

Summary
Contaminación durante la secuenciación de organismos microscópicos sigue siendo un gran problema. A continuación, os mostramos un método para secuenciar el genoma de un tardígrado de una sola muestra con tan poco como 50 pg de ADN genómico sin amplificación del genoma entero para minimizar el riesgo de contaminación.
Abstract
Tardígrados son animales microscópicos que entran en un estado ametabólico anhydrobiosis llamados frente a la desecación y puede volver a su estado original cuando se suministra agua. La secuenciación genómica de los animales microscópicos como la contaminación bacteriana de tardígrados riesgos que a veces conduce a interpretaciones erróneas, por ejemplo, en relación con el grado de transferencia horizontal del gene en estos animales. Aquí, ofrecemos un método de entrada ultrabajo para secuenciar el genoma de tardígrado, Hypsibius dujardini, de una sola muestra. Empleando la rigurosa exclusión de lavado y contaminante junto con una extracción eficiente del 50 ~ 200 pg de ADN genómico de una sola persona, construimos una biblioteca ordenada con un instrumento de la secuencia de ADN. Estas bibliotecas fueron altamente reproducible y objetiva, y un análisis de informática de la Lee secuenciado con otros genomas de H. dujardini mostró un mínimo de contaminación. Este método puede aplicarse a unculturable tardígrados que no podrían ser secuenciados utilizando los métodos anteriores.
Introduction
Tardígrados son animales microscópicos que pueden entrar en un estado ametabólico llamado anhydrobiosis frente a la desecación. Se recuperan por la absorción de agua1,2. En el estado de ametabólica, los tardígrados son capaces de tolerar ambientes extremos distintos, que incluyen temperaturas extremas3 y presiones de4,5, una alta dosis de luz ultravioleta6, rayos x y rayos gamma 7 , 8y9de espacio cósmico. Datos genómicos están un fundamento indispensable para el estudio de mecanismos moleculares de anhydrobiosis.
Los intentos anteriores para secuenciar el genoma de tardígrados han mostrado señales de contaminación bacteriana10,11,12,13,14. Secuenciación de estos pequeños organismos requiere una gran cantidad de animales y es propenso a la contaminación bacteriana; por lo tanto, previamente hemos establecido un protocolo de secuenciación utilizando un método ultra bajo de la entrada a partir de una única muestra de tardígrado, para minimizar el riesgo de contaminación15. Con estos datos, hemos realizado más un resecuenciación de alta calidad y el ensamblado del genoma de H. dujardini16,17. Aquí describimos en detalle este método de secuenciación de un solo individuo tardigrade ()figura 1). La validación de este método de secuenciación es más allá del enfoque de este trabajo y ya se ha discutido exhaustivamente en nuestro anterior informe16.
Este método se compone de dos partes: el aislamiento de un tardígrado solo con el más bajo posible de la contaminación y la extracción de alta calidad de niveles de pictograma de ADN. El tardígrado es hambre y enjuagarse con agua, así como antibióticos y observado bajo un microscopio con 500 aumentos para asegurar la eliminación de cualquier contaminación bacteriana. Mediciones y estimaciones previas muestran que un solo individuo de tardígrado contiene aproximadamente 50-200 pg de genomic DNA16, que se extrae por agrietar el exoesqueleto de quitina por ciclos de hielo-deshielo u homogeneización manual. Este ADN genómico es enviado a la construcción de la biblioteca y ordenado en un instrumento de la secuencia de ADN. Un análisis de informática adicional muestra la secuencia de alta calidad, así como bajos niveles de contaminación en comparación con anteriores proyectos de secuenciación tardigrade.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. preparación
- Preparar el gel de agarosa al 2% con agua destilada (DW) como solvente en una placa de cultivo de plástico de 90 mm y 10 mL de un 1% penicilina/estreptomicina cóctel con DW. El gel puede almacenarse durante 2-3 semanas en una incubadora a 18 ° C.
Nota: Evite cualquier almacenamiento de información de gel por debajo de 10 ° C, para la baja temperatura reducirá el gel de agarosa, resultando en un espacio minúsculo entre el gel y la pared del plato de cultura en la que los tardígrados pueden quedar atrapados.
2. muestra preparación y la exclusión de contaminantes
- Recoger un tardígrado sola, coloque sobre la placa preparada de agar y lavarlo 2 x - 3 x con DW para eliminar cualquier partícula restante.
- Incubar el tardígrado a 18-22 ° C durante 24 h para eliminar cualquier exceso de comida de los intestinos.
- Coloque el tardígrado hambrienta en antibióticos de penicilina/estreptomicina durante 2-6 h para retirar cualquier contaminación bacteriana y colocar el animal descontaminado en un vidrio portaobjetos limpio con una pipeta P10.
- Observar el tardígrado bajo un microscopio con 500 aumentos y confirmar que no hay ningunas bacterias restantes.
Nota: La alta magnificación con un estereomicroscopio es óptima, pero un microscopio óptico puede utilizarse alternativamente sin aplicar un cubreobjetos. - Recoger al individuo utilizando una pipeta P10 con un máximo de 5 μl de líquido, colóquelo en un tubo de reacción en cadena (PCR) polimerasa de baja obligatoria y eliminar todo el exceso de líquido posible.
Nota: Los microtubos, tubos PCR y las puntas de pipeta todos debe enlace baja para reducir al mínimo la pérdida de ADN.
Nota: Realizar una homogeneización tan pronto como sea posible, para la rápida desecación o muerte del animal puede dañar significativamente la DNA genomic.
3. homogeneización y extracción de ADN
- Homogeneizar el animal para obtener su ADN genómico (gDNA) con uno de los métodos siguientes.
Nota: Es fundamental para utilizar el equipo especificado que se muestra en la Tabla de materiales en los siguientes pasos de extracción de ADN, para otros equipos (ya sea columna y base del grano) no son eficaces en esta entrada muy baja. El tampón de lisis genómica ha sido suministrado con beta-mercaptoetanol 0,5% antes del uso.- Homogeneizar el animal con ciclos de congelación y descongelación18.
- Inmediatamente después de paso 2.5, añadir 100 μl de tampón de lisis en el tubo PCR que contiene el tardígrado.
- Coloque el tubo PCR en nitrógeno líquido durante 10 minutos y, durante 10 minutos, mover a un bloque de calor calentado a 37 ° C. Repetir este paso 2 x.
Nota: Este paso puede repetirse cuando la homogeneización no es suficiente o puede realizarse después de la trituración manual.
- Machacar manualmente el animal.
- Inmediatamente después de paso 2.5, bajo un estereomicroscopio, aplastar al individuo con la punta de la pipeta de P10 presionando el animal contra la pared del tubo PCR y añadir 100 μl de tampón de lisis.
Nota: Es fundamental observar este procedimiento bajo un estereomicroscopio, porque fácilmente puede escaparse el tardígrado, y machacando ineficiente dará como resultado un fracaso para extraer gDNA. Asegúrese de que la cutícula tardigrade es rota para que el tampón de lisis puede infiltrarse en el organismo.
- Inmediatamente después de paso 2.5, bajo un estereomicroscopio, aplastar al individuo con la punta de la pipeta de P10 presionando el animal contra la pared del tubo PCR y añadir 100 μl de tampón de lisis.
- Homogeneizar el animal con ciclos de congelación y descongelación18.
- Incubar el tubo durante 30 min a temperatura ambiente por lisis ocurrir.
Nota: Un mínimo de 30 minutos es necesario para una lisis eficiente, pero la incubación puede ser más larga. - Transferir el volumen completo (100 μL) de la mezcla de lisis en un microtubo de enlace de baja limpio de 1,5 mL.
- Añadir 100 μl de tampón de lisis en el tubo PCR bajo Unión que fue usado para la homogeneización y ahora está vacía y después de haber efectuado la mezcla, ponerlo en el microtubo de enlace de baja 1,5 mL utilizado en el paso 3.3. Repetir este paso 2 x.
- Como en el paso 3.4, añadir 300 μL de tampón de lisis en el tubo PCR de baja obligatoria, y, después de su uso, pasar la mezcla a los microtubos de 1,5 mL enlace de baja. Los microtubos de 1,5 mL baja unión deben contener 600 μl de la mezcla después de este paso.
Nota: Estos pasos son minimizar la pérdida de gDNA atado a la pared del tubo PCR, lavando la muestra varias veces. - Añadir el total de 600 μl de mezcla de lisis a la columna de vuelta colocada en un tubo de colección y centrifugar a 10.000 x g durante 1 minuto.
- Vuelva a aplicar el flujo a través de la columna y centrifugar a 10.000 x g durante 1 minuto.
Nota: Este paso es vital para asegurar que la mayoría de lo gDNA está enlazado a la columna. - Añadir 500 μl de tampón de lavado a la columna de spin y centrifugar a 10.000 x g durante 1 minuto transferencia la columna de la vuelta a un microtubo limpio de 1,5 mL.
- Aplicar 20 μl de 10 mM Tris-HCl, pH 8,5, a la columna de centrifugado, espere 5 minutos a temperatura ambiente y centrifugar a 10.000 x g durante 1 minuto.
Nota: El tampón de elución no debe contener EDTA, porque interfiere con las enzimas de la preparación de biblioteca. - Vuelva a aplicar el flujo a través de la columna de vuelta y después de 5 minutos de incubación a temperatura ambiente, centrifugue durante 1 min a 10.000 x g.
Nota: Este paso es crítico para asegurar la máxima elución del gDNA enlazado a la columna.
4. Biblioteca construcción secuencia
-
Fragmentación del ADN
- Transferencia 15 μl del eluant gDNA en un microtubo de 15 μl de fragmentación del ADN y centrifugar el tubo durante 1 min, utilizando una microcentrífuga de sobremesa.
Nota: El microtubo especificado que se muestra en la Tabla de materiales es óptimo para una entrada. La fragmentación con volumen bajo sonicación es fundamental, y esto no se puede sustituir con la fragmentación enzimática debido a la muy baja concentración de ADN. - Fragmento del gDNA a 550 bp.
Nota: El 550 bp hemos utilizado con los siguientes parámetros: energía incidente máxima = 30 W, factor de servicio = 20%, ciclos por explosión = 50, tiempo de tratamiento = 23 s. - Después de un cuidadoso pipeteado, transferir 10 μl de la mezcla de ADN fragmentada a un tubo PCR limpia baja obligatoria.
Nota: El experimento se puede parar aquí. Preservar el ADN a 4 º C ó -20 ° C.
- Transferencia 15 μl del eluant gDNA en un microtubo de 15 μl de fragmentación del ADN y centrifugar el tubo durante 1 min, utilizando una microcentrífuga de sobremesa.
-
Secuencia de construcción de biblioteca
Nota: Es imprescindible utilizar el juego especificado en la Tabla de materiales en los siguientes procedimientos, debido a la baja cantidad de ADN de entrada.- Añadir 2 μl de la solución de la preparación de plantilla y 1 μl de la enzima de preparación de la plantilla y homogeneizar con una pipeta.
- Realizar la reacción de preparación de la plantilla en un termociclador con las siguientes condiciones: 22 ° C por 25 min, 55 ° C por 20 min, hold de 4 ° C y una tapa caliente en 101-105 ° C. Una vez completada la reacción, proceder al siguiente paso.
- Añadir 1 μl del buffer de síntesis biblioteca y 1 μl de la enzima de síntesis de biblioteca a producto de la reacción de preparación de plantilla e incubar la mezcla a 22 ° C durante 40 minutos (con una espera de 4 ° C). Una vez completada la reacción, proceder al siguiente paso.
- Añadir 30 μl de la mezcla principal biblioteca amplificación (25 μl del buffer de amplificación de biblioteca, 1 μl de la enzima de la amplificación de la biblioteca y 4 μL de agua libre de nucleasas) y 5 μl del reactivo de indexación.
- Mezclar todo bien con una pipeta y centrifugar brevemente la mezcla con una centrífuga de mesa.
- Realizar una PCR con las condiciones presentadas en la tabla 1.
| Temperatura | Tiempo | Ciclos de |
| 72 ° C | 3 minutos | |
| 85 ° C | 2 minutos | |
| 98 ° C | 2 minutos | |
| 98 ° C | 20 segundos | 4 ciclos |
| 67 ° C | 20 segundos | |
| 72 ° C | 40 segundos | |
| 98 ° C | 20 segundos | 16 ciclos |
| 72 ° C | 50 segundos | |
| 4 ° C | Mantenga |
Tabla 1: Condiciones PCR.
-
Purificación de los productos PCR
Nota: Este paso puede ser sustituido con otros métodos de purificación. Si se utiliza un método alternativo, asegúrese de comprobar la pureza de ADN resultante usando un espectrofotómetro. Relación de absorbancia 260/280 y 260/230 debe estar por encima de 1,8.- Añadir 50 μl de bolas magnéticas, pipetear la solución 10 x y centrifugar brevemente con una microcentrífuga de sobremesa.
- Incubar la solución por 2 min a temperatura ambiente.
- Incubar en un soporte magnético para 5 min o hasta que la solución esté completamente claro y eliminar el sobrenadante.
- Añadir 200 μL de etanol al 80% recién preparada para el tubo PCR de baja obligatoria en un soporte de imán, esperar 30 s y eliminar el sobrenadante. Repetir este paso 2 x. No molestes a los granos.
- Brevemente Centrifugue el tubo PCR de baja obligatoria con una centrifugadora de mesa y retire cualquier exceso etanol en soporte magnético. Secar los granos, pero evitar el secado excesivo.
- Resuspender los granos con 15 μl de 10 mM Tris-HCl, pH 8.5 Fondo pipetear la solución para que los granos magnéticos se distribuyen homogéneamente, incubar el tubo durante 2 min a temperatura ambiente y después de una breve centrifugación, incubar en un soporte magnético de 2 min hasta que la solución es clara.
Nota: El tampón de elución no debe contener EDTA, pues interfiere con la química de secuenciación. - Transferir el sobrenadante, sin perturbar el pellet, a un nuevo tubo PCR de baja obligatoria.
Nota: El experimento se puede parar aquí. Preservar el ADN a 4 º C o a-20 ° C para el almacenamiento de largo plazo.
5. calidad verificación, cuantificación y secuencia de la DNA
Nota: No se realiza un control de calidad antes de este paso debido a la baja cantidad de ADN.
-
Validación de la distribución del tamaño de ADN biblioteca
Nota: Pueden utilizarse otros sistemas de electroforesis de alta sensibilidad con una divulgación digital de distribución de tamaño.- Regresa el agente reactivo tampón de electroforesis y gel de cartucho (25-1.000 bp en gama) a temperatura ambiente.
- Agregar 3 μl del reactivo del tampón de electroforesis con 1 μl de la biblioteca de la secuencia y mezclar bien durante 1 minuto con un vórtex y brevemente de centrífuga con una centrífuga de mesa.
- Realizar la electroforesis y validar la distribución de tamaño de la biblioteca con el software asociado. El pico principal del fragmento debe ser ampliamente desde cerca de 300 a 1.000 bp.
-
Cuantificación de ADN
Nota: Otros métodos basados en fluorescencia o PCR cuantitativa puede también ser utilizado, pero espectrofotometría debe evitarse, ya que no es lo suficientemente precisa como para la cuantificación de las bibliotecas de la secuencia.- Añadir 796 μl de solución tampón y 4 μL de reactivo fluorescente y mezclar bien. Dispensar 190 μl de la solución de trabajo a los dos tubos de ensayo y 197 μl a un tubo de ensayo.
- Añada 10 μl de estándares con concentraciones conocidas de ADN en cada tubo de ensayo que contiene 190 μl de la solución de trabajo y 3 μl de la biblioteca preparada para el tubo de ensayo que contiene 197 μl de la solución de trabajo.
- Vortex brevemente los tubos y centrifugar en una centrifugadora de mesa. Cuantificar el ADN utilizando un fluorómetro con configuración de 3 μl.
-
Secuencia de ADN biblioteca
Nota: La plataforma de secuenciación debe ser compatible con el kit de construcción de la biblioteca especificada.- Preparar la biblioteca de secuencia basada en el protocolo del fabricante.
- Establecer la célula del cassette y del flujo de reactivos en el instrumento de la secuencia y entrar en la secuencia de funcionamiento información siguiendo el protocolo del fabricante.
- Ejecutar la secuencia.
Nota: Hemos realizado dos carreras de la secuencia: una muestra/carrera, así como cuatro muestras multiplexadas en una carrera.
6. Computación Análisis
- Base de llamada y, si es necesario, demultiplexar la Lee.
- Validar la calidad de los datos de la secuencia con FastQC19.
Nota: Para una validación más profunda de los datos obtenidos, véase nuestro anterior informe16.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Exclusión de contaminantes:
Este protocolo trata de un lavado exhaustivo de una esterilización con tratamiento de antibióticos para minimizar la contaminación y el tardígrado. También implica un proceso de control visual para asegurar la integridad de estos procesos. Una imagen de microscopio en la validación (paso 2.4 del Protocolo) se muestra en la figura 2. Cuando se observa un aumento de X 500, las células bacterianas pueden verse como pequeñas partículas que se mueven alrededor del tardígrado individual.
Validación de la calidad de la biblioteca de ADN:
La cantidad total de la biblioteca de DNA Seq construida es aproximadamente 109,5 ng (7,3 ng/μl x 15 μl)16. Para validar la distribución de la longitud de la fragmentación, un patrón de electroforesis debe ser similar a la figura 3. Como establecemos el tamaño de la fragmentación a 550 bp con un sistema de corte de ADN, la biblioteca debe ser bp de 550-600, incluyendo los adaptadores de secuencia. Se puede observar que la mayoría de la biblioteca de secuencia está contenida entre 200-1000 bp y es coherente entre repeticiones (N1 - N4).
Análisis de datos la secuencia:
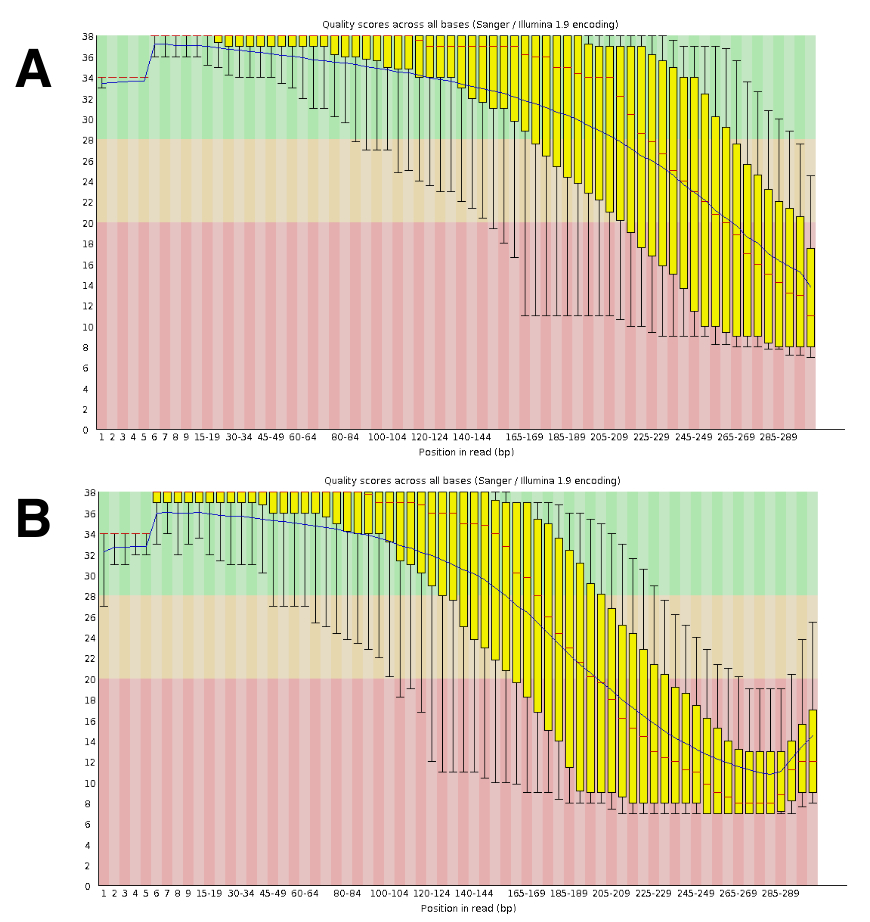
La secuencia de la DNA genera lecturas emparejadas aproximadamente 20 a 25 M por ejecutar. La validación de la calidad se realizó con FastQC (figura 4). La distribución de la calidad a lo largo de la lectura secuencial es típica de un funcionamiento sincronizado de 300 bp.

Figura 1: flujo de trabajo de este protocolo. Esta figura muestra un resumen de este protocolo. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2 : Foto representativa de un tardígrado libre de bacterias. Esta figura muestra imágenes de un tardígrado (izquierda) contaminado y tardígrado (derecha) limpio (Hypsibius dujardini), junto con más imágenes magnificadas (abajo). Las células bastoncillos alrededor el tardígrado son contaminantes y se indican con una flecha. La barra de escala indica 100 μm. haga clic aquí para ver una versión más grande de esta figura.

Figura 3 : Validación de la distribución de longitud de fragmento de la biblioteca de ADN construida. Este panel muestra la distribución del tamaño de la biblioteca de la secuencia. Las líneas púrpura y verdes indican los marcadores superiores e inferiores a 1.500 y 25 PB, respectivamente. L = escalera, S = 1 muestra/run, N1 - N4 = 4 repeticiones y correr. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4 : Ejemplo de la validación de la calidad del ADN-Seq con FastQC. Datos de DNA Seq fueron sometidos a FastQC para validar el rendimiento de la secuencia. Un resultado representativo para DRR055040 por la calidad de la secuencia de bases se muestra (DDBJ secuencia leer archivo DRA00445516). (A) este panel muestra las lecturas avance (R1). (B) este panel muestra la lectura inversa (R2). Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
La contaminación bacteriana es una amenaza para la secuenciación de organismos microscópicos. Mientras que estudios anteriores sobre la secuenciación del genoma tardigrade han filtrado contaminación empleando métodos de informática amplia12,20, hemos secuenciado el genoma de un solo individuo para minimizar el riesgo de contaminación. Puesto que un tardígrado individual contiene aproximadamente 50-200 pg de ADN genómico16 y es encerrado en una gruesa capa de exoesqueleto de quitina, la exclusión de contaminantes y extracción de ADN de alta calidad son los puntos críticos de este protocolo. Culturas de tardígrados existentes no son asépticas y los recogidos de los salvaje llevan una gran cantidad de contaminantes en la superficie, así como los restos de comida en sus intestinos. Anteriores proyectos de secuenciación de genoma de tardígrados han secuenciado a 10.000-100.000 individuos colectivamente como una muestra de12,14, que significa que los resultados son muy probables ser influenciado por bacterias contaminantes. En su informe, Boothby et al. recogidos a individuos de H. dujardini usando su comportamiento negativo phototaxis del14, y el grupo de no emplear a ningún método anti-bacterial.
Para examine visualmente si hay contaminantes, se incubó el tardígrado de antibióticos (penicilina/estreptomicina) y examinó al individuo bajo un microscopio de X 500. Al aislar a un solo individuo y cuidadosamente la inspección de cualquier contaminantes, reducen al mínimo el riesgo de posibles contaminaciones. Bajos niveles de contaminación fueron confirmados de los datos de secuenciación como bien16. En cuanto a la extracción de ADN, se empleó la homogeneización manual, así como la homogenización térmica18. Sometiendo al individuo tardigrade nitrógeno líquido y 37 ° C, grietas fueron inducidas en el exoesqueleto de quitina, y el tampón de lisis fue capaz de penetrar el cuerpo y lisar las células. Cuando el rendimiento de ADN sigue siendo inferior al previsto, homogeneización térmica y manual puede llevarse a cabo para maximizar el rendimiento.
El método indicado en este artículo tiene varias limitaciones. En primer lugar, se aplicó la homogeneización por ciclos de congelación y descongelación de un estudio sobre nematodos; así, el método sólo puede ser eficaz contra ecdysozoa. En segundo lugar, debido a la amplificación de fragmentos de ADN durante la fase de biblioteca de secuencia de ADN, la posibilidad de errores de la PCR no se puede ignorar. Así, los datos de la secuencia no se recomiendan para el análisis que requiere lecturas de alta precisión (es decir, análisis SNP). Además, como hemos indicado en el protocolo, el uso del kit de SNA-Seq especificado que se muestra en la Tabla de los materiales es imprescindible, debido a la baja cantidad de ADN de entrada. Este kit de construcción de biblioteca de ADN ligates secuencias de Illumina adaptador antes de la amplificación; por lo tanto, esta biblioteca no se puede aplicar para la secuencia larga de leer usando tecnología PacBio o Nanopore. Por último, una verificación de la calidad de la biblioteca de ADN construida durante este protocolo ocurre solamente una vez, después de la construcción de la biblioteca de la secuencia. Esto es debido a la entrada de ADN desde la mayoría cuantificación de ADN y métodos de electroforesis no pueden detectar 50-200 pg de ADN. Por lo tanto, nos hemos llevado a cabo controles de calidad, como la electroforesis (figura 1) y cuantificaciones basadas en fluorescencia, solamente después de la amplificación por PCR.
Una discusión completa de Bioinformática análisis de estos datos está fuera del alcance de este artículo; sin embargo, hemos indicado brevemente varios análisis que hemos realizado. Una verificación de la calidad de los datos de secuenciación con FastQC19 calcula las cualidades por la base, duplicación de la secuencia, etc. datos de la secuencia que han sido validados pueden ser presentadas a la Asamblea del genoma. Han montado un genoma de 132 Mb con MaSuRCA v3.1.321 y han comparado las estadísticas de asignación calculadas con BWA22 y23 de la QualiMap de esta biblioteca de secuenciación de ADN con otros de asambleas del genoma de H. dujardini 16. Además, también han utilizado estos datos de secuenciación de ADN para la exclusión de contaminantes en nuestro estudio17y han observado que las lecturas secuenciadas se distribuyen uniformemente por todo el genoma.
Mayoría de los proyectos en organismos no-modelo partir cultivo suficiente material de muestra, como fue el caso de los tardígrados24. Avances en las técnicas de cultivo han permitido altas cantidades de cultura tardigrade, pero los métodos actuales de cultivo no son asépticos y tardígrados mayoría son todavía unculturable en labs, ha sido casi imposible llevar a cabo genoma o secuenciación de transcriptoma. Este método de secuenciación de ADN de un solo individuo hace posible analizar especies tardigrade, incluyendo las especies marinas que se han estudiado menos. Mediante la realización de genómica comparativa en un área más amplia filéticas, un mayor entendimiento de mecanismos anhydrobiosis en tardígrados puede lograrse.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
Los autores agradecen a Nozomi Abe Yuki Takai y Nahoko Ishii por su apoyo técnico en la secuencia genomic. Este trabajo fue apoyado por subvenciones de la sociedad japonesa para la promoción de la ciencia (JSPS) investigador KAKENHI subvenciones para jóvenes científicos (No.22681029) y KAKENHI subvenciones para investigación científica (B), no. 17 H 03620 desde las JSP, por un Subvención para proyectos básicos de investigación de ciencia de la Fundación de Sumitomo (No.140340) y en parte por fondos de investigación del gobierno de la Prefectura Yamagata y ciudad de Tsuruoka, Japón. Chlorella vulgaris utilizado para alimentar los tardígrados fue cortesía de Chlorella Industry LTD. Co.
Materials
| Name | Company | Catalog Number | Comments |
| SZ61 microscope | OLYMPUS | ||
| BactoAgar | Difco Laboratories | 214010 | |
| Penicillin Streptomycin (10,000 U/mL) | Gibco by life technologies | 15140-148 | |
| VHX-5000 System | Keyence | ||
| 0.2mL Silicone coating tube | Bio Medical Science | BC-bmb20200 | |
| Quick-DNA Microprep Kit | ZYMO Research | D3021 | Use of this kit is absolutey critical; see step 3.1 |
| 1.5 mL microtube | greiner bio-one | 616-201 | See 4.1.1 |
| HIgh speed refrigerated micro centrifuge | TOMY | MX-307 | |
| Covaris M220 | Covaris Inc. | 4482277 | |
| ThruPLEX DNA-Seq kit | Rubicon Genomics | CAT. NO. R400406 | Use of this kit is absolutey critical; see step 4.2 |
| Thermal Cycler | Bioer Technology | TC-96GHbC | |
| AMPure XP reagent | BECKMAN COULTER Life Science | A63881 | |
| Ethanol | Wako | 054-027335 | |
| EB buffer | QIAGEN | 19086 | |
| 2200 TapeStation | Agilent | G2965AA | |
| D1000 Reagents | Agilent | 5067-5583 | |
| D1000 ScreenTape | Agilent | 5067-5582 | |
| Qubit dsDNA BR Buffer/Reagent | ThermoFisher Scientific | Q32850 | |
| Cubee Mini-Centrifuge | RecenttecGenereach | R5-AQBD01aqbd | |
| MiSeq 600 cycle v3 | Illumina Inc. | MS-102-3003 | |
| MiSeq Sequencer | Illumina Inc. | SY-410-1003 |
References
- Crowe, J. H., Hoekstra, F. A., Crowe, L. M. Anhydrobiosis. Annual Review of Physiology. 54 (1), 579-599 (1992).
- Mobjerg, N., et al. Survival in extreme environments - on the current knowledge of adaptations in tardigrades. Acta Physiologica. 202 (3), 409-420 (2011).
- Becquerel, P. La suspension de la vieau dessous de 1/20 K absolu par demagnetization adiabatique de L'alun de fer dans le vide les plus eléve. Comptes Rendus de l'Académie des Sciences. 231, 261-264 (1950).
- Ono, F., et al. Effect of ultra-high pressure on small animals, tardigrades and Artemia. Cogent Physics. 3 (1), 1167575 (2016).
- Horikawa, D. D., et al. Tolerance of anhydrobiotic eggs of the Tardigrade Ramazzottius varieornatus to extreme environments. Astrobiology. 12 (4), 283-289 (2012).
- Horikawa, D. D., et al. Analysis of DNA repair and protection in the Tardigrade Ramazzottius varieornatus and Hypsibius dujardini after exposure to UVC radiation. PLoS One. 8 (6), e64793 (2013).
- Horikawa, D. D., et al. Radiation tolerance in the tardigrade Milnesium tardigradum. International Journal of Radiation Biology. 82 (12), 843-848 (2006).
- May, R. M., Maria, M., Gumard, J. Action différentielle des rayons x et ultraviolets sur le tardigrade Macrobiotus areolatus, a L'état actif et desséché. Bulletin Biologique de la France et de la Belgique. 98, 349-367 (1964).
- Jonsson, K. I., Harms-Ringdahl, M., Torudd, J. Radiation tolerance in the eutardigrade Richtersius coronifer. International Journal of Radiation Biology. 81 (9), 649-656 (2005).
- Bemm, F., Weiss, C. L., Schultz, J., Forster, F. Genome of a tardigrade: Horizontal gene transfer or bacterial contamination? Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3054-E3056 (2016).
- Delmont, T. O., Eren, A. M. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ. 4, e1839 (2016).
- Koutsovoulos, G., et al. No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proceedings of the National Academy of Sciences of the United States of America. 113 (18), 5053-5058 (2016).
- Boothby, T. C., Goldstein, B., et al. Reply to Bemm et al. and Arakawa: Identifying foreign genes in independent Hypsibius dujardini genome assemblies. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3058-E3061 (2016).
- Boothby, T. C., et al. Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 112 (52), 15976-15981 (2015).
- Arakawa, K. No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3057 (2016).
- Arakawa, K., Yoshida, Y., Tomita, M. Genome sequencing of a single tardigrade Hypsibius dujardini individual. Scientific Data. 3, 160063 (2016).
- Yoshida, Y., et al. Comparative genomics of the tardigrades Hypsibius dujardini and Ramazzottius varieornatus. PLoS Biology. 15 (7), e2002266 (2017).
- He, F. Total RNA Extraction from C. elegans. Bio-protocol. Bio101, e47 (2011).
- Andrews, S. FastQC a quality-control tool for high-throughput sequence data. , http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2015).
- Hashimoto, T., et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature Communications. 7, 12808 (2016).
- Zimin, A. V., et al. The MaSuRCA genome assembler. Bioinformatics. 29 (21), 2669-2677 (2013).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Okonechnikov, K., Conesa, A., Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32 (2), 292-294 (2016).
- Horikawa, D. D., et al. Establishment of a rearing system of the extremotolerant tardigrade Ramazzottius varieornatus: a new model animal for astrobiology. Astrobiology. 8 (3), 549-556 (2008).

{kind=link}