

Summary
Bakterienzellen sind räumlich sehr organisiert. Um diese Organisation im Laufe der Zeit in den langsam wachsenden Myxococcus Xanthus Zellen zu folgen, wurde ein Setup für Fluoreszenz live Cell Imaging mit hoher räumlich-zeitliche Auflösung über mehrere Generationen hinweg entwickelt. Mit dieser Methode konnte raumzeitliche Dynamik wichtige Proteine für Chromosom Segregation und Zellteilung bestimmt werden.
Abstract
Fluoreszenz live Cell Imaging von bakteriellen Zellen ist eine wichtige Methode in der Analyse der räumlichen und zeitlichen Dynamik von Proteinen und Chromosomen zentrale Zellzyklus Ereignisse zugrunde liegen. Jedoch Bildgebung dieser Moleküle in langsam wachsenden Bakterien stellt eine Herausforderung wegen Immunofluoreszenz Fluorophore und Phototoxizität während der Bildaufnahme. Hier beschreiben wir ein einfaches Protokoll zur Umgehung dieser Einschränkungen bei Myxococcus Xanthus (die eine Generationszeit von 4-6 h). Zu diesem Zweck M. Xanthus Zellen sind auf eine starke Nährstoff-haltigen Agar Auflage in einer feuchten Umgebung mit kontrollierter Temperatur angebaut. Unter diesen Bedingungen ermitteln wir die Verdopplungszeit der einzelnen Zellen durch Anschluss an das Wachstum einzelner Zellen. Darüber hinaus Prozesse wichtige zelluläre, wie Chromosom Segregation und Zellteilung von Fluoreszenz live Cell Imaging von Zellen mit relevanten eindringmittel beschrifteten markerproteine wie Nachwuchscontest-YFP FtsZ-GFP und mCherry-PomX über mehrere abgebildet werden können Zelle Zyklen. Anschließend werden die aufgenommenen Bilder verarbeitet, um Montagen und/oder Filme zu erzeugen.
Introduction
Bakterienzellen sind räumlich sehr viele Proteine asymmetrisch Lokalisierung innerhalb von zellulären Kompartimenten1,2,3,4organisiert. Diese Lokalisierung ist oft sehr dynamisch und ändert sich im Laufe der Zeit als Reaktion auf Zellzyklus Cues oder externe Signale. Ebenso ist das bakterielle Chromosom räumlich hoch mit einzelnen Loci zu bestimmten subzellulären Orten positioniert, vor und während der Trennung Prozess5organisiert. Diese dynamische räumliche Organisation ist wichtig für Wachstum, Teilung, Zellzyklus-Verordnung, Differenzierung, Motilität, Signaltransduktion sowie Chromosom Organisation und Trennung; So wirkt sich dies im Wesentlichen alle Aspekte der bakterielle Funktion.
Die räumlich-zeitliche Dynamik dieser zelluläre Prozesse werden in einer Vielzahl von verschiedenen Bakterienarten mit Escherichia coli, Bacillus Subtilis, Vibrio Choleraeund Caulobacter Crescentus dienen als wichtige analysiert Modellorganismen. Aber diese vier Arten decken nur ein kleines Spektrum der bakteriellen Vielfalt und vielleicht nicht überraschend angesichts der großen phylogenetische Entfernung zwischen diesen Arten, zelluläre Organisation und Polarisation Mechanismen unterscheiden sich in diesen Bakterien. Dies wirft die Notwendigkeit für zusätzliche Bakterienarten zu studieren, schließlich allgemeinen Grundsätze für die räumlich-zeitliche Dynamik der Bakterienzellen extrahieren zu können.
Die Gram-negativen Delta-proteobakterium M. Xanthus ist ein Modellorganismus in der Studie von sozialen Verhaltensweisen und Zusammenarbeit in Bakterien6. M. Xanthus ist eine strenge aeroben und in Anwesenheit von Nährstoffen, bildet Kolonien, in denen Zellen nach außen in eine sehr koordinierte, schwärmen Mode und Beute auf andere Mikroorganismen7verbreiten. In Erwiderung auf Nährstoff Hunger, initiieren Zellen eine Entwicklungs Programm, die Ergebnisse in die Bildung der Fruchtkörper, die besteht aus Tausenden von Zellen, und im Inneren, die die stabförmige bewegliche Zellen differenzieren zu kugelförmig diploiden Sporen8. Beide Arten von Verhalten, d. h., schwärmen und Fruchtkörper Bildung sind nur von Zellen, die auf einer festen Oberfläche ausgeführt. Darüber hinaus engagieren unter beiden Nährstoffverhältnisse Zellen in Prozessen, bei denen unter anderem der Austausch der äußeren Membran Lipoproteine, die Motilität zu stimulieren oder fungieren als Giftstoffe in den Empfänger9,10 direkten Zell-Zell-Kontakte , der Austausch von LPS11, Anregung der Motilität von exopolysaccharide auf benachbarte Zellen12und interzellulären Signale von einer Zelle Oberfläche verankerte Protein13,14-Signalisierung.
Vor kurzem, M. Xanthus inzwischen auch ein Modellorganismus für die Untersuchung der Mechanismen, die Motilität und seine Verordnung15, Zellteilung16,17,18, Chromosom Organisation19 ,20,21. Kritische Schritte in der M. Xanthus Zellzyklus wurden analysiert im Detail durch Fluoreszenz-Mikroskopie mit Snap-Shot Bilder oder kurze Zeitraffer Aufnahmen auf Stämme tragen relevanten eindringmittel markierte Proteine16, 17,18,19,20. Im Idealfall sollte viele Zellen mit einzelligen Auflösung Fluoreszenz live Cell imaging für mindestens eine volle Zellzyklus, robuste quantitative Daten über Zellzyklus-Parameter folgen. Dies ist jedoch eine Herausforderung bei der M. Xanthus aufgrund seiner relativ langen Generationszeit von 4-6 h unter standard Laborbedingungen und Immunofluoreszenz Fluorophore und Phototoxizität während der Bildaufnahme.
Hier beschreiben wir ein Protokoll folgen M. Xanthus Zellen mit einzelnen Zelle Auflösung von Fluoreszenz-Leben-Cell imaging für mindestens 24 Stunden und umfasst mehrere Zelle Zyklen. Wichtig ist, während das gesamte Protokoll Zellen werden auf eine Agar-Pad und in engem Kontakt ermöglicht die Kontakt-abhängige Aktivitäten für die soziale Lebensweise der M. Xanthus. Das Protokoll erlaubt auch Benutzern, Monitor-Form, Größe, Divisionen und fluoreszierende Sonden mit einer hohen zeitlichen Auflösung und mit einer einzelnen Zelle Auflösung und ermöglicht somit die Quantifizierung von Zelle zu Zelle Variabilität und Zusammenhänge des Zellzyklus-Veranstaltungen.
Protocol
1. Vorbereitung und das Wachstum des M. Xanthus Stämme
Hinweis: Siehe Tabelle 1 und Tabelle 2.
- Bereiten Sie 1 % Casitone Brühe (CTT) Wachstum Mittel-1 % (w/V) Pankreas verdauen von Kasein (z.B. Bacto Casitone), 10 mM Tris-HCl pH 8.0, 1 mM KH2PO4 pH 7.6, 8 mM MgSO422, ergänzt mit Kanamycin (50 µg/mL) oder OXYTETRACYCLINE (10 µg/mL). Hinzufügen von Gentamycin (10 µg/mL) für alle Medien zur Verringerung des Risikos einer Kontamination mit anderen Bakterien, da M. Xanthus Zellen sind natürliche Resistenz gegen es.
- 5 mL von 1 % zu impfen CTT mit den entsprechenden Antibiotika mit einer einzigen frisch gewachsenen Kolonie von Wild geben (WT) DK1622 23, SA4420 (ΔMglA)24, SA4797 (ΔMglA, ΔPomX/pPomZ mCherry-PomX )16, SA8241 (ΔMglA, FtsZ+/pNatFtsZ-Gfp), oder SA4749 (ΔMglA, Nachwuchscontest+/pNatNachwuchscontest-Yfp) in der am Morgen des 1. Tages.
- Ein einzelnes Aufschwemmen M. Xanthus Kolonie in 500 µL 1 % CTT mit Antibiotika in ein steriles Röhrchen ergänzt und übertragen die gesamte Federung auf einen 50-mL-Erlenmeyerkolben mit 5 mL 1 % CTT.
Hinweis: Verwenden Sie einen Erlenmeyerkolben mit 10mal das Volumen der Kultur ausreichend Aeriation und optimales Wachstum zu gewährleisten.
- Ein einzelnes Aufschwemmen M. Xanthus Kolonie in 500 µL 1 % CTT mit Antibiotika in ein steriles Röhrchen ergänzt und übertragen die gesamte Federung auf einen 50-mL-Erlenmeyerkolben mit 5 mL 1 % CTT.
- Wachsen Sie die Zellen seit acht Generationen (ca. 40-48 h mit einer Generationszeit von 4-6 h) bei 32 ° C, schütteln bei 220 u/min, im Dunkeln. Zellen in der exponentiellen Wachstumsphase zu erhalten (OD550 < 1.2) und sie daran hindern, die stationäre Phase zu erreichen. Bei Bedarf verdünnen die Zellen in frischen 1 % CTT Medium mit den entsprechenden Antibiotika zu einem OD-550 von 0,1 - 0,2.
Hinweis: Eine optimale OD550 für eine einzelne Zelle-Mikroskopie ist 0,5 - 0,7. Bei dieser OD550ist eine ausreichende Anzahl von Zellen pro Bild zur Quantifizierung sowie statistische Analyse der zellulären Parameter erlauben.
2. Vorbereitung der Mikroskopie-Proben
Hinweis: Zellen von Mikroskopie eingesehen werden auf ein Mikroskop-Deckglas gesetzt und dann durch eine Agarose-Pad mit Nährstoffen abgedeckt. Das Deckglas ist auf einem Kunststoff geklebt oder Metallrahmen mechanische zu unterstützen. In Vorbereitung für die Mikroskopie sollte einen großen Block von 1 % agarose/TPM/0.2% CTT wie in 2.1-2.3 beschrieben vorbereitet werden. Siehe auch die Tabelle der Materialien für bestimmte Produkte, die hier verwendet.
- 500 mL des TPM-Puffer (10 mM Tris-HCl pH 7.6, 1 mM KH2PO4 pH 7.6, 8 mM MgSO4) bereiten und Autoklaven oder Filter sterilisieren mit einer Flasche Top Filter.
Hinweis: Der sterile Puffer kann mehrere Monate bei Raumtemperatur aufbewahrt werden. - Bereiten Sie 1 % Agarose Mikroskopie Lösung mit 0,2 % CTT (Mix 1 g Agarose mit 80 mL des TPM-Puffer und 20 mL 1 % CTT Medium). Erhitzen Sie in der Mikrowelle, bis die Agarose geschmolzen ist.
Hinweis: Die 0,2 % CTT ist ausreichend, um Zellen zu wachsen und Hunger zu verhindern. Höhere Konzentrationen von CTT in der Mikroskopie Medium führt zu hohen Hintergrundfluoreszenz. - Füllen Sie eine Petrischale mit geschmolzenen Agarose bis zu einer Dicke von 0,5 cm (für eine 11,5 x 11,5 cm quadratische Petrischale, ca. 60 mL flüssige Agarose erforderlich ist) und lassen Sie auf Raumtemperatur abkühlen.
Hinweis: Die Agarose-Pad kann bei 4 ° C in einer feuchten Umgebung bis zu 2 Tage aufbewahrt werden.- Vorwärmen der 1 % agarose/TPM/0.2% CTT Pad bei 32 ° C für mindestens 15 Minuten vor dem Gebrauch.
Hinweis: Um die Zellen für die Mikroskopie vorzubereiten, führen Sie Schritte 2.4-2.8.
- Vorwärmen der 1 % agarose/TPM/0.2% CTT Pad bei 32 ° C für mindestens 15 Minuten vor dem Gebrauch.
- Legen Sie ein steriles Glas Deckglas (60 mm x 22 mm, Dicke: 0,7 mm) auf einen aus Kunststoff oder Metall Rahmen, ein Loch in der Mitte (Abb. 1A); der Rahmen dient als mechanische Stütze für die dünnen Deckglas und hilft, um Drift während Mikroskopie zu reduzieren. Fixieren Sie das Deckglas auf den Rahmen mit Klebeband.
- Um den Rahmen vorzubereiten, schneiden Sie einen 75 mm × 25 mm Rahmen aus einer 1 mm dicken Metallplatte, dann schneiden Sie eine entsprechend dimensionierte Loch (20 × 30 mm in diesem Experiment) in der Mitte.
- Fügen Sie 10-20 µL exponentiell gewachsenen M. Xanthus Zellen auf das Deckglas.
- Fügen Sie fluoreszierende 0,5 µm Mikrosphären als kugelmarker zu den Zellen, Verfolgung von Zellen oder Proteine im Zeitraffer-Aufnahmen zu vereinfachen.
- Die Mikrosphären 1: 100 im TPM-Puffer verdünnen und Lagerung bei 4 ° C für bis zu mehreren Monaten. Vor Gebrauch gründlich schütteln und die Zellen ca. 5-10 µL verdünnten Mikrosphären hinzufügen.
Hinweis: Hier sind Mikrosphären, die fluoreszierende in allen gemeinsamen blau, grün, gelb und rot fluoreszierende Kanäle dienten.
- Die Mikrosphären 1: 100 im TPM-Puffer verdünnen und Lagerung bei 4 ° C für bis zu mehreren Monaten. Vor Gebrauch gründlich schütteln und die Zellen ca. 5-10 µL verdünnten Mikrosphären hinzufügen.
- Schneiden Sie ein kleines Polster etwa so groß wie das Deckglas des großen vorgewärmten 1 % agarose/TPM/0.2% CTT Pads und legen Sie es auf die Zellen (Abbildung 1B). Legen Sie ein Deckglas auf dem 1 % agarose/TPM/0.2% CTT Agarose Pad um Verdunstung zu verhindern und zu Zellen in einer feuchten Umgebung zu pflegen.
Hinweis: Das Deckglas allein verhindert erhebliche Verdunstung für mindestens 2 h. Für mehr Zeitraffer Aufnahmen sollte die 1 % agarose/TPM/0.2% CTT Pad und Deckglas Sandwich mit Paraffin Film um Verdunstung zu verhindern abgedichtet werden. - Inkubieren Sie die Mikroskopie-Probe bei 32 ° C für 15-20 min, die Zellen an der Unterseite des Agarose-Pads befestigen lassen. Dann beginnen Sie die Zeitraffer-Mikroskopie-Aufnahmen.
(3) Mikroskop-Set-up und Zeitraffer-Übernahme
Hinweis: Die hier beschriebene Protokoll wurde entwickelt für eine invertierte Weitfeld-Mikroskop mit Autofokus, ein 100 X / 1,30 NA Öl PH3 Ziel, ein X, Y motorisierte Bühne, ein sCMOS Kamera, einer Lichtquelle, Filter für grün-fluoreszierende, rot-fluoreszierend oder gelb fluoreszierend Proteine und einer Temperatur-kontrollierten Inkubation-Kammer. Diese Kammer hält Zellen geschützt vor Licht und bei konstanter Temperatur.
- Vorheizen der Inkubation Kammer und das Mikroskop auf 32 ° C für ~ 1-2 h vor Beginn der Mikroskopie.
Hinweis: Je nach Mikroskop, kann die Heizung länger dauern. Vorheizen ist unerlässlich, Drift zu senken und stabilisiert die Autofokus-Steuerung. - Schalten Sie das Mikroskop und starten Sie die Mikroskop-Steuerungs-Software zu. Wählen Sie das richtige Ziel und die richtigen Spiegel und Filter zu erwerben phase kontrastreiche Bilder sowie Bilder von grün-fluoreszierende, rot-fluoreszierend oder gelb fluoreszierende Proteine.
Hinweis: Ein Mikroskop ist in der Regel mit einer bevorzugten Software für Steuerung und Bild Erwerb Mikroskop geliefert. Hier eine kommerziell verfügbare Software (siehe die Tabelle der Materialien) diente mit dem Mikroskop und Bild zu steuern. - Geben Sie einen Tropfen des hochwertigen Immersionsöl auf die Linse des Objektivs und an der Unterseite der Probe vorab bei 32 ° c inkubiert Legen Sie das Ziel an der niedrigsten möglichen Z-Position zu beschädigen das Objektiv, wenn die Probe auf den Mikroskoptisch gelegt wird. Legen Sie den Metallrahmen mit der Probe auf der Bühne Mikroskop und mit dem"Loch" in Richtung des Ziels. Befestigen Sie die Probe in der Bühne Halterung.
- Konzentrieren Sie sich auf die Zellen durch Verschieben der Bühne in Z-Richtung näher zum Ziel. Verschieben Sie die Phase langsamer, wenn die Öltropfen an der Unterseite der Probe und das Objektiv Stellen Kontakt. Verschieben Sie die Phase, in der X / Y-Richtung bis mehrere Einzelzellen sind sichtbar in der Region der Ansicht, wenn Zellen in der Brennebene. Stellen Sie sicher, dass mindestens ein fluoreszierenden Mikrosphären in der Region der Ansicht ist, um später die erworbenen Bilder ausrichten.
Hinweis: Unter optimalen Bedingungen sollte eine Zelldichte von 15-30 Zellen pro Region der Ansicht (2.048 x 2.048 Pixel "oder" 133.1 x 133.1 µm) erreicht werden. - Öffnen Sie den Multi-Dimensional Übernahme -Assistenten der Steuerungssoftware Mikroskop eine Zeit verfallen Experiment einrichten, die das Mikroskop Bilder bei mehreren Wellenlängen und Bühne Positionen zu erwerben, bei Bedarf ermöglicht.
- Aktivieren Sie in der Registerkarte "Main" Timelapse und Mehreren Wellenlängen. Zusätzliche Registerkarten erscheinen auf der linken Seite des Fensters.
- Klicken Sie auf die Registerkarte " Speichern " und Wählen Sie Verzeichnis um einen leeren Ordner auf der Festplatte des Computers zum Speichern der erworbenen Bilder auszuwählen. Inkrement Basisnamen wenn Datei vorhanden ist um sicherzustellen, dass die aufeinander folgenden Datensätzen nicht älteren überschreiben aktivieren. Dann benennen Sie dem Experiment mit Datum und der Stamm Name oder Titel des Experiments.
- Klicken Sie auf die Registerkarte " Zeitraffer " die Zeitraffer-Parameter anpassen. Dauer bis 24 h und Zeitintervall auf eingestellt 20 min. Die Anzahl von Zeitpunkten ändert sich automatisch.
Hinweis: Die optimale Zeitspanne richtet sich auf das Experiment und die zelluläre Funktion analysiert werden. Häufiges Bild Akquisitionen verursachen Immunofluoreszenz. Daher muss ein Kompromiss zwischen zeitlicher Auflösung und Immunofluoreszenz empirisch gefunden werden. Bei einem Verdopplungszeit von 4-6 h können Bilder leicht in einem Intervall von 5 Minuten (oder sogar kleinere Intervalle, falls gewünscht) für Phasenkontrastmikroskopie erworben werden. Fluoreszenz-Mikroskopie über einen zeitlichen Verlauf der 24 h Wunsch Bilder sollten in einem Abstand von ca. 15-30 min aufgezeichnet werden. - Klicken Sie auf die Registerkarte " Wellenlängen " wählen Sie die Anzahl der Wellenlängen, für jedes Bild zu jedem Zeitpunkt zu erwerben, indem Sie die Anzahl ändern.
Hinweis: für jede Wellenlänge eine neue Registerkarte wird angezeigt, auf der linken Seite von der Multi-Dimensional Erwerb " Assistenten und Wellenlängen werden in der Reihenfolge von oben nach unten erworben werden. Für jede Wellenlänge können die Übernahme Einstellungen separat geändert werden. - Klicken Sie auf der ersten Registerkarte der Wellenlänge von oben. Phasenkontrast in der Beleuchtung Dropdown-Liste auswählen. Wählen Sie 100 ms für Belichtung und Jeden Zeitpunkt in der Dropdown-Liste erwerben . Deaktivieren Sie Auto aussetzen , indem Sie nie in der Dropdown Liste auswählen.
- Wiederholen Sie Schritt 3.5.5 für jede Wellenlänge, die zu jedem Zeitpunkt erworben werden muss. Für den Versuchsaufbau und Gewebekulturen markierte Proteine beschrieben hier, verwenden die folgenden Parameter für Exposition: 250 ms für mCherry Fusionsproteine, 200 ms für YFP Fusionsproteine und 1.000 ms für GLP-Fusionsproteinen.
Hinweis: Die optimale Ausleuchtung-Einstellungen für jede Belastung und fluoreszierende Protein sollte im Voraus bestimmt werden, indem die Intensität der Lampe und die Bild-Erfassungszeit für jede Wellenlänge. Zu lange Bild Erfassungszeiten werden die phototoxische Wirkung zu erhöhen und letztlich zu Wachstum Verhaftung und Zelle Tod führen. Daher sollte ein Kompromiss zwischen Bild Qualität und Zelle Tragfähigkeit erreicht werden. - Abrufen von Bildern aus mehreren Bühne Positionen zur Erhöhung der Zahl der Zellen im gleichen Experiment aufgenommen.
- Bilder aus mehreren Bühne Positionen erwerben, aktivieren Sie Mehrere Phase-Positionen in der Registerkarte " Main ". Dann klicken Sie auf die Registerkarte " Phase " und klicken Sie auf die Live -Taste, um das Blickfeld betrachten.
- Verschieben Sie die Phase in der X/Y-Richtung, bis eine Region of Interest (ROI) in das Blickfeld ist. Speichern der X - und Y-Koordinaten durch Anklicken bewegen das "+" in die Registerkarte " Bühne " die Bühne wieder in X/Y-Richtung bis ein neue ROI gefunden wird und Speichern der Koordinaten wieder durch Klicken auf das "+". Weitergehen Sie, bis die gewünschte Anzahl von Regionen gespeichert wird.
Hinweis: Bei Fluoreszenz Bildaufnahme, stellen sicher, dass die Regionen von Interesse (ROIs) sind nicht zu nah an einander, Phototoxizität zu minimieren.
- Überprüfen Sie noch einmal, dass die Zellen im Fokus durch Klicken auf die verschiedenen gespeicherten X - und Y-Positionen und starten den Hardware-Autofokus mit einem Klick AFC halten , um die gespeicherte Z-Position im Laufe des Experiments konstant zu halten.
- Starten Sie die Zeitraffer-Aufnahmen in der Mikroskop-Steuerungssoftware Acquire im Multi-Dimensional Übernahme -Assistenten.
Hinweis: Erscheint ein Fenster für jede Wellenlänge, die erworben wird und ein zusätzliches Fenster erscheint, das zeigt die Anzahl der erworbenen Zeitpunkte und die Zeit bis zum nächsten Bild-Erwerb. - Überprüfen Sie, dass die Zellen noch im Fokus nach den ersten paar Zeitpunkten in den Zeitraffer-Aufnahmen zu maximieren Sie die Qualität der Bilder und bei Bedarf zu konzentrieren.
4. Generation von Zeitraffer-Filme und Bildausrichtung
Hinweis: Mehrere kommerzieller und freier Software-Pakete sind verfügbar für Bildaufnahme und Bildanalyse. Wir verwenden eine handelsübliche Software (siehe die Tabelle der Materialien) mit mehreren vorinstallierten Plugins und zusätzliche Tools.
- Speichern Sie die einzelnen Bilder von Zeitraffer-Aufnahmen auf einen Computer, der die Analyse/Bildverarbeitungs-Software installiert hat.
- Starten Sie die Software und Bilder als Stapel öffnen, indem Multi-Dimensional Bewertungsdaten | Wählen Sie Base-Datei | Wählen Sie Verzeichnis. Öffnen Sie den Ordner mit den multi-dimensionalen Daten. Überprüfen Sie das Dataset, und klicken Sie auf Ansicht; Das Dataset wird als einzelne Bilder aus der Zeit bis zum Ende Punkt angezeigt. Aktivieren Sie die Wellenlänge (für die Erstellung eines Stapels) zu, wählen Sie alle Bilder, die im Stapel werden sollte, und klicken Sie auf Last Bild(er). Wiederholen Sie diesen Schritt für alle Wellenlängen und retten Sie fertigen Stapel zu.
- (Optional) Alle Bilder müssen für den Film mit Datei öffnen | Offene.
Hinweis: Es empfiehlt sich, Bilder zu öffnen durch einen erworbenen Wellenlänge gleichzeitig, um den Computer nicht verlangsamen, wenn Rechenleistung begrenzt ist. Wenn bestimmte Teile des Zeitraffer-Aufnahmen, z.B., Anfang, Ende oder mehreren Zeitpunkten übersprungen werden sollen, kann dies in den fertigen Film eingestellt werden. - Aktivieren Sie den Stapel von Bildern, die für Drift korrigiert werden muss. Öffnen Sie das Alignment-Tool von Apps | Auto-Align... Check Stack als Quelle für die Bilder und das erste Flugzeug/Zeitpunkt , als die Referenzebene. Wählen Sie den Stapel mit der Source-Stack -Taste und klicken Sie auf anwenden.
Hinweis: Die automatische Ausrichtung dauert einige Zeit und Rechenleistung aber ist ein guter Weg, um bigstacks für Drift des Mikroskop-Set-up zu korrigieren. Diese automatische Anpassung funktioniert gut, wenn Mikrosphären gehören aber auch ohne sie funktionieren könnte. - Speichern Sie den ausgerichteten Stapel.
- ROIs zu verwenden.
Hinweis: Fluoreszierende Zeitraffer Mikroskopie erstellt einfach große Mengen von Datendateien, die nehmen viel Rechenleistung und verlangsamen die Weiterverarbeitung dieser Filme. Wir empfehlen daher dringend zum ROIs zu identifizieren und Isolieren von Zellen mit kleineren Dateien arbeiten.- Rechteckigen Bereich auswählen. Erstellen Sie einen ROI um die Zellen von Interesse, indem manuell zeichnen einen ROI im Phase Kontrast Bild. Stellen Sie sicher, dass die Zellen von Interesse sichtbar und scharf über den gesamten Zeitraffer Film sind.
- Öffnen Sie den Zeitraffer-Film der zweiten Wellenlänge des gleichen Datensatzes. Übertragen den ROI aus der Phase Kontrast Bilder auf die Fluoreszenz Bilder der zweiten Wellenlänge verwenden das Transfer-Regionen -Werkzeug mit Regionen | Regionen zu übertragen. Wählen Sie die Phase Kontrast Dataset als Quell-Image und das zweite Wellenlänge Dataset als Zielbild. Wählen Sie Alle Regionen und drücken Sie "OK".
- Wiederholen Sie Schritt 4.6.2 für jede Wellenlänge für das gleiche Dataset erworben.
- Wählen Sie den ROI und Duplizieren Sie es als einen Stapel mit Bearbeiten | Duplizieren Sie | Stack... oder drücken Sie UMSCHALT + STRG + D . Speichern Sie den duplizierten Stapel mit Datei | Speichern Sie in den gleichen Ordner wie die ursprünglichen Daten.
- Wiederholen Sie Schritt 4.6.4 für jedes ROI von jeder Wellenlänge für das gleiche Dataset erworben
- Um einen Film in MOV oder AVI Formate zu erzeugen, öffnen Sie die Film -Funktion über Stack | Film. Wählen Sie die Zeitraffer-Aufnahmen mit der Taste Source-Stack . Wählen Sie das Ausgabeformat, die Frame-Rate, die Anzahl der Frames, und klicken Sie auf Speichern.
Representative Results
M. Xanthus ist eine langsam wachsende Bakterium, das bewegt sich auf festen Oberflächen. Um unsere Versuchsanordnung zu testen, haben wir eine Zeitraffer-Experiment mit bewegliche DK1622 WT-Zellen durchgeführt. Phase kontrastreiche Bilder wurden im Abstand von 5 min für 24 h (Abbildung 2A, B) erworben. Der Großteil der Zellen in Gruppen ausgerichtet. Wie erwartet, Zellen angezeigt Motilität und überwiegend in Gruppen verschoben. Wir beobachten weiter, dass Zellen gelegentlich Bewegungsrichtung umgekehrt. Diese Ergebnisse legen nahe, dass WT-Zellen unter den getesteten Bedingungen in Bezug auf die Zelle Motilität normal Verhalten. Jedoch selbst wenn Zellen alle 5 Minuten aufgezeichnet werden, ist die Identifizierung der einzelnen Zellen schwer. Darüber hinaus da bewegliche Zellen viele Zellen zu entkommen oder geben Sie das Sichtfeld macht es schwierig, Zellen über einen längeren Zeitraum verfolgen.
Um die gleiche Spur M. Xanthus Zellen über mehrere Runden des Zellzyklus von live Cell Imaging, einzelne Stämme können gelöscht werden, für das MglA -gen, welches für Motilität25unerlässlich. Dadurch wird verhindert, dass Zellen während der Bildgebung Protokolls aus dem Blickfeld verschieben. In-Frame Deletionen entstehen wie von Shi Et al.beschrieben. 26
Wie erwartet, in Phase Kontrast live Cell Imaging mit nicht-bewegliche ΔMglA Zellen (Abbildung 3), wurden die Zellen nicht aktiven Bewegung angezeigt. Wir waren in der Lage, das Wachstum und die Teilung der einzelnen Zellen während Microcolony Bildung zu folgen. Basierend auf die Zeitraffer-Aufnahmen in denen Bilder im Abstand von 5 min für 24 h erworben wurden, war es möglich, die interdivision Zeit (die Zeit zwischen zwei Ereignissen der Zellteilung) mit einer einzelnen Zelle Auflösung zu quantifizieren. Zellen des ΔMglA mutierten hatten eine Inter-Abteilung Zeit 235 ± 50 min (n = 97 Zellen). Mit ca. 4 h ist die interdivision Zeit ähnlich wie die Verdopplungszeit in Suspensionskulturen für WT-Zellen gemessen. Dadurch haben die Beweise dafür, dass M. Xanthus Zellen wachsen optimal unter diesen Versuchsbedingungen.
Um zu untersuchen, ob unsere Aufstellung Zellen, normalerweise zu wachsen, während tracking YFP-markierten Proteine über einen längeren Zeitraum ermöglicht, führten wir Fluoreszenz Zeitraffer Bildgebung mit M. Xanthus Zellen, die ein YFP-markierte Protein Ausdrücken. Zu diesem Zweck sind wir Nachwuchscontest-YFP als Marker für den Ursprung der Replikation (Ori) gefolgt. Nachwuchscontest ist als Bestandteil des Systems der ParABS M. Xanthus und bindet an die ParS Websites proximal zu den Ori; Daher kann die Herkunft Doppelarbeit und Chromosom Segregation gefolgt19,20,21. Erwerb (Phasenkontrast und Fluoreszenz, 200 ms Erfassungszeit YFP Kanal) Bild mit alle 20 min, Zellen wuchsen, unterteilt und Wachstum auch in 24 h (Abb. 4A) angezeigt. Zu Beginn der Aufnahmen gebildet Nachwuchscontest-YFP zwei Clustern in den subpolaren Regionen in den meisten Zellen (Abb. 4A). Kurz vor oder nach der Zellteilung die subpolaren Nachwuchscontest-YFP cluster an der alten Zelle Pole dupliziert. Eines der beiden Cluster blieb an der alten Zelle Pole während der zweiten Kopie der neuen Zelle Pol, erreichen die subpolaren Endstellung nach circa 40-60 min (Abb. 4A, B) umgesiedelt. Diese Beobachtungen sind in Übereinstimmung mit früheren Daten aus kurzen Zeitraffer-Aufnahmen mit dünnen Agar Pads19generiert. Wir schlussfolgern, dass diese Versuchsanordnung Zeitraffer Fluoreszenzmikroskopie Chromosom Trennung über mehrere Zyklen der Zelle in den langsam wachsenden verfolgen kann M. Xanthus Zellen, ohne störenden Zellwachstum oder die Chromosom-Segregation-Maschinen.
In einem ähnlichen Experiment haben wir versucht, die Markierungen für die Zellteilung von Time-Lapse fluoreszenzmikroskopische Untersuchungen folgen. Ähnlich wie fast alle anderen Bakterien M. Xanthus erfordert FtsZ, eine bakterielle Tubulin-ähnlichen GTPase für Zellteilung16,17,18. FtsZ bildet eine ringförmige Struktur im Midcell, den sogenannten Z-Ring, der hilft, um alle anderen Proteine notwendig für Zellteilung27,28zu rekrutieren. In M. Xanthus, die Bildung von Z-Ring und seine Positionierung im Midcell wird durch die drei PomXYZ Proteine16,17stimuliert. Diese drei Proteine bilden einen Chromosom verbunden sind komplex, der von der Website der Zellteilung in der "" Mutterzelle bis zur Mitte des Nucleoid in zwei Tochterzellen über die Nucleoid überträgt. Mitte der Nucleoid deckt sich mit Midcell vor dem Chromosom Segregation und hier die PomXYZ komplexe Rekruten FtsZ und stimuliert die Bildung von Z-Ring.
Hier folgten wir zunächst nicht bewegliche Zellen FtsZ-Gfpzum Ausdruck zu bringen. Da FtsZ-GFP insgesamt zeigt eine schwächere Fluoreszenz-signal als Nachwuchscontest-YFP wir erhöht die Belichtungszeit 5-fach 1 s in der GFP-Kanal. Wie erwartet, starke Anhäufung von FtsZ-GFP wurde nur bei Midcell beobachtet und diese Lokalisierung bestimmt die Position der Zellteilung Verengung (Abb. 5A). FtsZ-GFP überwiegend einen Cluster an Midcell in längeren Zelle gebildet. Es zeigte sich auch, dass diese Cluster in ihrer Intensität im Laufe der Zeit erhöht. Nach der Zellteilung beobachteten wir, dass FtsZ-GFP erneut am Midcell in die zwei Tochter gesammelt ca. 2 h später (Abb. 5B) Zellen. Dies steht im Einklang mit der Feststellung, dass etwa 50 % der Zellen in einer Population FtsZ Lokalisierung im Midcell basierend auf Snap-Shot Analyse16,17angezeigt.
In einem zweiten Experiment folgten wir nicht bewegliche ΔMglA Zellen für 24 h, die mCherry-PomXzum Ausdruck bringen. Als Teil des PomXYZ Systems hilft PomX, Z-Ringbildung und Positionierung, führen dabei anregende Zellteilung bei midcell16. Das Fluoreszenzsignal des mCherry-PomX ist stark und ermöglicht eine Belichtungszeit in den Fluoreszenz-Kanal von 250 Ms. wichtig ist, alle Zellen wuchs in der Größe und einer Zellteilung Ereignis im Laufe des Experiments, bilden mikrokolonien nach 24 h ( angezeigt Abbildung 6A). Wie bereits berichtet16enthalten fast alle Zellen ein mCherry-PomX-Clusters. Die meisten davon an Midcell und Clustern von Midcell umgesiedelt, Midcell im Laufe des Experiments lokalisiert. Während Zellteilungen wurden mCherry-PomX-Cluster mit jede tochterzelle erhält einen Cluster aufgeteilt. Im Gegensatz zu FtsZ-GFP mCherry-PomX midcell 80-90 % des Zellzyklus lokalisiert und diese Position erreicht, bald nach der Zellteilung (Abb. 6B).
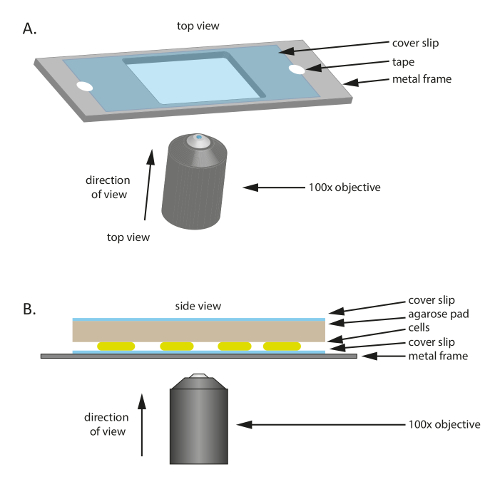
Abbildung 1 : Schema der Versuchsanordnung, die in dieser Studie verwendet. (A) A Metall oder Kunststoff Rahmen dient als Unterstützung für die Probe. Ein Deckglas wird mit Klebeband, Bewegung der Probe zu reduzieren an den Metallrahmen befestigt. (B) Seitenansicht des experimentellen Probe Set-up. Zellen werden auf das Deckglas in (A) gezeigt montiert. Die Agarose-Pad, das liefert Nährstoffe und Feuchtigkeit in den Zellen wird über die Zellen platziert. Die Agarose-Pad fällt eine zusätzliche deckgläschen um die Verdunstung zu reduzieren. Für Bilder in hoher Qualität ist ein 100 X Öl Immersion Phase Kontrast Ziel verwendet. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2 : Phase Kontrast Zeitraffer Mikroskopie von WT M. Xanthus Zellen. Zellen wurden 24 h verfolgt und Bilder alle 5 min. (A) erworben wurden repräsentative Bilder von der gleichen Sichtfeld alle 5 min. angezeigt werden. Farbige Pfeile zeigen Direktionalität der Bewegung der einzelnen Zellen. Die gleiche Farbe markiert die gleiche Zelle im Laufe der Zeit. Zahlen geben die Zeit in Minuten. Maßstab: 5 µm. (B) Bilder von der gleichen Sichtfeld nach jeder Stunde angezeigt werden. Beachten Sie, dass die gleiche Sichtfeld gezeigt aber da Zellen bewegen, Zellen sind ständig betreten und verlassen das Sichtfeld. Zahlen geben die Zeit in Stunden. Maßstab: 5 µm. PH: phase Kontrast. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 3 : Phase Kontrast Zeitraffer Mikroskopie von nicht-bewegliche M. Xanthus Zellen. ΔMglA Zellen folgten für 24 Std. Bilder alle 5 min. erworben wurden und repräsentative Bilder nach jeder Stunde angezeigt werden. Ausgewählten Zellteilung Verengungen sind mit orangefarbenen Pfeile gekennzeichnet. Zahlen geben die Zeit in Stunden. PH: Phasenkontrast. Bitte klicken Sie hier für eine größere Version dieser Figur.
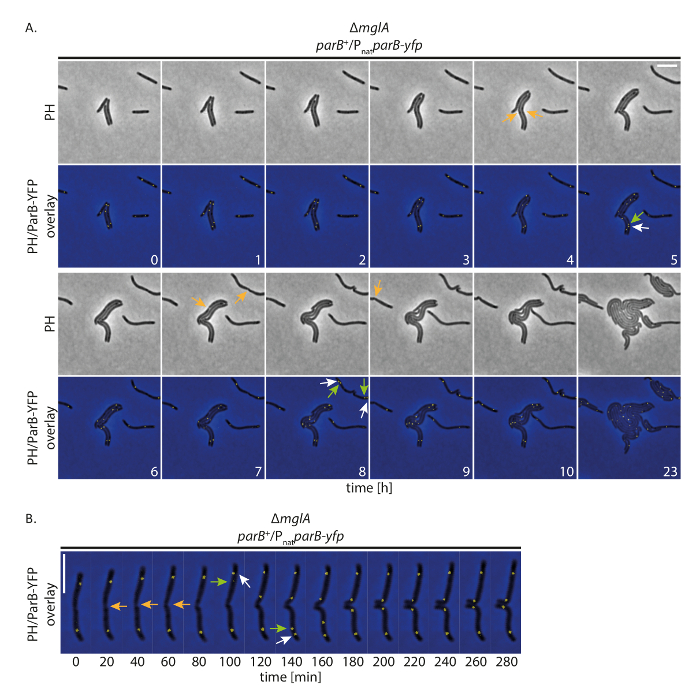
Abbildung 4 : Time-Lapse Fluoreszenzmikroskopie von Nachwuchscontest-YFP in nicht-bewegliche M. Xanthus Zellen. Zellen von einer ΔMglA mutantmit dem Ausdruck ihrer Nachwuchscontest-Yfp in Anwesenheit von native Nachwuchscontest (SA4749; ΔMglA; Nachwuchscontest +/PNatNachwuchscontest-Yfp) folgten Phase Kontrast und Fluoreszenz-Mikroskopie für 24 h. (A) Bilder erworben wurden alle 20 min und Vertreter Bilder stündlich bis 10 h gezeigt, zusammen mit den gleichen Zellen nach 24 h. Bilder im Phasenkontrast (PH) angezeigt werden und wie Überlagerung der Phasenkontrast und die YFP signal. Ausgewählten Zellteilungen sind mit orangefarbenen Pfeile gekennzeichnet. Weiße und grüne Pfeile zeigen Nachwuchscontest-YFP Cluster Vervielfältigung Veranstaltungen, mit den grünen Pfeilen markieren die translocating Cluster. Zahlen geben die Zeit in Stunden. Maßstab: 5 µm. (B) Bilder wie in (A) erworben wurden, sondern auf höhere zeitliche Auflösung gezeigt werden. Zahlen geben die Zeit in Minuten. Pfeile sind wie in (A). Maßstab: 5 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 5 : Time-Lapse Fluoreszenzmikroskopie der FtsZ-GLP in nicht-bewegliche M. Xanthus Zellen. Zellen von einer ΔMglA Mutant mit dem Ausdruck FtsZ-Gfp in Anwesenheit von native FtsZ (SA8241; ΔMglA; ftsZ +/PNatFtsZ-Gfp) folgten Phase Kontrast und Fluoreszenz-Mikroskopie für 24 h. (A) Bilder erworben wurden alle 20 min und repräsentative Bilder stündlich bis 10 h angezeigt werden, zusammen mit den gleichen Zellen nach 24 Uhr Bilder im Phasenkontrast (PH) und als Überlagerung von Phasenkontrast und GFP Signal angezeigt werden. Ausgewählten Zellteilungen sind mit orangefarbenen Pfeile gekennzeichnet. Weiße Pfeile zeigen FtsZ-GFP-Clustern auf Midcell. Zahlen geben die Zeit in Stunden. Maßstab: 5 µm. (B) Bilder wie in (A) erworben wurden, sondern auf höhere zeitliche Auflösung gezeigt werden. Zahlen geben die Zeit in Minuten. Grüne und weiße Pfeile markieren bzw. FtsZ-GFP-Cluster in den Zellen nach links und rechten. Orange Pfeile zeigen Zellteilungen. Maßstab: 5 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 6 : Time-Lapse Fluoreszenzmikroskopie von mCherry-PomX in nicht-bewegliche M. Xanthus Zellen. Non-bewegliche ΔPomX Zellen ansammeln mCherry-PomX (SA4797; ΔMglA; ΔPomX/pPomZ mCherry-PomX) folgten Phase Kontrast und Fluoreszenz-Mikroskopie alle 20 min. (A) für 24 h repräsentative Bilder stündlich bis 10 h angezeigt werden, zusammen mit den gleichen Zellen nach 24 Uhr Bilder angezeigt werden, im Phasenkontrast (PH) und als Überlagerung von Phasenkontrast und mCherry Signal. Ausgewählten Zellteilungen sind mit orangefarbenen Pfeile gekennzeichnet. Weißen und grünen Pfeile zeigen mCherry-PomX-Cluster, vor und nach Veranstaltungen, bzw. teilen. Zahlen geben die Zeit in Stunden. Maßstab: 5 µm. (B) Bilder wie in (A) erworben wurden und werden mit höheren zeitlichen Auflösung angezeigt. Pfeile sind wie in (A). Maßstab: 5 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.
| Bakterienstamm | Entsprechenden Genotyp1 | Referenz |
| DK1622 | Wildtyp | 23 |
| SA4420 | ΔmglA | 24 |
| SA4749 | ΔMglA; Nachwuchscontest+/AttB:: PNatNachwuchscontest-Yfp (pAH7) | Diese Studie |
| SA4797 | ΔMglA; ΔPomX / AttB::PPomZ mCherry-PomX (pAH53) | 16 |
| SA8241 | ΔMglA; FtsZ+/ mxan18-19::PNatFtsZ-GLP (pDS150) | Diese Studie |
| Plasmide in Klammern angegebenen gen Fusionen enthalten und wurden an den angegebenen Standorten über das Genom intergated. Plasmide integriert am Standort AttB oder die mxan18-19 -intergenetischer-Region wurden von ausgedrückt Ihre native Promotor (PNat) oder die einheimischen Förderer der PomZ (PPomZ). |
||
Tabelle 1: Liste der in dieser Studie verwendeten Bakterienstämme.
| Plasmide | Relevanten Merkmale | Referenz |
| pAH7 | PNatNachwuchscontest-Yfp; Mx8 AttP; TetR | 19 |
| pAH53 | P-PomZ - mCherry-PomX; Mx8 AttP ; KmR | 16 |
| pDS150 1 | PNatFtsZ-Gfp ; mxan18-19 ; TetR | Diese Studie |
| pMR3691 | Plasmid vanillate induzierbaren Genexpression | 18 |
| pKA51 | PNatFtsZ-Gfp ; Mx8 AttP; TetR | 17 |
| 1 pDS150: pDS150 ist ein Derivat des pKA51, in denen die Mx8 AttP Website wurde mit der mxan18-19 -intergenetischer-Region ersetzt. Dafür wurde die mxan18-19 -intergenetischer-Region verstärkt von pMR3691 mit Mxan18-19 fwd BsdRI Primer (GCGATCATTGCGCGCCAGACGATAACAGGC) und Mxan18-19 Rev BlpI (GCGGCTGAGCCCGCGCCGACAACCGCAACC) und geklont in pKA51. |
||
Tabelle 2: Liste der Plasmide, die in dieser Studie verwendet.
Discussion
Fluoreszenz live Cell Imaging ist ein mächtiges Werkzeug für die räumlich-zeitliche Dynamik der Bakterienzellen studieren geworden. Time-Lapse Fluoreszenzmikroskopie bewegliche und langsam wachsende Bakterien wie z. B. M. Xanthus, jedoch wurde schwierig und wurde nur für kurze Zeitdauer durchgeführt. Hier präsentieren wir eine einfach zu bedienende und robuste Methode für live Cell Imaging von M. Xanthus von Zeitraffer-Fluoreszenz-Mikroskopie. Diese Methode ermöglicht dem Benutzer, Zellen und Gewebekulturen markierte Proteine über mehrere Runden des Zellzyklus mit einzelligen Auflösung zu folgen.
Es gibt mehrere Voraussetzungen, die Einfluss auf den Erfolg von live-Cell Imaging der langsam wachsenden M. Xanthus Zellen, einschließlich: 1) eine feste Oberfläche für Zellhaftung; (2) die Verfügbarkeit von Nährstoffen und Sauerstoff; (3) ständige Feuchtigkeit und Temperatur; und 4) die Optimierung der experimentellen Bedingungen wie Belichtung Zeit und Bild-Übernahme-Frequenz.
In unserer Versuchsanordnung verwenden wir Dicke Agarose-Pads mit Nährstoffen ergänzt. Mit dicken Agarose Pads im Gegensatz zu mikrofluidischen Geräten, um einzelne Zellen zu folgen, hat einige grundlegende Vorteile aber auch einige Nachteile. Erstens die Agarose-Pad bietet nicht nur eine Oberfläche für M. Xanthus Zell-Anlage und Bewegung, aber auch ausreichend Nährstoffe für das Wachstum von mindestens 24 h. Zweitens, snap Shot Analysen häufig verwendet, um intrazelluläre Lokalisation der Gewebekulturen markierte Proteine zu studieren war bisher auf die gleiche Art von Agarose-Pads16,17,29. Daher können Daten aus Snap Shot Analysen direkt mit mit dem hier beschriebenen Verfahren gewonnenen Daten verglichen werden. Drittens Agarose-Pads können leicht modifiziert und ergänzt mit Antibiotika oder andere Ergänzungen wie CuSO4 und vanillate, sind weit verbreitet für Gen Ausdruck Induktion18,30. Schließlich, weil Zellen im Laufe eines Experiments Form mikrokolonien berechtigt sind, ermöglicht dieses Set-up auch Untersuchung der Wirkung von direkten Zell-Zell-Interaktionen auf die bestimmten Parameter analysiert wird. Dieser Aspekt ist besonders wichtig im Falle von M. Xanthus weil dieses Bakterium mehrere Kontakt-abhängige Interaktionen angezeigt. Der größte Nachteil dieser Methode ist, dass die experimentellen Bedingungen für die Dauer eines Experiments voreingestellt sind. Hingegen erlaubt mikrofluidischen Geräten im allgemeinen Versuchsbedingungen im Laufe eines Experiments zu ändern, indem man zum Beispiel Antibiotika31.
Kostenlose Software-Pakete (z.B., MicrobeJ, Oufti) stehen automatisch das Wachstum einzelner Zellen und Protein Lokalisierung innerhalb einzelner Zellen zu analysieren. Diese Programme sind jedoch nur gut geeignet für die Analyse von Einzelzellen oder kleine Gruppen von Zellen. So bleibt es eine Herausforderung für die 24 h-Aufnahmen, die hier beschriebenen generierten Daten automatisch zu analysieren.
Zusammenfassend lässt sich sagen, wir beschrieben, eine einfach zu bedienende und reproduzierbare Protokoll ausführen live Cell Imaging mit langsam wachsenden M. Xanthus Bakterien. Wir zeigen, dass einfache Nährstoff ergänzt Agarose-Pads ausreichend sind, um nachhaltiges Wachstum für mindestens 24 Stunden und für die Beobachtung und Analyse von Protein-Lokalisierung und Wachstum mit einer einzelnen Zelle Auflösung über mehrere Generationen hinweg zu ermöglichen.
Disclosures
Die Autoren erklären, dass sie keine finanziellen Interessenkonflikte.
Acknowledgments
Diese Arbeit wurde unterstützt durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Transregio 174 "räumlich-zeitliche Dynamik von bakteriellen Zellen" und der Max-Planck-Gesellschaft.
Materials
| Name | Company | Catalog Number | Comments |
| DMI6000B with AFC | Leica microsystems | 11888945 | Automated inverted widefield fluorescence microscope with adaptive focus control |
| Universal mounting frame | Leica microsystems | 11532338 | Stage holder for different sample sizes |
| HCX PL FLUOTAR 100x/1.30 oil PH3 | Leica microsystems | 11506197 | Phase contrast objective |
| Orca Flash 4.0 camera | Hamamatsu | 11532952 | 4.0 megapixel sCMOS camera for picture aquisition |
| Filter set TXR ET, k | Leica microsystems | 11504170 | Fluorescence filter set, Ex: 560/40 Em: 645/75 |
| Filter set L5 ET, k | Leica microsystems | 11504166 | Fluorescence filter set, Ex: 480/40 Em: 527/30 |
| Filter set YFP ET, k | Leica microsystems | 11504165 | Fluorescence filter set, Ex: 500/20 Em: 535/30 |
| ProScan III | Prior | H117N1, V31XYZEF, PS3J100 | Microscope automation controller with interactive control center |
| EL 6000 light source | Leica microsystems | 11504115 | External fluorescence light source |
| Incubator BLX Black | Pecon | 11532830 | Black incubation chamber surrounding the microscope |
| Tempcontrol 37-2 digital | Leica microsystems | 11521719 | Automated temperature control for incubation chamber |
| Gentmycin sulphate | Carl Roth | 0233.4 | Gentamycin |
| Oxytetracylin dihydrate | Sigma Aldrich | 201-212-8 | Oxytetracyclin |
| Kanamycin sulphate | Carl Roth | T832.3 | Kanamycin |
| Filtropur BT25 0.2 bottle top filter | Sarstedt | 831,822,101 | Bottle top filter for sterilization of buffers |
| Deckgläser | VWR | 630-1592 | Glass cover slip (60 x 22 mm, thickness: 0.7 mm) |
| Seakem LE agarose | Lonza | 50004 | Agarose for microscopy slides |
| Leica Metamorph AF | Leica microsystems | 11640901 | Microscope control software and software for picture analysis |
| Tetraspeck Microsperes, 0.5 µm | ThermoFisher | T7281 | Fluorescent microspheres |
| petri dish | Greiner Bio-one | 688102 | 120 mm x 120 mm x 17 mm squared petri dish for agarose pads |
| BD Bacto Casitone | Becton Dickinson | 225930 | Casitone |
| Parafilm M | VWR | 291-1213 | Parafilm |
| Tris(hydroxymethyl)-aminomethane | Carl Roth | AE15.2 | Tris |
| Magnesium sulphate heptahydrate | Carl Roth | P027.2 | Magnesium sulphate |
| Potassium dihydrogen phosphate p.a. | Carl Roth | 3904.1 | Potassium dihydrogen phosphate |
| 1% CTT medium: 1 % (w/v) BD Bacto™ casitone, 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Cultivation medium for M.xanthus | ||
| TPM buffer: 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Buffer for preparation of microscopy slides for M.xanthus |
References
- Shapiro, L., McAdams, H. H., Losick, R. Why and how bacteria localize proteins. Science. 326 (5957), 1225-1228 (2009).
- Treuner-Lange, A., Søgaard-Andersen, L. Regulation of cell polarity in bacteria. J Cell Biol. 206 (1), 7-17 (2014).
- Laloux, G., Jacobs-Wagner, C. Spatiotemporal control of PopZ localization through cell cycle-coupled multimerization. J Cell Biol. 201, 827-841 (2013).
- Rudner, D. Z., Losick, R. Protein subcellular localization in bacteria. Cold Spring Harb. Perspect. Biol. 2 (4), 000307 (2010).
- Badrinarayanan, A., Le, T. B. K., Laub, M. T. Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol. 31 (1), 171-199 (2015).
- Munoz-Dorado, J., Marcos-Torres, F. J., Garcia-Bravo, E., Moraleda-Munoz, A., Perez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front Microbiol. 7, 781 (2016).
- Berleman, J. E., Kirby, J. R. Deciphering the hunting strategy of a bacterial wolfpack. FEMS Microbiol Rev. 33 (5), 942-957 (2009).
- Konovalova, A., Petters, T., Søgaard-Andersen, L. Extracellular biology of Myxococcus xanthus. FEMS Microbiol. Rev. 34, 89-106 (2010).
- Nudleman, E., Wall, D., Kaiser, D. Cell-to-cell transfer of bacterial outer membrane lipoproteins. Science. 309, 125-127 (2005).
- Vassallo, C. N., et al. Infectious polymorphic toxins delivered by outer membrane exchange discriminate kin in myxobacteria. eLife. 6, 29397 (2017).
- Vassallo, C., et al. Cell rejuvenation and social behaviors promoted by LPS exchange in myxobacteria. Proc Natl Acad Sci USA. 112 (22), 2939-2946 (2015).
- Li, Y., et al. Extracellular polysaccharides mediate pilus retraction during social motility of Myxococcus xanthus. Proc. Natl. Acad. Sci. USA. 100, 5443-5448 (2003).
- Kim, S. K., Kaiser, D. Cell alignment required in differentiation of Myxococcus xanthus. Science. 249, 926-928 (1990).
- Lobedanz, S., Søgaard-Andersen, L. Identification of the C-signal, a contact dependent morphogen coordinating multiple developmental responses in Myxococcus xanthus. Genes Dev. 17, 2151-2161 (2003).
- Schumacher, D., Søgaard-Andersen, L. Regulation of cell polarity in motility and cell division in Myxococcus xanthus. Annu Rev Microbiol. 71 (1), 61-78 (2017).
- Schumacher, D., et al. The PomXYZ proteins self-organize on the bacterial nucleoid to stimulate cell division. Dev Cell. 41 (3), 299-314 (2017).
- Treuner-Lange, A., et al. PomZ, a ParA-like protein, regulates Z-ring formation and cell division in Myxococcus xanthus. Mol Microbiol. 87 (2), 235-253 (2013).
- Iniesta, A. A., Garcia-Heras, F., Abellon-Ruiz, J., Gallego-Garcia, A., Elias-Arnanz, M. Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl-beta-D-thiogalactopyranoside or vanillate. J Bacteriol. 194 (21), 5875-5885 (2012).
- Harms, A., Treuner-Lange, A., Schumacher, D., Søgaard-Andersen, L. Tracking of chromosome and replisome dynamics in Myxococcus xanthus. reveals a novel chromosome arrangement. PLoS Genet. 9 (9), 1003802 (2013).
- Iniesta, A. A. ParABS system in chromosome partitioning in the bacterium Myxococcus xanthus. PLoS One. 9 (1), 86897 (2014).
- Lin, L., Osorio Valeriano, M., Harms, A., Søgaard-Andersen, L., Thanbichler, M. Bactofilin-mediated organization of the ParABS chromosome segregation system in Myxococcus xanthus. Nat Commun. 8 (1), 1817 (2017).
- Hodgkin, J., Kaiser, D. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc Natl Acad Sci U S A. 74 (7), 2938-2942 (1977).
- Kaiser, D. Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc Natl Acad Sci USA. 76 (11), 5952-5956 (1979).
- Miertzschke, M., et al. Structural analysis of the Ras-like G protein MglA and its cognate GAP MglB and implications for bacterial polarity. EMBO J. 30 (20), 4185-4197 (2011).
- Hodgkin, J., Kaiser, D. Genetics of gliding motility in Myxococcus xanthus. (Myxobacterales): Two gene systems control movement. Mol Gen Genet. 171, 177-191 (1979).
- Shi, X., et al. Bioinformatics and experimental analysis of proteins of two-component systems in Myxococcus xanthus. J Bacteriol. 190 (2), 613-624 (2008).
- Bi, E. F., Lutkenhaus, J. FtsZ ring structure associated with division in Escherichia coli. Nature. 354 (6349), 161-164 (1991).
- Lutkenhaus, J., Pichoff, S., Du, S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton. 69 (10), 778-790 (2012).
- McLoon, A. L., et al. MglC, a Paralog of Myxococcus xanthus GTPase-Activating Protein MglB, Plays a Divergent Role in Motility Regulation. J Bacteriol. 198 (3), 510-520 (2015).
- Gomez-Santos, N., et al. Comprehensive set of integrative plasmid vectors for copper-inducible gene expression in Myxococcus xanthus. Appl Environ Microbiol. 78 (8), 2515-2521 (2012).
- Treuner-Lange, A., et al. The small G-protein MglA connects to the MreB actin cytoskeleton at bacterial focal adhesions. J Cell Biol. 210 (2), 243-256 (2015).
