

Summary
Kontamination während der Genom-Sequenzierung von mikroskopisch kleinen Organismen bleibt ein großes Problem. Hier zeigen wir eine Methode um das Genom der Bärtierchen aus einem einzigen Exemplar mit so wenig wie 50 Pg genomic DNA ohne Verstärkung des gesamten Genoms zur Minimierung des Risikos einer Kontamination zu sequenzieren.
Abstract
Bärtierchen sind mikroskopisch kleine Tiere, die einen Ametabolic Staat eingeben genannt Anhydrobiosis gegenüber Austrocknung und kann in ihren ursprünglichen Zustand zurück, wenn Wasser zugeführt wird. Die Genom-Sequenzierung von mikroskopisch kleinen Tieren wie Bärtierchen Risiken bakterielle Kontamination, die manchmal führt zu falschen Interpretationen, zum Beispiel über den Umfang der horizontalen Gentransfer bei diesen Tieren. Hier bieten wir eine Frequenzverschiebungen Eingabemethode um das Genom der Bärtierchen Hypsibius Dujardini, aus einem einzigen Exemplar zu sequenzieren. Durch den Einsatz von strengen waschen und Schadstoffbelastung Ausgrenzung sowie eine effiziente Extraktion der 50 ~ 200 Pg genomische DNA aus einer einzelnen Person, errichteten wir eine Bibliothek mit einem DNA-Sequenzierung sequenziert. Diese Bibliotheken wurden hoch reproduzierbare und unvoreingenommene und eine Informatik-Analyse des sequenzierten lautet mit anderen H. Dujardini Genome zeigte eine minimale Menge an Verunreinigungen. Diese Methode kann auf unculturable Bärtierchen angewendet werden, die mit bisherigen Methoden nicht sequenziert werden könnte.
Introduction
Bärtierchen sind mikroskopisch kleine Tiere, die einen Ametabolic Staat namens Anhydrobiosis gegenüber Austrocknung eingeben können. Sie wiederherstellen, indem Sie die Absorption von Wasser1,2. Im Ametabolic Zustand sind Bärtierchen in der Lage, verschiedene extreme Umgebungen, die auch extremen Temperaturen3 und Druck4,5, eine hohe Dosierung von UV Licht6, Röntgenstrahlen und Gammastrahlen zu dulden 7 , 8und kosmischen Raum9. Genomdaten ist eine unabdingbare Grundlage für die Erforschung der molekularen Mechanismen der Anhydrobiosis.
Frühere Versuche, das Genom der Bärtierchen sequentiell zeigten Anzeichen einer bakteriellen Kontamination10,11,12,13,14. Genom-Sequenzierung von solchen kleinen Organismen erfordert eine große Anzahl von Tieren und ist anfällig für bakterielle Kontamination; Daher haben wir vorher eine Sequenzierung-Protokoll unter Verwendung einer Frequenzverschiebungen Eingabemethode, ausgehend von einem einzigen Exemplar der Bärtierchen, zur Minimierung des Risikos von Kontaminationen15etabliert. Mithilfe dieser Daten haben wir eine qualitativ hochwertige Neuanordnung und Remontage des Genoms von H. Dujardini16,17weiter durchgeführt. Hier beschreiben wir im Detail dieser Methode für die genomische Sequenzierung von tardigrade einzelnen ()Abbildung 1). Die Validierung dieser Sequenzierung Methode ist darüber hinaus im Mittelpunkt dieses Papiers und ist bereits in unserem vorherigen Bericht16gründlich diskutiert worden.
Diese Methode besteht aus zwei Teilen: die Isolation von einem einzigen Bärtierchen mit möglichst geringen Verunreinigungen und die qualitativ hochwertige Gewinnung von Piktogramm Ebenen der DNA. Die Bärtierchen ist verhungert und gründlich mit Wasser sowie Antibiotika gespült und beobachtet unter dem Mikroskop mit 500 X Vergrößerung um die Beseitigung der bakteriellen Verunreinigungen zu gewährleisten. Frühere Schätzungen und Messungen zeigen, dass ein einzelner Bärtierchen ca. 50-200 Pg genomische DNA-16, enthält, die durch das Chitin-Exoskelett knacken, durch Frost-Tau-wechseln oder durch manuelle Homogenisierung extrahiert wird. Diese genomische DNA ist Bibliotheksbau vorgelegt und auf einem Instrument DNA-Sequenzierung sequenziert. Eine zusätzliche Informatik-Analyse zeigt qualitativ hochwertige Sequenzierung sowie geringe Mengen an Verunreinigungen im Vergleich zu früheren tardigrade Sequenzierung Projekten.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Vorbereitung
- Bereiten Sie 2 % Agarose-Gel mit destilliertem Wasser (DW) als Lösungsmittel in einer 90-mm-Kunststoff-Kulturschale und 10 mL einer 1 % Penicillin/Streptomycin cocktail mit DW. Das Gel kann 2-3 Wochen im Brutkasten gesetzt bei 18 ° c gelagert werden
Hinweis: Vermeiden Sie jede Lagerung des Gels unter 10 ° C, für die niedrige Temperatur schrumpft Agarosegel, was in einem winzigen Spalt zwischen dem Gel und der Kultur Gericht Wand, in der die Bärtierchen gefangen werden können.
2. Probieren Sie Vorbereitung und Schadstoff-Ausschluss
- Ein einzelnes Bärtierchen zu sammeln, legen Sie sie auf die vorbereiteten nährbodenplatte und waschen Sie es 2 x - 3 X mit DW, alle verbleibenden Partikel zu entfernen.
- Inkubieren Sie die Bärtierchen bei 18-22 ° C für 24 h, jede überschüssige Nahrung aus dem Darm zu entfernen.
- Legen Sie die ausgehungerten Bärtierchen in Penicillin/Streptomycin Antibiotika für 2-6 h, bakterielle Verunreinigungen zu entfernen und legen das Dekontaminierte Tier auf eine saubere Folie Glas mit einer Pipette P10.
- Beobachten Sie die Bärtierchen unter dem Mikroskop bei 500 X Vergrößerung und bestätigen Sie, dass es keine verbleibenden Bakterien.
Hinweis: Die hohe Vergrößerung mit einem Stereomikroskop ist optimal, aber ein optisches Mikroskop verwendet werden, alternativ ohne Anwendung ein Deckglas. - Sammeln Sie das Individuum mit einer P10-Pipette mit einem Maximum von 5 µL Flüssigkeit zu, legen Sie sie in ein Low-Bindung Polymerase-Kettenreaktion (PCR) Rohr und entfernen Sie so viel überschüssige Flüssigkeit wie möglich.
Hinweis: Die mikroröhrchen, PCR-Röhrchen und Pipettenspitzen sollte alle geringe Bindung an den Verlust der DNA zu minimieren.
Hinweis: Durchführen eine Homogenisierung so bald wie möglich für die schnelle Austrocknung oder Tod des Tieres kann die genomische DNA erheblich beeinträchtigen.
3. Homogenisierung und DNA-Extraktion
- Das Tier um seine genomische DNA (gDNA) zu erhalten mit einer der folgenden Methoden zu homogenisieren.
Hinweis: Es ist entscheidend für das angegebene Kit gezeigt in der Tabelle der Materialien in der DNA-Extraktion folgendermaßen verwenden für andere Kits (Spalte und Wulst basiert) nicht bei diesen extrem niedrigen Eingang wirksam sind. Die genomische Lyse-Puffer wurde mit 0,5 % Beta-Mercaptoethanol vor dem Gebrauch bereitgestellt.- Das Tier mit Frost-Tau-Zyklen18zu homogenisieren.
- Unmittelbar nach Schritt 2.5, 100 µL der Lyse Puffer hinzu kommt die PCR-Röhrchen mit den Bärtierchen.
- Verschieben Sie das PCR-Rohr in flüssigem Stickstoff für 10 min und 10 min statt ihn auf einem Heizblock auf 37 ° c erwärmt Wiederholen Sie diesen Schritt 2 X.
Hinweis: Dieser Schritt kann wiederholt werden, wenn die Homogenisierung reicht nicht aus oder werden, nachdem die manuelle Zerkleinerung durchgeführt kann.
- Zerdrücken Sie manuell das Tier.
- Unmittelbar nach Schritt 2.5, unter einem Stereomikroskop crush das Individuum mit der P10 Spitze der Pipette durch drücken das Tier gegen die Rohrwandung PCR, und 100 µL der Lyse Puffer sofort hinzufügen.
Hinweis: Es ist wichtig, dieses Verfahren unter einem Stereomikroskop zu beobachten, weil die Bärtierchen leicht wegrutschen kann und ineffiziente Zerkleinerung führt zu mangelnder gDNA extrahieren. Stellen Sie sicher, dass die tardigrade Kutikula gebrochen ist, so dass der Lyse-Puffer des Organismus infiltrieren kann.
- Unmittelbar nach Schritt 2.5, unter einem Stereomikroskop crush das Individuum mit der P10 Spitze der Pipette durch drücken das Tier gegen die Rohrwandung PCR, und 100 µL der Lyse Puffer sofort hinzufügen.
- Das Tier mit Frost-Tau-Zyklen18zu homogenisieren.
- Inkubieren Sie das Rohr für 30 min bei Raumtemperatur für Lyse auftreten.
Hinweis: Ein Minimum von 30 min ist für eine effiziente Lyse erforderlich, aber die Inkubation kann auch länger sein. - Übertragen Sie das komplette Volumen (100 µL) der Lyse Mischung auf eine saubere 1,5 mL niedrig-Bindung reaktionscup.
- Die Low-Bindung-PCR-Röhre, die diente für die Homogenisierung und ist jetzt leer 100 µL der Lyse Puffer hinzu, und nach der Mischung pipettieren, überträgt es auf die 1,5 mL niedrig-Bindung reaktionscup in Schritt 3.3 verwendet. Wiederholen Sie diesen Schritt 2 X.
- Wie in Schritt 3.4 niedrig-Bindung PCR Rohr 300 µL Puffer Lyse hinzu, und nach dem pipettieren, bewegen Sie die Mischung, die 1,5 mL niedrig-Bindung reaktionscup. 1,5 mL niedrig-Bindung reaktionscup sollte nach diesem Schritt 600 µL der Mischung enthalten.
Hinweis: Diese Schritte sind für den Verlust von gDNA gebunden an die Rohrwandung PCR durch Waschen der Probenmaterials mehrmals zu minimieren. - Die Spin-Spalte platziert in einem Sammelrohr insgesamt 600 µL Lyse-Mischung hinzufügen und bei 10.000 x g für 1 min zentrifugieren.
- Erneut bewerben der Durchströmung der Spalte und bei 10.000 x g für 1 min zentrifugieren.
Hinweis: Dieser Schritt ist wichtig, um sicherzustellen, dass ein Großteil der gDNA ist verpflichtet, die Spalte. - Der Spin-Spalte 500 µL Waschpuffer hinzu und Zentrifugieren sie bei 10.000 x g für 1 min. Transfer der Spin-Spalte, um eine saubere 1,5 mL reaktionscup.
- Gelten Sie 20 µL 10 mM Tris-HCl, pH 8,5, bis der Spalte "Spin" Wartezeit für 5 min bei Raumtemperatur und bei 10.000 x g für 1 min zentrifugieren.
Hinweis: Der Elution Buffer muss EDTA, nicht enthalten, denn es die Bibliothek Vorbereitung Enzyme stört. - Den Fluss durch die Spin-Säule und nach 5 min Inkubation bei Raumtemperatur erneut bewerben, für 1 min bei 10.000 x g zentrifugieren.
Hinweis: Dieser Schritt ist entscheidend, um die maximale Elution von den gDNA gesprungen, um die Spalte zu gewährleisten.
(4) Bibliothek Bauablauf
-
DNA-Fragmentierung
- 15 µL gDNA Laufmittel auf einem 15 µL reaktionscup DNA-Fragmentierung übertragen und das Rohr für 1 min mit einer Tischplatte Microcentrifuge Zentrifugieren.
Hinweis: Die angegebenen reaktionscup gezeigt in der Tabelle der Materialien ist optimal für eine low-Input. Die Fragmentierung mit geringem Volumen Beschallung ist von entscheidender Bedeutung, und diese kann nicht wegen der extrem niedrigen Konzentration von DNA mit enzymatische Fragmentierung ersetzt werden. - Fragment der gDNA bis 550 bp.
Hinweis: Die 550 bp-Einstellungen, die wir gewohnt sind wie folgt: einfallende Spitzenleistung = 30 W, Einschaltdauer = 20 %, Zyklen pro platzen = 50, Behandlungszeit = 23 s. - Übertragen Sie nach einer gründlichen pipettieren 10 µL der fragmentierten DNA-Mischung zu einem sauberen Low-Bindung PCR Schlauch
Hinweis: Das Experiment kann hier eingestellt werden. Bewahren Sie die DNA bei 4 ° C oder-20 ° C.
- 15 µL gDNA Laufmittel auf einem 15 µL reaktionscup DNA-Fragmentierung übertragen und das Rohr für 1 min mit einer Tischplatte Microcentrifuge Zentrifugieren.
-
Bibliothek-Bauablauf
Hinweis: Es ist absolut entscheidend für den angegebenen Kit in die Tabelle der Materialien in den folgenden Verfahren aufgrund der geringen Menge von input-DNA zu verwenden.- Fügen Sie 2 µL des Puffers Vorlage Vorbereitung und 1 µL des Enzyms Vorlage Vorbereitung und mischen Sie sie gründlich mit einer Pipette.
- Führen Sie die Vorlage Vorbereitung Reaktion auf ein Thermocycler mit den folgenden Bedingungen: 22 ° C für 25 min, 55 ° C für 20 min, 4 ° C halten und eine beheizte Deckel bei 101-105 ° C. Sobald die Reaktion abgeschlossen ist, fahren Sie mit dem nächsten Schritt fort.
- Das Reaktionsprodukt Vorlage Vorbereitung 1 µL des Puffers Bibliothek Synthese und 1 µL des Synthese Enzyms Bibliothek hinzufügen und die Mischung bei 22 ° C für 40 min. (mit einer 4 ° C halten) inkubieren. Sobald die Reaktion abgeschlossen ist, fahren Sie mit dem nächsten Schritt fort.
- Fügen Sie 30 µL der Bibliothek Verstärkung master-Mix (25 µL des Puffers Bibliothek Verstärkung, 1 µL des Enzyms Bibliothek Verstärkung und 4 µL Nuklease-freies Wasser) und 5 µL der Indizierung Reagenz.
- Mischen Sie alles gründlich mit einer Pipette und Zentrifugieren Sie die Mischung kurz mit einer Tischplatte Zentrifuge.
- Führen Sie eine PCR mit den Bedingungen, die in Tabelle 1dargestellt.
| Temperatur | Zeit | Zyklen |
| 72 ˚C | 3 Minuten | |
| 85 ° C | 2 Minuten | |
| 98 ° C | 2 Minuten | |
| 98 ° C | 20 Sekunden | 4 Zyklen |
| 67 ° C | 20 Sekunden | |
| 72 ˚C | 40 Sekunden | |
| 98 ° C | 20 Sekunden | 16 Zyklen |
| 72 ˚C | 50 Sekunden | |
| 4 ° C | Halten |
Tabelle 1: PCR-Zustände.
-
Reinigung der PCR Produkte
Hinweis: Dieser Schritt kann mit anderen Reinigungsverfahren ersetzt werden. Wenn eine alternative Methode verwendet wird, stellen Sie sicher, für die sich daraus ergebenden DNA Reinheit mit einem Spektralphotometer zu überprüfen. Extinktion Verhältnisse von 260/280 und 260/230 muss sowohl über 1,8.- Fügen Sie 50 µL der magnetischen Beads, pipette die Lösung 10 X, und Zentrifugieren Sie es kurz mit einem Tabletop Microcentrifuge.
- Inkubieren Sie die Lösung für 2 min bei Raumtemperatur.
- Auf einem Magnetstativ für 5 min inkubieren Sie oder bis die Lösung vollständig wird klar, und den überstand zu entfernen.
- Das Low-Bindung PCR Rohr auf einem Magnet Ständer 200 µL von frisch zubereiteten 80 % Ethanol hinzu, warten Sie 30 s und Entfernen der Überstand. Wiederholen Sie diesen Schritt 2 X. Stören Sie die Perlen nicht.
- Kurz Zentrifuge Low-Bindung PCR-Röhrchen mit einer Tischplatte Zentrifuge und entfernen Sie alle überschüssige Ethanol auf die Magnetstativ. Trocknen Sie die Perlen an der Luft, aber vermeiden Sie übertrocknetes.
- Aufschwemmen der Perlen mit 15 µL 10 mM Tris-HCl, pH 8,5, gründlich pipette die Lösung, so dass die magnetische Beads homogen verteilt sind, brüten die Röhre für 2 min bei Raumtemperatur und nach einer kurzen Zentrifugation, inkubieren Sie es auf einem Magnetstativ für 2 min, bis die Lösung klar ist.
Hinweis: Der Elution Puffer sollte EDTA, nicht enthalten, denn es die Sequenzierung Chemie stört. - Übertragen Sie den überstand, ohne zu stören das Pellet, zu einem neuen Low-Bindung PCR Schlauch.
Hinweis: Das Experiment kann hier eingestellt werden. Bewahren Sie die DNA bei 4 ° C oder bei längerer Lagerung-20 ° C.
5. Qualitätsprüfung, Quantifizierung und die DNA-Sequenz
Hinweis: Eine Qualitätsprüfung erfolgt nicht vor diesem Schritt aufgrund der geringen Menge an DNA.
-
Validierung der Größenverteilung DNA-Bibliothek
Hinweis: Andere High-Sensitive-Elektrophorese-Systeme mit einer digitalen Berichterstattung der Größenverteilung können verwendet werden.- Rückkehr der Elektrophorese Puffer Reagenz und gel-Kartusche (25-1.000 bp im Bereich) auf Raumtemperatur.
- Fügen Sie 3 µL Reagenz Elektrophorese Puffer mit 1 µL der Sequenzierung Bibliothek hinzu und mischen Sie sie gründlich für 1 min mit einem Wirbel und Zentrifugieren sie kurz mit einer Tischplatte Zentrifuge.
- Durchführen Sie Elektrophorese und überprüfen Sie die Größenverteilung der Bibliothek mit der zugehörigen Software. Die wichtigsten Fragment Spitze sollte werden weitgehend von bis zu etwa 300 1.000 bp.
-
DNA-Quantifizierung
Hinweis: Andere Fluoreszenz basierende Methoden oder quantitative PCR kann auch verwendet werden, aber Spektralphotometrie sollte vermieden werden, da es nicht genau genug, zur Quantifizierung der Sequenzierung Bibliotheken ist.- 796 µL Lösung Puffer und 4 µL fluoreszierende Reagenz hinzufügen und gründlich mischen. 190 µL Arbeitslösung zu zwei Test-Tubes und 197 µL zu einem Assay Rohr zu verzichten.
- Fügen Sie 10 µL Standards mit bekannten Konzentrationen von DNA zu jedem Assay-Röhrchen mit 190 µL der Arbeitslösung und 3 µL der vorbereiteten Bibliothek an der Assay-Röhrchen mit 197 µL der Arbeitslösung.
- Wirbel die Rohre kurz und Zentrifugieren sie auf einer Tischplatte Zentrifuge. Quantifizieren Sie die DNA mit Hilfe einer Fluorometer mit 3-µL-Einstellungen.
-
DNA-Sequenz Bibliothek
Hinweis: Die Sequenzierung Plattform muss mit der angegebenen Bibliothek Baukasten kompatibel.- Bereiten Sie die Sequenzierung Bibliothek auf der Hersteller-Protokoll basiert.
- Die Sequenzierung Instrument eingelassen Sie die Reagenz Kassette und Flow Zelle und geben Sie die Sequenzierung führen Sie Protokoll des Herstellers nach Informationen.
- Führen Sie die Sequenzierung.
Hinweis: Wir haben zwei Läufe der Sequenzierung durchgeführt: ein Beispiel/ausführen, sowie vier Proben in einem Durchgang gemultiplext.
(6) computergestützte Analyse
- Basis-Call und ggf. demultiplex liest.
- Überprüfen Sie die Qualität von Sequenzdaten mit FastQC19.
Hinweis: Für eine tiefer gehende Validierung der erhaltenen Daten, siehe unsere vorherigen Bericht16.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Schadstoff-Ausschluss:
Dieses Protokoll beinhaltet ein gründliches Waschen der Bärtierchen und eine Sterilisation mit Antibiotika-Behandlung um Kontamination zu minimieren. Dazu gehört auch eine visuelle Überprüfung um die Vollständigkeit dieser Prozesse sicherzustellen. Ein Mikroskopbild gemacht während der Validierung (Schritt 2.4 des Protokolls) ist in Abbildung 2dargestellt. Wenn bei einer 500 X Vergrößerung beobachtet, Bakterienzellen als kleine Partikel ersichtlich, die um die einzelnen Bärtierchen bewegen.
Validierung der DNA Bibliothek Qualität:
Der Gesamtbetrag der konstruierten DNA-Seq-Bibliothek ist ca. 109,5 ng (7.3 ng/µL 15 µL)16. Um die Längenverteilung der Fragmentierung überprüfen, eine Elektrophorese Muster sollte ähnlich sein Abbildung 3. Wie wir die Fragmentierung Größe bis 550 bp mit einer DNA System Scheren, die Bibliothek soll 550-600 bp, einschließlich die Sequenzierung-Adapter. Es ist festzustellen, dass die Mehrheit der Sequenz Bibliothek sich zwischen 200-1000 bp befindet und konsequent auf Wiederholungen (N1 - N4 ist).
Sequenz-Datenanalyse:
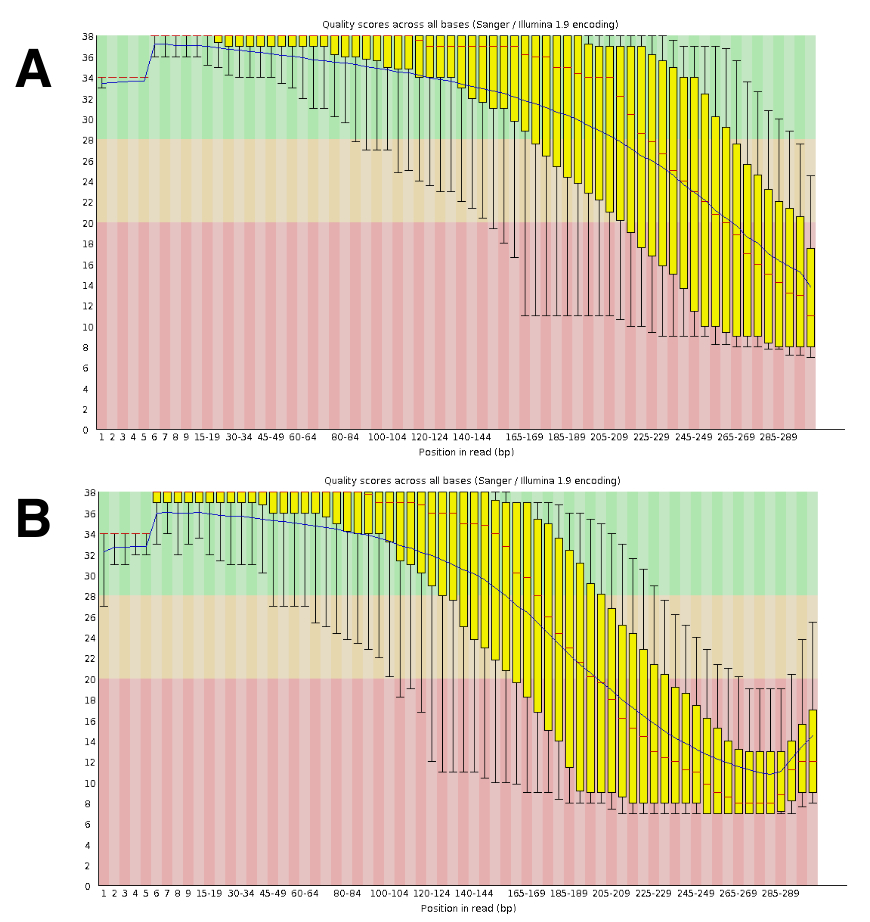
Die DNA-Sequenzierung erzeugt etwa 20 bis 25 M gekoppelten liest pro Durchlauf. Die Validierung der Qualität wurde mit FastQC (Abbildung 4) durchgeführt. Die Verteilung der Qualität entlang der sequenzierten lesen ist typisch für eine 300-bp gekoppelten laufen.

Abbildung 1: Workflow dieses Protokolls. Diese Abbildung zeigt eine Zusammenfassung dieses Protokolls. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2 : Repräsentatives Foto eine keimfreie Bärtierchen. Diese Abbildung zeigt Bilder eines kontaminierten Bärtierchen (links) und gereinigt (rechts) Bärtierchen (Hypsibius Dujardini), zusammen mit weiteren vergrößerte Bilder (unten). Stäbchenförmigen Zellen um die Bärtierchen sind Verunreinigungen und sind mit einem Pfeil gekennzeichnet. Der Maßstab zeigt 100 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 3 : Validierung von dem Fragment Längenverteilung der konstruierten DNA Bibliothek. Dieses Fenster zeigt die Verteilung der Sequenzierung bibliotheksgröße. Die lila und grünen Linien zeigen die oberen und unteren Marker bei 1.500 und 25 bp, beziehungsweise. L = Leiter, S = 1 Probe/Run, N1 - N4 = 4 Wiederholungen/ausführen. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 4 : Beispiel für die Validierung von DNA-Seq-Qualität mit FastQC. DNA-Seq Daten abgegeben FastQC Sequenz Leistung zu beurteilen. Ein repräsentatives Ergebnis für DRR055040 pro basensequenz Qualität ist (DDBJ Reihenfolge lesen Archiv DRA00445516) gezeigt. (A) dieses Panel zeigt nach vorne liest (R1). (B) dieses Panel zeigt die umgekehrte liest (R2). Bitte klicken Sie hier für eine größere Version dieser Figur.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Bakterielle Kontamination stellt eine Bedrohung für die Genom-Sequenzierung von mikroskopischen Organismen. Während frühere Studien zu tardigrade Genomsequenzierung, Kontamination mit umfangreichen Informatik Methoden12,20herausgefiltert haben, sequenziert wir das Genom von einer einzelnen Person das Risiko von Kontaminationen zu minimieren. Da eine einzelne Bärtierchen enthält ungefähr 50-200 Pg genomische DNA16 und ist umhüllt von einer dicken Schicht aus Chitin Exoskelett, der Ausschluss von Verunreinigungen und qualitativ hochwertige DNA-Extraktion sind die kritischen Punkte in diesem Protokoll. Bestehende Bärtierchen Kulturen sind nicht aseptisch, und diejenigen aus der wild tragen eine Menge von Verunreinigungen auf der Oberfläche, als auch die Reste der Nahrung in den Darm gesammelt. Vorherigen Genom-Sequenzierung-Projekte der Bärtierchen haben 10.000-100.000 Personen gemeinsam als eine Probe12,14, sequenziert, was, dass die Ergebnisse sehr wahrscheinlich bedeutet beeinflusst werden durch bakterielle Verunreinigungen sind. In ihrem Bericht Boothby Et al. H. Dujardini Einzelpersonen mithilfe ihrer negative Phototaxis Verhalten14gesammelt, und die Gruppe keine Anti-bakteriellen Methoden beschäftigen.
Um visuell zu prüfen gibt es Verunreinigungen, wir die Bärtierchen in Antibiotika (Penicillin/Streptomycin) inkubiert und die einzelnen unter dem 500 X Mikroskop untersucht. Durch eine einzelne Person zu isolieren und sorgfältig inspizieren es für sämtliche Verunreinigungen minimiert wir das Risiko von möglichen Verunreinigungen. Geringe Mengen an Verunreinigungen wurden aus den Sequenzierungsdaten als gut16bestätigt. Wie DNA-Extraktion beschäftigten wir manuelle Homogenisierung, sowie thermische Homogenisierung18. Mit dem Absenden der tardigrade einzelne Flüssigstickstoff und 37 ° C, Risse wurden in das Chitin-Exoskelett induziert, und der Lyse Puffer war in der Lage, in den Körper eindringen und lösen Sie die Zellen. Wenn die DNA-Ausbeute niedriger bleibt als erwartet, kann sowohl thermische als auch manuelle Homogenisierung durchgeführt werden, um den Ertrag zu maximieren.
Die Methode in diesem Artikel genannten hat mehrere Einschränkungen. Zunächst wurde aus einer Studie über Nematoden Homogenisierung durch Frost-Tau-Zyklen; So kann die Methode nur wirksam gegen Ecdysozoa sein. Zweitens kann nicht durch die Verstärkung von DNA-Fragmenten in der DNA-Sequenz Bibliothek Phase, PCR-Fehler ignoriert werden. Damit die Sequenzdaten empfiehlt sich nicht für die Analyse, die hochpräzise liest erfordert (d.h., SNP-Analyse). Darüber, wie wir im Protokoll erwähnt haben, ist die Verwendung des angegebenen SNA-Seq Kit gezeigt in der Tabelle der Materialien absolut kritisch, aufgrund der geringen Menge von input-DNA. Diese DNA-Bibliothek-Baukasten ligates Illumina Adapter Sequenzen vor der Verstärkung; Daher kann diese Bibliothek für lange lesende Sequenzierung PacBio oder Nanopore Technologie angewendet werden. Schließlich tritt eine Qualitätsprüfung der konstruierten DNA Bibliothek während dieses Protokoll nur einmal nach der Sequenzierung Bibliotheksbau. Dies ist aufgrund der low-Input von DNA seit den meisten DNA-Quantifizierung und Elektrophorese Methoden können nicht erkennen, 50-200 Pg DNA. Daher führten wir Qualitäts-Checks, wie z. B. die Elektrophorese (Abbildung 1) und Fluoreszenz-basierte Quantifizierungen, erst nach der PCR-Amplifikation.
Eine vollständige Beschreibung der Bioinformatik Analysen dieser Daten sprengt den Rahmen dieses Artikels; Wir haben jedoch mehrere Analysen, die wir durchgeführt haben, kurz gesagt. Eine Qualitätsprüfung der Sequenzierungsdaten mit FastQC19 berechnet, dass die pro-Base Qualitäten, Sequenz Vervielfältigung, etc. -Sequenzdaten, die validiert wurden die Genom-Versammlung vorgelegt werden können. Wir haben eine 132 Mb Genom mit MaSuRCA v3.1.321 zusammengestellt und haben im Vergleich der Mapping-Statistiken mit BWA22 und QualiMap23 dieser DNA-Sequenzierung-Bibliothek mit anderen H. Dujardini Genom Baugruppen16berechnet. Darüber hinaus wir auch habe diese DNA-Sequenzierung-Daten für den Ausschluss von Verunreinigungen in unserer Studie17und habe beobachtet, dass die sequenzielle Lesevorgänge des Genoms gleichmäßig verteilt sind.
Die meisten Projekte auf nicht-Modellorganismen starten von Kultivierung genügend Probenmaterial wie bei Bärtierchen24der Fall war. Technische Fortschritte in der Kulturtechniken haben hohe Mengen an tardigrade Kultur, ermöglicht aber aktuelle Kulturmethoden noch nicht aseptische und da die meisten Bärtierchen noch unculturable in den Labors sind, wurde es fast unmöglich, Genom durchzuführen oder Transkriptom Sequenzierung. Diese DNA-Sequenzierung-Methode aus einer einzelnen Person macht es möglich, seltene tardigrade Arten, einschließlich der marine Arten, die weniger untersucht wurden zu analysieren. Durch die Durchführung von Vergleichende Genomforschung an ein größeres phyletic Gebiet, kann eine weitere Verständnis der Anhydrobiosis Mechanismen in Bärtierchen erreicht werden.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben nichts preisgeben.
Acknowledgments
Die Autoren danken Nozomi Abe, Yuki Takai und Nahoko Ishii für ihre technische Unterstützung bei der Genom-Sequenzierung. Diese Arbeit wurde unterstützt von Beihilfe für die japanische Gesellschaft für Promotion of Science (JSPS) Research Fellow, KAKENHI Beihilfe für junge Wissenschaftler (No.22681029) und KAKENHI Beihilfe für wissenschaftliche Forschung (B), Nr. 17 H 03620 von JSPS, durch eine Zuschuss für grundlegende Wissenschaft Forschungsprojekte der Sumitomo Foundation (No.140340) und andererseits durch Forschungsgelder von der Regierung der Präfektur Yamagata und Tsuruoka City, Japan. Chlorella Vulgaris verwendet, um die Bärtierchen ernähren wurde mit freundlicher Genehmigung von Chlorella Industry Co. LTD. zur Verfügung gestellt.
Materials
| Name | Company | Catalog Number | Comments |
| SZ61 microscope | OLYMPUS | ||
| BactoAgar | Difco Laboratories | 214010 | |
| Penicillin Streptomycin (10,000 U/mL) | Gibco by life technologies | 15140-148 | |
| VHX-5000 System | Keyence | ||
| 0.2mL Silicone coating tube | Bio Medical Science | BC-bmb20200 | |
| Quick-DNA Microprep Kit | ZYMO Research | D3021 | Use of this kit is absolutey critical; see step 3.1 |
| 1.5 mL microtube | greiner bio-one | 616-201 | See 4.1.1 |
| HIgh speed refrigerated micro centrifuge | TOMY | MX-307 | |
| Covaris M220 | Covaris Inc. | 4482277 | |
| ThruPLEX DNA-Seq kit | Rubicon Genomics | CAT. NO. R400406 | Use of this kit is absolutey critical; see step 4.2 |
| Thermal Cycler | Bioer Technology | TC-96GHbC | |
| AMPure XP reagent | BECKMAN COULTER Life Science | A63881 | |
| Ethanol | Wako | 054-027335 | |
| EB buffer | QIAGEN | 19086 | |
| 2200 TapeStation | Agilent | G2965AA | |
| D1000 Reagents | Agilent | 5067-5583 | |
| D1000 ScreenTape | Agilent | 5067-5582 | |
| Qubit dsDNA BR Buffer/Reagent | ThermoFisher Scientific | Q32850 | |
| Cubee Mini-Centrifuge | RecenttecGenereach | R5-AQBD01aqbd | |
| MiSeq 600 cycle v3 | Illumina Inc. | MS-102-3003 | |
| MiSeq Sequencer | Illumina Inc. | SY-410-1003 |
References
- Crowe, J. H., Hoekstra, F. A., Crowe, L. M. Anhydrobiosis. Annual Review of Physiology. 54 (1), 579-599 (1992).
- Mobjerg, N., et al. Survival in extreme environments - on the current knowledge of adaptations in tardigrades. Acta Physiologica. 202 (3), 409-420 (2011).
- Becquerel, P. La suspension de la vieau dessous de 1/20 K absolu par demagnetization adiabatique de L'alun de fer dans le vide les plus eléve. Comptes Rendus de l'Académie des Sciences. 231, 261-264 (1950).
- Ono, F., et al. Effect of ultra-high pressure on small animals, tardigrades and Artemia. Cogent Physics. 3 (1), 1167575 (2016).
- Horikawa, D. D., et al. Tolerance of anhydrobiotic eggs of the Tardigrade Ramazzottius varieornatus to extreme environments. Astrobiology. 12 (4), 283-289 (2012).
- Horikawa, D. D., et al. Analysis of DNA repair and protection in the Tardigrade Ramazzottius varieornatus and Hypsibius dujardini after exposure to UVC radiation. PLoS One. 8 (6), e64793 (2013).
- Horikawa, D. D., et al. Radiation tolerance in the tardigrade Milnesium tardigradum. International Journal of Radiation Biology. 82 (12), 843-848 (2006).
- May, R. M., Maria, M., Gumard, J. Action différentielle des rayons x et ultraviolets sur le tardigrade Macrobiotus areolatus, a L'état actif et desséché. Bulletin Biologique de la France et de la Belgique. 98, 349-367 (1964).
- Jonsson, K. I., Harms-Ringdahl, M., Torudd, J. Radiation tolerance in the eutardigrade Richtersius coronifer. International Journal of Radiation Biology. 81 (9), 649-656 (2005).
- Bemm, F., Weiss, C. L., Schultz, J., Forster, F. Genome of a tardigrade: Horizontal gene transfer or bacterial contamination? Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3054-E3056 (2016).
- Delmont, T. O., Eren, A. M. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ. 4, e1839 (2016).
- Koutsovoulos, G., et al. No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proceedings of the National Academy of Sciences of the United States of America. 113 (18), 5053-5058 (2016).
- Boothby, T. C., Goldstein, B., et al. Reply to Bemm et al. and Arakawa: Identifying foreign genes in independent Hypsibius dujardini genome assemblies. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3058-E3061 (2016).
- Boothby, T. C., et al. Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 112 (52), 15976-15981 (2015).
- Arakawa, K. No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3057 (2016).
- Arakawa, K., Yoshida, Y., Tomita, M. Genome sequencing of a single tardigrade Hypsibius dujardini individual. Scientific Data. 3, 160063 (2016).
- Yoshida, Y., et al. Comparative genomics of the tardigrades Hypsibius dujardini and Ramazzottius varieornatus. PLoS Biology. 15 (7), e2002266 (2017).
- He, F. Total RNA Extraction from C. elegans. Bio-protocol. Bio101, e47 (2011).
- Andrews, S. FastQC a quality-control tool for high-throughput sequence data. , http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2015).
- Hashimoto, T., et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature Communications. 7, 12808 (2016).
- Zimin, A. V., et al. The MaSuRCA genome assembler. Bioinformatics. 29 (21), 2669-2677 (2013).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Okonechnikov, K., Conesa, A., Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32 (2), 292-294 (2016).
- Horikawa, D. D., et al. Establishment of a rearing system of the extremotolerant tardigrade Ramazzottius varieornatus: a new model animal for astrobiology. Astrobiology. 8 (3), 549-556 (2008).

{kind=link}