

Summary
We demonstrate three different tissue preparation techniques for immunohistochemical visualization of rat retinal microvascular pericytes, i.e., cryo-sections, whole-mounts, and hypotonic isolation of the vascular network.
Abstract
Retinal pericytes play an important role in many diseases of the eye. Immunohistochemical staining techniques of retinal vessels and microvascular pericytes are central to ophthalmological research. It is vital to choose an appropriate method of visualizing the microvascular pericytes. We describe retinal microvascular pericyte immunohistochemical staining in cryo-sections, whole-mounts, and hypotonic isolated vasculature using antibodies for platelet-derived growth factor receptor β (PDGFRβ) and nerve/glial antigen 2 (NG2). This allows us to highlight advantages and shortcomings of each of the three tissue preparations for the visualization of the retinal microvascular pericytes. Cryo-sections provide transsectional visualization of all retinal layers but contain only a few occasional transverse cuts of the microvasculature. Whole-mount provides an overview of the entire retinal vasculature, but visualization of the microvasculature can be troublesome. Hypotonic isolation provides a method to visualize the entire retinal vasculature by the removal of neuronal cells, but this makes the tissue very fragile.
Introduction
Retinal pericytes are the focus of many research laboratories as these cells play a major role in the integrity of the vasculature. Pathological conditions such as diabetic retinopathy1, ischemia2, and glaucoma3 have vascular characteristics that involve the function of pericytes. Pericytes are found in the inner retinal capillary plexuses. The central retinal artery that supplies the inner retina branches into two layers of capillary plexuses. The inner vascular bed is situated between the ganglion cell and inner nuclear layers. The deeper layer is more dense and complex and is localized between the inner and outer nuclear layers4,5. In addition, some parts of the retina also contain a third network termed the radial parapapillary capillaries. These are long, straight capillaries that lie among the nerve fibers and rarely anastomose with one another or the other two plexuses6. Within the capillary wall, pericytes are embedded in the basement membrane and line the abluminal side of vascular endothelial cells.
To this date, there is no unique biological marker of these pericytes that can differentiate them from other vascular cells. Platelet-derived growth factor receptor β (PDGFRβ) and nerve/glial antigen 2 (NG2) are commonly used markers which both present on pericytes but also other vascular cells. Identification of pericytes is further complicated by the existence of pericyte subsets that vary in morphology and protein expression7. Currently, the best identification relies on a combination of protein markers and the characteristic positioning of the pericyte in the vascular wall. We demonstrate here three different tissue preparation techniques for immunohistochemical PDGFRβ/NG2 staining of rat retinal microvascular pericytes, i.e., cryo-sections, whole-mounts, and hypotonic isolation of the vascular network.
With cryo-sections, the retina and sclera are cut through the optic nerve. This allows for the visualization of all layered structures of neurons. The distinct ten layers of the retina are apparent as interchanging nuclear and axonal/dendritic structures that can be visualized with stains such as hematoxylin/eosin or fluorescent nuclear 4',6-diamidino-2-phenylindole (DAPI)8. The metabolic requirements differ between the layers9 and it provides a method to determine the thickness or total absence of a specific layer (e.g., the loss of retinal ganglion cells is one of the hallmarks of retinal ischemia10,11). The vasculature is evident as transverse cuts through the retina, making it possible to separately study the capillary plexuses within the respective retinal layers12,13.
More traditionally, the investigations of the retinal vasculature network are performed in retinal whole-mounts. With this tissue preparation, the retina is cut and flattened as a flower-shaped structure. The method is a relatively fast tissue preparation technique that can highlight the overall architecture retinal vasculature and it is therefore often applied in the investigation of neovascularization in the murine retina. Successful visualization of the microvasculature in whole-mounted retinas is also reported in the developing neonatal mouse and rat retina14,15,16,17,18,19. These studies reveal a more defined pericytic activity with larger capillary-free areas in the adult compared to the neonatal retina14.
Another way of visualizing is the retinal microvasculature after hypotonic isolation. This tissue preparation technique results in retinal blood vessels and capillaries being freed of the neuronal cells. This type of two-dimensional imaging of the isolated retinal vascular network is usually performed after retinal trypsin digestion20 and used to assess the vascular abnormalities of diabetic retinopathy including pericyte loss and capillary degeneration20,21,22. The hypotonic isolation method offers the investigations of retinal vascular gene and protein regulatory responses as they has been done with RT-PCR and western blotting23,24,25. We provide here a protocol for the free-float immunohistochemical staining of the hypotonic isolated retinal vasculature as an alternative to trypsin digestion to examine the microvascular pericytes.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
The protocol was optimized and demonstrated on adult male albino rats. In all experimental procedures, animals were treated according to the regulations in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Animals were euthanized by carbon dioxide and subsequent cervical dislocation.
1. Rat Retinal Tissue Preparations
- Cryo-section
- Make posterior and anterior ~0.5 cm slits of in the rat eyelid with a scalpel.
- Optional: Using a diathermy burner, mark the eye at the inner angle to orientate the eye in a uniform fashion during embedding and allow for vertical cryostat sectioning through the optic nerve.
- Grab the eye with forceps and tilt carefully to the side to expose the surrounding tissue. Enucleate the eye by making cuts with dissection scissors in the connective and muscular tissue.
CAUTION: Do not pull the eye too hard as excessive pressure on the optic nerve may cause retinal detachment. - Put the eye briefly in 4% formaldehyde in phosphate-buffered saline (PBS) to stabilize before making an initial hole at the corneal limbus by applying light pressure with the tip of a scalpel.
- Under a microscope, cut along the corneal limbus with dissection scissors to remove the cornea and remove the lens with forceps before submerging in 4% formaldehyde in PBS for 2–4 h.
- Rinse sequentially in Sörensen's phosphate buffer with 10% sucrose and 25% sucrose.
- Embed in Yazulla medium prepared with 3% gelatin from porcine skin and 30% albumin from chicken egg white.
NOTE: The protocol can be paused here with tissue storage at -20 °C.
- Make posterior and anterior ~0.5 cm slits of in the rat eyelid with a scalpel.
- Whole-mount
- Make posterior and anterior ~0.5 cm slits in the rat eyelid with a scalpel.
- Grab the eye with forceps and tilt carefully to the side to expose the surrounding tissue. Enucleate the eye by making cuts with dissection scissors in the connective and muscular tissue.
CAUTION: Do not pull the eye too hard as excessive pressure on the optic nerve may cause retinal detachment. - Put the eye briefly in 4% formaldehyde in PBS to stabilize before making an initial hole at the corneal limbus by applying light pressure with the tip of a scalpel.
- Under a microscope, cut along the corneal limbus with dissection scissors to remove the cornea and remove the lens with forceps.
- Separate the retina from the retinal pigment epithelium towards the optic nerve with forceps using small opening movements to avoid major tearing.
- Free the retina at the optic nerve with dissection scissors and make four slits of a few millimeters length from the retinal periphery towards the optic nerve head.
- Spread the retina onto a glass slide and allow drying for 5–10 min.
- Fix in 4% formaldehyde for 20–30 min by dripping formaldehyde onto the retina.
CAUTION: Do not apply directly on the retina as it may detach from the glass. - Rinse with PBS. For optimal results, immuno-stain directly after rinsing.
- Hypotonic isolation
- Make posterior and anterior ~0.5 cm slits in the rat eyelid with a scalpel.
- Grab the eye with forceps and tilt carefully to the side to expose the surrounding tissue. Enucleate the eye by making cuts with dissection scissors in the connective and muscular tissue.
CAUTION: Do not pull the eye too hard as excessive pressure on the optic nerve may cause retinal detachment. - Make an initial hole at the corneal limbus by applying light pressure with the tip of a scalpel.
- Under a microscope, cut along the corneal limbus with dissection scissors to remove the cornea and remove the lens with forceps.
- Separate the retina from the retinal pigment epithelium towards the optic nerve head with forceps using small opening movements to avoid major tearing.
- Free the retina at the optic nerve with dissection scissors, place the retina in 1 mL of deionized water in a 24-well plate and shake at 200 rpm with a 1.5 mm vibration orbit for 1 h at room temperature.
NOTE: Hereafter, the retina will appear less defined at the edges. - Add 200 U DNAse 1 to dissociate the lysed cell debris from the retinal vasculature and shake for another 30 min at room temperature.
NOTE: Debris might start to form in the wells. - Rinse minimum 3 times in deionized water for 5 min with shaking at 150–300 rpm to remove neuronal cell debris. The retina should become more transparent with each rinse indicative of the removal of neuronal cellular debris.
- Use a dark background to look into the 24-well plate to clearly see the diaphanous isolated retinal vasculature.
- (Optional): If the vasculature does not appear free of neuronal layers (semi-transparent) at this point either add more rinse steps, increase the shaking speed or use a pipet to aspirate liquid onto the vasculature.
CAUTION: Either of the optional steps may damage the vasculature.
- Fix 10 min in 1 mL of 4% paraformaldehyde in PBS at room temperature and rinse 3 times in PBS.
NOTE: The protocol can be paused here with tissue storage at 4 °C.
2. Immunohistochemistry
- Staining of cryo-sections
- Cut 10 µm cryo-sections of the gelatin-embedded retina as vertical sectioning through the optic nerve and place the cryo-sections on a glass slide and let dry (minimum 1 h).
- Submerge the glass slide in PBS with 0.25 % Triton X-100 (PBS-T) for 15 min.
- Drip 1:100 PDGFRβ and 1:500 NG2 primary antibodies diluted in PBS-T + 1% BSA onto the cryo-section and incubate in incubation chambers at 4 °C overnight.
- Submerge the glass slide 2 times in PBS-T for 15 min and drip 1:100 anti-mouse Alexa Fluor 594-linked and 1:100 anti-rabbit FITC-linked secondary antibodies diluted in PBS-T with 3% BSA onto the cryo-sections.
- Incubate the glass slide 1 h at room temperature in the dark.
- Rinse the glass slide in PBS-T 2x 15 min.
NOTE: Optional: For double and triple immunofluorescent staining, sequential staining can be performed by repeating the procedure from 2.1.3 to 2.1.6 two and three times, respectively. - Mount the stained cryo-sections with anti-fading mounting medium containing DAPI and a coverslip.
- Staining of whole-mount
- Drip PBS-T onto the whole-mount and incubate at room temperature for 15 min.
- Pour off and drip 1:100 PDGFRβ and 1:500 NG2 primary antibodies diluted in PBS-T + 1% BSA and incubate in moist chambers at 4 °C overnight.
- Pour off and drip on PBS-T to rinse the glass slide in 2x 15 min.
- Pour off and drip on 1:100 anti-rabbit Cy2- and 1:100 anti-mouse Cy3-linked secondary antibody diluted in PBS-T with 3% BSA to incubate 1 h in a moist chamber at room temperature in the dark.
- Pour off and drip on PBS-T to rinse in 2x 15 min in the dark.
NOTE: Optional: For double and triple immunofluorescent staining, sequential staining can be performed by repeating the procedure from 2.2.2 to 2.2.5 two and three times, respectively. - Mount the stained whole-mount with anti-fading mounting medium containing DAPI and a coverslip.
- Staining of hypotonic isolated vasculature
- Block the hypotonic isolated vasculature 1 h with shaking at 100 rpm and room temperature with 500 µL/well of 10 % donkey serum diluted in PBS.
- Incubate overnight at room temperature and shake at 100 rpm with 600 µL/well of 1:100 PDGFRβ and 1:500 NG2 primary antibodies diluted in 10% donkey serum in PBS.
- Rinse the retinal network 3x in PBS for 5 min and incubate in 1:100 anti-mouse Alexa Fluor 594-linked and 1:100 anti-rabbit FITC-linked secondary antibodies diluted in 10% donkey serum in PBS shaking at 100 rpm and room temperature for 1 h in the dark.
- Rinse in PBS-T for 5 min and incubate in 0.2 ng/mL DAPI in PBS-T for 15 min followed by 3x 5 min rinses in PBS-T in the dark.
- Cut the tip of a plastic Pasteur pipet, moisten it with PBS-T and use it to transfer the retinal network to a 4-well glass chamber slide.
NOTE: The moistening step is important to avoid the retinal vasculature sticking to the inside of the Pasteur pipet. - Unfold the retinal vasculature. Avoid touching the retinal vasculature with forceps as this can cause the vasculature to tangle.
NOTE: Unfolding can be done by tilting the chamber slide back and forth or aspirating drops of liquid onto the retinal vasculature. - Remove the medium from the wells. The surface tension of the liquid will flatten the vasculature onto the bottom of the slide.
- Ensure correct unfolding under a microscope before removing the plastic wells from the chamber slide.
- Mount the stained vasculature with anti-fading mounting medium and a coverslip.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
The successful protocols provide three different retinal preparations for visualizing microvascular pericytes. Each of these methods uses the PDGFRβ and NG2 immunoreactivity co-localization and the unique position of the pericytes that wrap around the capillary endothelium foridentification.
With cryo-sections, the neuronal layers can be identified by the fluorescent density of DAPI-labelled nuclei and the inner and deep capillary plexuses contain pericytes that display PDGFRβ and NG2 immunoreactivity (Figure 1). The unique position of pericytes in the vascular wall can be seen as circular or horseshoe-shaped immunoreactivity in the periphery of the vessels (Figure 1, arrows) or occasional longitudinally cut vessels (Figure 1, arrowhead).
Successful visualization of the microvascular pericytes in whole-mount preparation of adult rat can be challenging14,20. PDGFRβ immunoreactivity in the whole-mounted adult retina resulted only in a very weak signal (not shown). NG2 staining outlined the inner vascular network with intense staining in abluminal pericytes (Figure 2).
To obtain an overview of the vascular network of the adult rat retina, immunohistochemistry of hypotonic isolated rat retinal vascular network provides an alternative method to whole-mount. When successfully performed, this tissue preparation technique provides an overview of the entire retinal microvasculature including inner and deep capillary plexuses. Immunohistochemical staining with pericytic markers results in the entire microvascular network showing NG2 immunoreactivity with an intense response in specific vascular cells (Figure 3). Some of these cells also display PDGFRβ immunoreactivity (Figure 3).
A careful unfolding of the tissue is important to obtain a good overview of the retinal vasculature. Insufficient unfolding can result in the vasculature being arranged in multiple focus layers which makes fluorescent imaging difficult (Figure 4). Unfolding can be facilitated by gentle shaking, tilting the chamber slide back and forth or aspirating drops of liquid onto the retinal vasculature. It should be noted that the anatomy of the retinal capillary plexuses will result in some overlay of the vascular beds. Hence, it is problematic to distinguish the inner from the deeper capillary plexuses when flattened onto the glass slide.
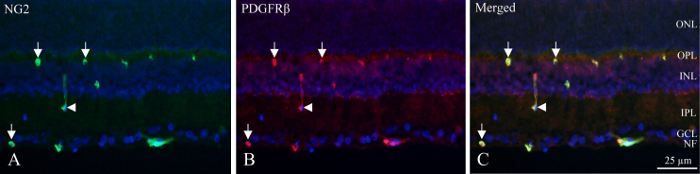
Figure 1: Double immunohistochemical pericyte staining of cryo-section of the inner rat retina. (A) NG2 (green) immunohistochemistry reveals positive vessels within the inner part of the retina. Arrows indicate circular and horseshoe-shaped immunoreactivity in the vessels. The arrowhead points out a longitudinally cut vessel. (B) PDGFRβ (red) immunohistochemistry show reactivity similar to NG2 immunoreactivity. Arrows and arrowhead point at the same structures as for NG2 staining. (C) The image shows a merge of NG2, PDGFRβ and DAPI (blue) by superimposing the three different filters. It is revealed that the two antibodies are co-localized (green + red = yellow). NF: nerve fiber layer, GCL: ganglion cell layer, IPL: inner plexiform layer, INL: inner nuclear layer, OPL : outer plexiform layer, ONL: outer nuclear layer. Please click here to view a larger version of this figure.

Figure 2: Immunohistochemical pericyte staining of whole-mounted rat retina. NG2 (red) immunoreactivity is visible along the superficial vascular networkwith intense staining in abluminal pericytes. The images show merged NG2 and DAPI (blue). The neuronal cells are visible as DAPI-blue nuclei between the NG2-stained microvasculature. PDGFRβ immunoreactivity in the whole-mounted adult retina resulted only in a very weak signal and is therefore not included in this figure. Please click here to view a larger version of this figure.

Figure 3: Double immunohistochemical pericyte staining of hypotonic isolated rat retinal vasculature. (A). The image shows hypotonic isolated rat retinal vascular network immunohistochemical stained with NG2 (green) and PDGFRβ (red). The complete network showed NG2 immunoreactivity. However, PDGFRβ immunoreactivity was found in cell somas. The insert of A is an overview of retinal vasculature network stained with DAPI (blue). (B) The image shows the same specimen as (A) at a different location and higher magnification. It is clear that PDGFRβ staining is only displayed in cell somas in the vascular network indicating pericyte immunoreactivity. The insert of B is the image that shows high magnification of vasculature stained with NG2 (green), PDGFRβ (red) and merged with DAPI. Including DAPI staining together with NG2 and PDGFRβ reveals three pericytes (PDGFRβ-positive) and two unidentified vessel wall cells. Please click here to view a larger version of this figure.

Figure 4: Example of an inadequate unfolded hypotonic isolated retinal vasculature. The careful unfolding of a hypotonic isolated vasculature is important as insufficient unfolding can result in the vasculature being stacked. The pictures show DAPI-stained retinal vasculature at the same site but taken in different focus layers. (A) The DAPI-stained cells in one focus layer show an approximately 25 µm thick blood vessel in the retinal vasculature. (B) The DAPI-stained cells in a second focus layer elucidate thin branching capillaries at the same site that has been stacked on the larger blood vessel displayed in (A). Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
We present three retinal preparation techniques that can be applied in the study of microvascular retinal pericytes. Below, we provide a comparison between each of the methods and highlight critical steps in the protocols.
With cryo-sectioning, the retina is cut in sagittal sections and hence, it is possible to obtain numerous specimens from the same retina. The numeral sections resulting from this method make it an ideal choice for antibody specificity and titration testing as it prevents unnecessary animal sacrifice. The preparatory work is decisive for good results. It is crucial while dissecting the eye not to pull the eye but to cut all the muscles around the eye. There is a risk of retinal detachment if force is used. Because the eye cup is thin, one should not fix too long. Immersion with 4% paraformaldehyde in PBS for 2–4 h will fix the tissue. If fixed longer, there is a risk of destroying the antigen of interest. In order to generate the best possible immunostainings, it is critical for each section to be as flat as possible against the glass. Because the sections contain only a few occasional transverse cuts of the microvasculature, this tissue preparations technique offers only intermittent visualization of the microvasculature. Therefore, this method is not suitable for the studies of the overall vascular architecture and quantifiable measures of pericytes.
The overall vasculature can be stained after whole-mounting to provide an overview of the vasculature. When mastering this technique, it can be a fast tissue preparation procedure that requires relatively little workload. However, there are a few points to be particularly aware of. The retina is fragile and it is paramount to handle them with care and avoid rips and cuts. Furthermore, while carefully handling the retina, it is of key importance to avoid folds and other crevasses during mounting that may affect the immunohistochemical imaging later on. The mounted retina has a thickness of 250-300 µm and antibody penetration of the neuronal layers might be problematic. In regard to pericyte visualization, the NG2 staining in the whole-mount protocol could only be achieved with a Cy3-linked secondary antibody, and PDGFRβ immunoreactivity in the whole-mounted adult retina resulted in such a very weak signal that it was not shown in the figure. Hence, the usefulness of the whole-mount staining technique is strongly hampered by the successful staining depending mainly on the specific primary and secondary antibodies used. Further suggestions to optimize the protocol for visualized microvascular plexuses of an adult rat retina is by the incubation in primary antibody for a longer period of time and/or changing the incubation temperature26.
Due to the thickness of the whole-mounted, it is important to be aware of the direction of the retina after making the four incisions from the retinal periphery towards the optic nerve head to avoid accidental mounting with the inner retina containing the capillaries facing down. Still, detailed visualization of the deeper microvascular plexuses and the pericytes can be troublesome with this method14,20. As whole-mount staining of the adult retina is difficult in the deeper capillary plexus, this might result in the retina appearing avascular in some parts. The immunohistochemical staining of whole-mounted adult retina provides only a fragmented overview of the vasculature with pericytes lining the vessels in the superficial capillary plexus and therefore, this method can lead to false negative results in the visualization of the rat retinal vasculature.
Trypsin digestion has long been considered the gold standard technique for isolation and visualization of the retinal vasculature20. Hypotonic isolation provides an alternative method for the visualization of the entire complex three-dimensional vascular network and faces many of the same challenges working with very fragile tissue which is difficult to handle. Due to the resemblance in the retinal vascular product between the two methods, the hypotonic isolation faces many of the same challenges as trypsin digestion20 including careful dissection to prevent major tears and cautious handling by avoiding pipetting and touching with forceps. An important difference between the methods is the fixation. Trypsin digestion is typically done after fixation and can be applied successfully on preserved retinas that have been in fixative for several years20. Hypotonic isolation is performed before fixation and thus allows for the implementation in various other assays as previously described in literature23,24,25. The trypsin digestion is performed over two days20, whereas the hypotonic isolation can be completed within hours. Furthermore, the hypotonic isolation protocol does not include the risk of over-digestion and enzymatic cleavage of membrane markers required for pericyte recognition, e.g. PDGFRβ and NG2, making this method preferred for subsequent immunohistochemical microvascular staining.
Finally, although out of the immediate scope of this manuscript, there is a less widely known but very useful technique for isolating large, intact microvascular plexi from rodent out of the retina27. The retina is positioned in a special glass chamber and coverslip. Upon the removal of the coverslip, the microvasculature remains attached to the coverslip to create a fully alive (≈ 98% of cells) tissue print. The biggest advantage of this method is that it allows for the physiological study of the vessels and subsequent fixation for immunostaining. An obvious limitation of the method is that it does not provide the visualization/isolation of the entire microvascular network as retina flat mount and hypotonic preparations do. Still, pericytes can be visualized by application of this method27.
The three tissue preparation techniques described here are complementary in the sense that each method comprises advantages and shortcomings for visualization of the microvascular pericytes. The assessment of each methods potentials and weaknesses is important to select the optimal method for investigating the microvascular pericytes under specific pathological conditions. The demonstrated methods can be expanded to staining not only for pericyte markers but also for the visualization of microvascular structures in general. Eventually, it is for the individual researcher to select the appropriate visualization method based on the research hypothesis in question.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The research was funded by The Lundbeck Foundation, Denmark.
Materials
| Name | Company | Catalog Number | Comments |
| Geletin from porcine skin | Sigma-Aldrich | G2625-500G | |
| Albumin from chicken egg white | Sigma-Aldrich | A5253-500G | |
| Deoxyribonuclease (DNAse) I from bovine pancreas | Sigma-Aldrich | D5025-15KU | Dissolved in 0.15 M NaCl |
| Bovine serum albumin (BSA) | VWR | 0332-100G | |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121, lot 129348 | |
| Rabbit anti-PDGFRβ | Santa Cruz | sc-432 | 1:100 |
| Mouse anti-NG2 | Abcam | ab50009 | 1:500 |
| Alexa Fluor 594 AffiniPure Donkey Anti-Rabbit IgG | Jackson ImmunoResearch | 711-585-152 | 1:100 |
| Fluorescein (FITC) AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | 715-095-151 | 1:100 |
| Cy2 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | 711-225-152 | 1:100 |
| Cy3 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | 715-165-150 | 1:100 |
| 4',6-diamidino-2-phenylindole (DAPI) | Sigma-Aldrich | D9542-1MG | Dissolved in DMSO |
| Anti-fading mounting medium | Vector Laboratories | H-1000 | |
| Anti-fading mounting medium with DAPI | Vector Laboratories | H-1200 | |
| Nunc Lab-Tek II 4-well chamber slide | Thermo Fisher Scientific | 154526 |
References
- Eshaq, R. S., Aldalati, A. M. Z., Alexander, J. S., Harris, N. R. Diabetic retinopathy: Breaking the barrier. Pathophysiology. , (2017).
- Cai, W., et al. Pericytes in Brain Injury and Repair After Ischemic Stroke. Translational Stroke Research. , (2016).
- Trost, A., et al. Brain and Retinal Pericytes: Origin, Function and Role. Frontiers in Cellular Neuroscience. 10, 20 (2016).
- Ramos, D., et al. The Use of Confocal Laser Microscopy to Analyze Mouse Retinal Blood Vessels. Confocal Laser Microscopy - Principles and Applications in Medicine, Biology, and the Food Sciences. Lagali, N. , InTech. (2013).
- Moran, E. P., et al. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. American Journal of Physiology. Heart and Circulatory Physiolog. 311, H738-H749 (2016).
- Henkind, P. Microcirculation of the peripapillary retina. Transactions - American Academy of Ophthalmology and Otolaryngology. 73, 890-897 (1969).
- Attwell, D., Mishra, A., Hall, C. N., O'Farrell, F. M., Dalkara, T. What is a pericyte? Journal of Cerebral Blood Flow & Metabolism. 36, 451-455 (2016).
- Fernandez-Bueno, I., et al. Histologic Characterization of Retina Neuroglia Modifications in Diabetic Zucker Diabetic Fatty Rats. Investigative Ophthalmology & Visual Science. 58, 4925-4933 (2017).
- Yu, D. -Y., Yu, P. K., Cringle, S. J., Kang, M. H., Su, E. -N. Functional and morphological characteristics of the retinal and choroidal vasculature. Progress in Retinal and Eye Research. 40, 53-93 (2014).
- Allen, R. S., et al. Severity of middle cerebral artery occlusion determines retinal deficits in rats. Experimental Neurology. 254, 206-215 (2014).
- Kyhn, M. V., et al. Acute retinal ischemia caused by controlled low ocular perfusion pressure in a porcine model. Electrophysiological and histological characterisation. Experimental Eye Research. 88, 1100-1106 (2009).
- Blixt, F. W., Radziwon-Balicka, A., Edvinsson, L., Warfvinge, K. Distribution of CGRP and its receptor components CLR and RAMP1 in the rat retina. Experimental Eye Research. 161, 124-131 (2017).
- Sarlos, S., Wilkinson-Berka, J. L. The renin-angiotensin system and the developing retinal vasculature. Investigative Ophthalmology & Visual Science. 46, 1069-1077 (2005).
- Wittig, D., Jaszai, J., Corbeil, D., Funk, R. H. W. Immunohistochemical localization and characterization of putative mesenchymal stem cell markers in the retinal capillary network of rodents. Cells Tissues Organs. 197, 344-359 (2013).
- Tual-Chalot, S., Allinson, K. R., Fruttiger, M., Arthur, H. M. Whole mount immunofluorescent staining of the neonatal mouse retina to investigate angiogenesis in vivo. Journal of Visualized Experiments. , e50546 (2013).
- Park, D. Y., et al. Plastic roles of pericytes in the blood-retinal barrier. Nature Communications. 8, 15296 (2017).
- Hughes, S., Chan-Ling, T. Characterization of smooth muscle cell and pericyte differentiation in the rat retina in vivo. Investigative Ophthalmology & Visual Science. 45, 2795-2806 (2004).
- Lange, C., et al. Intravitreal injection of the heparin analog 5-amino-2-naphthalenesulfonate reduces retinal neovascularization in mice. Experimental Eye Research. 85, 323-327 (2007).
- Higgins, R. D., et al. Diltiazem reduces retinal neovascularization in a mouse model of oxygen induced retinopathy. Current Eye Research. 18, 20-27 (1999).
- Chou, J. C., Rollins, S. D., Fawzi, A. A. Trypsin digest protocol to analyze the retinal vasculature of a mouse model. Journal of Visualized Experiments. , e50489 (2013).
- Hazra, S., et al. Liver X receptor modulates diabetic retinopathy outcome in a mouse model of streptozotocin-induced diabetes. Diabetes. 61, 3270-3279 (2012).
- Zhang, L., Xia, H., Han, Q., Chen, B. Effects of antioxidant gene therapy on the development of diabetic retinopathy and the metabolic memory phenomenon. Graefe's Archive for Clinical and Experimental Ophthalmology. 253, 249-259 (2015).
- Dagher, Z., et al. Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes. 53, 2404-2411 (2004).
- Navaratna, D., McGuire, P. G., Menicucci, G., Das, A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes. 56, 2380-2387 (2007).
- Gustavsson, C., et al. Vascular cellular adhesion molecule-1 (VCAM-1) expression in mice retinal vessels is affected by both hyperglycemia and hyperlipidemia. PLoS One. 5, e12699 (2010).
- Kornfield, T. E., Newman, E. A. Regulation of blood flow in the retinal trilaminar vascular network. Journal of Neuroscience. 34, 11504-11513 (2014).
- Puro, D. G. Retinovascular physiology and pathophysiology: new experimental approach/new insights. Progress in Retinal and Eye Research. 31, 258-270 (2012).
