumina Inc. | MS-102-3003 | |
| MiSeq Sequencer | Illumina Inc. | SY-410-1003 |
References
- Crowe, J. H., Hoekstra, F. A., Crowe, L. M. Anhydrobiosis. Annual Review of Physiology. 54 (1), 579-599 (1992).
- Mobjerg, N., et al. Survival in extreme environments - on the current knowledge of adaptations in tardigrades. Acta Physiologica. 202 (3), 409-420 (2011).
- Becquerel, P. La suspension de la vieau dessous de 1/20 K absolu par demagnetization adiabatique de L'alun de fer dans le vide les plus eléve. Comptes Rendus de l'Académie des Sciences. 231, 261-264 (1950).
- Ono, F., et al. Effect of ultra-high pressure on small animals, tardigrades and Artemia. Cogent Physics. 3 (1), 1167575 (2016).
- Horikawa, D. D., et al. Tolerance of anhydrobiotic eggs of the Tardigrade Ramazzottius varieornatus to extreme environments. Astrobiology. 12 (4), 283-289 (2012).
- Horikawa, D. D., et al. Analysis of DNA repair and protection in the Tardigrade Ramazzottius varieornatus and Hypsibius dujardini after exposure to UVC radiation. PLoS One. 8 (6), e64793 (2013).
- Horikawa, D. D., et al. Radiation tolerance in the tardigrade Milnesium tardigradum. International Journal of Radiation Biology. 82 (12), 843-848 (2006).
- May, R. M., Maria, M., Gumard, J. Action différentielle des rayons x et ultraviolets sur le tardigrade Macrobiotus areolatus, a L'état actif et desséché. Bulletin Biologique de la France et de la Belgique. 98, 349-367 (1964).
- Jonsson, K. I., Harms-Ringdahl, M., Torudd, J. Radiation tolerance in the eutardigrade Richtersius coronifer. International Journal of Radiation Biology. 81 (9), 649-656 (2005).
- Bemm, F., Weiss, C. L., Schultz, J., Forster, F. Genome of a tardigrade: Horizontal gene transfer or bacterial contamination? Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3054-E3056 (2016).
- Delmont, T. O., Eren, A. M. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ. 4, e1839 (2016).
- Koutsovoulos, G., et al. No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proceedings of the National Academy of Sciences of the United States of America. 113 (18), 5053-5058 (2016).
- Boothby, T. C., Goldstein, B., et al. Reply to Bemm et al. and Arakawa: Identifying foreign genes in independent Hypsibius dujardini genome assemblies. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3058-E3061 (2016).
- Boothby, T. C., et al. Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 112 (52), 15976-15981 (2015).
- Arakawa, K. No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3057 (2016).
- Arakawa, K., Yoshida, Y., Tomita, M. Genome sequencing of a single tardigrade Hypsibius dujardini individual. Scientific Data. 3, 160063 (2016).
- Yoshida, Y., et al. Comparative genomics of the tardigrades Hypsibius dujardini and Ramazzottius varieornatus. PLoS Biology. 15 (7), e2002266 (2017).
- He, F. Total RNA Extraction from C. elegans. Bio-protocol. Bio101, e47 (2011).
- Andrews, S. FastQC a quality-control tool for high-throughput sequence data. , http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2015).
- Hashimoto, T., et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature Communications. 7, 12808 (2016).
- Zimin, A. V., et al. The MaSuRCA genome assembler. Bioinformatics. 29 (21), 2669-2677 (2013).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Okonechnikov, K., Conesa, A., Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32 (2), 292-294 (2016).
- Horikawa, D. D., et al. Establishment of a rearing system of the extremotolerant tardigrade Ramazzottius varieornatus: a new model animal for astrobiology. Astrobiology. 8 (3), 549-556 (2008).

{kind=link}