

Summary
Ici, nous présentons une approche combinatoire utilisant la microscopie à haute résolution, les outils de calcul, et l’étiquetage à une seule cellule dans les embryons vivants de C. elegans pour comprendre la dynamique de cellule unique pendant le neurodéveloppement.
Abstract
Caenorhabditis elegans (C. elegans) se distingue comme le seul organisme dans lequel le défi de comprendre les origines cellulaires d’un système nerveux entier peut être observé, avec une résolution de cellule unique, in vivo. Ici, nous présentons un protocole intégré pour l’examen du neurodéveloppement chez les embryons de C. elegans . Notre protocole combine l’imagerie, le ligage et le traçage neuroanatomique des cellules individuelles dans les embryons en développement. Nous atteignons l’imagerie à long terme en quatre dimensions (4D) des embryons vivants de C. elegans avec une résolution spatiale presque isotrope grâce à l’utilisation de la microscopie à double vue à plan sélectif inversé (dispim). Les noyaux et les structures neuronales dans les embryons de nématodes sont imagés et fusionné isotropiquement pour produire des images avec une résolution de ~ 330 nm dans les trois dimensions. Ces ensembles de données 4D à haute résolution minute par minute sont ensuite analysés pour corréler les identités de lignée cellulaire définitives avec l’expression génique et la dynamique morphologique à des niveaux de détail à une seule cellule et sous-cellulaire. Notre protocole est structuré pour permettre la mise en œuvre modulaire de chacune des étapes décrites et améliorer les études sur l’embryogenèse, l’expression génique ou le neurodéveloppement.
Introduction
C. elegans se distingue comme le seul organisme dans lequel chaque cellule de l’embryon peut être observée tout au long du neurodéveloppement. Avec l’ensemble de la lignée cellulaire connue et invariante1, et avec le développement de nouveaux outils qui permettent l’étiquetage et l’imagerie continue des cellules individuelles dans les embryons, les biologistes peuvent maintenant commencer à examiner différentes étapes dans le développement du nématode nerveux système de tous les angles-la naissance des cellules; migration et différenciation; la formation de neurites, l’excroissance ciblée et la fasciculation; formation de Synapse; et le réglage des circuits fonctionnels. La capture de la dynamique de l’excroissance neuronale dans l’embryon de C. elegans , en combinant des reporters exprimés de façon stable et la microscopie à fluorescence, est précieuse pour la communauté scientifique.
Les études de développement chez C. elegans tirent souvent parti de la lignée cellulaire invariante et des cartes de la cellule-destin de cette espèce pour augmenter la compréhension contextuelle au niveau de la cellule unique au sein de l’organisme intact1. Analyse d’auto-linéarité-utilisation de starrynite2,3,4 et acetree5,6,7,8 Software-avantages du contraste élevé, haute résolution images de noyaux fluorescents. Pour fonctionner de façon optimale, StarryNite/AceTree dépend également de l’orientation prévisible des embryons imagés pendant le développement. La microscopie confocale, faite en C. elegans embryons compressés entre deux lamelles, a été la méthode standard de microscopie auto-linéante depuis plus d’une décennie parce qu’elle fournit à la fois un contraste élevé/haute résolution et une contrainte prévisible orientation de l’embryon7,8. Nous avons décrit précédemment la construction et l’utilisation d’un microscope d’illumination à plan sélectif inversé à double vue à base de feuilles légères (dispim) pour l’imagerie d’échantillons vivants tel que C. elegans embryogenèse9,10 , le 11 , le 12 , 13. la microscopie en feuilles légères, en général, fournit une faible phototoxicité, une haute vitesse et une imagerie à long terme des spécimens 3D vivants14,15. La méthode diSPIM, en particulier, produit des images en quatre dimensions (4D) avec une résolution spatiale presque isotrope d’environ 330 nm9.
Comparé à la microscopie confocale, le diSPIM offre un signal-bruit et une vitesse plus élevés, une résolution spatiale plus isotrope, et est plus approprié pour l’imagerie in vivo à long terme16. Nous avons donc travaillé à adapter les données du diSPIM pour l’entrée dans StarryNite/AceTree et nous avons étudié si cela améliorerait les analyses de linéarité. Un obstacle majeur est que les spécimens de diSPIM ne peuvent pas être facilement contraints par la compression de coquille d’oeuf pour adopter les orientations attendues pour StarryNite/AceTree. L’orientation aléatoire des positions des cellules dans le volume analysé dégrade la précision de l’analyse d’auto-linéarité.
Nous avons donc employé CytoSHOW, une interface utilisateur guidée par le spectateur qui permet aux utilisateurs de sélectionner une orientation 3D précise des embryons lors du pré-traitement des images diSPIM, produisant des données d’image à la fois optimisées pour la qualité et prenant en compte le contexte pour l’entrée dans StarryNite /AceTree. Lors de la sélection des embryons imagés par l’utilisateur, CytoSHOW orchestre un pipeline automatisé de traitement des données. Les images d’embryons recadrées et en arrière-plan sont enregistrées dans les fichiers de pile TIFF pour chaque position, point de temps et vue. Cytoshow appelle alors itérativement le programme spimfusion pour co-enregistrer et décongestirer conjointement les deux vues prétraitées, en utilisant l’algorithme Richardson-Lucy17,18 pour produire des images volumétriques à haute résolution isotrope. Un ensemble de paramètres spécifique au diSPIM a été optimisé pour StarryNite pour régir son comportement lors de la segmentation d’image et du suivi des noyaux dans les images fusionnées isotropiquement. Les images fusionnées et les résultats d’alignement sont ensuite modifiés à l’aide d’AceTree, ce qui permet aux utilisateurs d’identifier et de corriger toutes les erreurs dans la trace de la lignée automatique générée par StarryNite. AceTree peut également présenter l’arbre de lignage et les rendus modélisés en 3D des noyaux suivis dans l’embryon. Nous constatons que la vitesse et la précision d’auto-linéarité sont nettement améliorées à l’aide d’images isotropiquement fusionnées, par rapport aux images brutes de l’une ou l’autre des caméras SPIM. Notre protocole, tout en étant optimisé pour l’application C. elegans décrite ici, pourrait être généralement adapté pour l’auto-ligage des données dispim produites pour d’autres espèces ou spécimens. S’il s’agit de l’utilisation prévue du protocole, veuillez noter qu’un réglage supplémentaire des paramètres starrynite sera probablement exigé pour les nouveaux spécimens, comme décrit3,4.
La mise en œuvre réussie de ce protocole aboutit à des images avec une résolution 4D-isotrope et permet aux biologistes de tracer les lignées cellulaires, tout en identifiant et analysant simultanément les neurones dans l’embryon de C. elegans en développement. En outre, en fusionnant plusieurs algorithmes de post-traitement-avec l’accélération matérielle étant le plus chronophage de ces derniers-, nous pouvons maintenant analyser à la fois les détails subcellulaires fins et les lignées cellulaires et les cellules-Fates d’embryons vivants en temps réel. Ce nouveau protocole permet une manipulation précise et éclairée du comportement cellulaire lors d’études de différenciation et de morphogenèse in vivo. Dans ce manuscrit, nous présentons une explication détaillée des protocoles améliorés que nous avons développés pour le lignage et le suivi cellulaire dans le développement des embryons de C. elegans , pour améliorer les études sur l’embryogenèse, l’expression génique ou le neurodéveloppement.
Protocol
1. assemblage de la chambre d’imagerie en acier dispim avec lamelle enduit poly-L-lysine
NOTE: les étapes ci-dessous sont toutes nécessaires pour optimiser et automatiser l’analyse de lignée d’embryons de nématodes par StarryNite/acetree. Plusieurs options (indiquées en tant que telles) peuvent être omises pour des expériences nécessitant le traçage de lignées cellulaires C. elegans .
- Dessinez un petit rectangle (2mm x 5mm) au centre d’une lèvre rectangulaire propre (n ° 1,5, 24 mm x 50 mm) avec un Sharpie (ou un stylo similaire).
- Retournez la lèvre sur le côté non marqué et placez 10 μL de poly-L-lysine (cat. no. P1524) sur le rectangle marqué.
NOTE: faire une concentration de 1 mg/mL de poly-L-lysine dissous dans de l’eau filtrée (ou équivalent). Pour le stockage à long terme, préparer 5 à 10 mL d’aliquotes et conserver à − 20 ° c. Une fois décongelés, les aliquotes peuvent être stockés à température ambiante (23 ° c) pendant 3 à 4 semaines. - Laisser la poly-L-lysine enduire la lamelle pendant 5 minutes (figure 1a). Poly-L-lysine est utilisé pour le revêtement des feuillets de couverture en verre où les embryons seront montés, permettant à la coquille d’oeuf d’embryon de coller fermement à la lèvre, même lorsque les objectifs sont immergés dans le milieu d’imagerie.
- Placer la lamelle revêtue de poly-L-lysine dans la moitié inférieure de la chambre d’imagerie en acier.
- Placer la moitié supérieure de la chambre d’imagerie en acier sur la moitié inférieure avec la lèvre et serrer avec les quatre vis associées à la chambre. Vérifier visuellement sur le côté pour s’assurer que la moitié supérieure est uniformément assise dans la moitié inférieure (figure 1b).
- Remplissez la chambre avec 7 – 8 mL de tampon M919, un tampon isotonique qui aide à empêcher les embryons de stade précoce de succomber à une pression osmotique anormale. 1-Cell, et même les embryons à 2 cellules sont osmotiquement sensibles et peuvent se développer anormalement sinon dans ce tampon isotonique. Les embryons ont également tendance à arrêter au stade 3 fois si M9 est substitué avec de l’eau.
2. préparation des échantillons d’embryons de C. elegans pour le montage
Remarque: environ 18 heures avant l’imagerie, cinq jeunes (1 jour depuis la mue finale) adultes et dix larves de stade 4 (L4) C. elegans sont prélevés sur une gélose à milieu de croissance des nématodes (NGM) ensemencée avec la souche OP50 de E. coli . Les pics de fil de platine sont utilisés pour déplacer les larves et les jeunes adultes C. elegans sans nuire à l’animal19.
- Préparer un 1% de méthylcellulose (cat. no. H7509-25G) solution dans le tampon M9.
Remarque: la cellulose méthylique doit être agitée au chaud M9 jusqu’à ce qu’elle soit dissoute. Une fois préparée, cette solution peut être stockée à température ambiante. - Ajouter 500 μL de solution de méthylcellulose-in-M9 à 1% dans la dépression d’une glissière de microscope concave. Ce tampon visqueux sera utilisé en deux occasions: 1) lors de la récolte des embryons précoces par dissection des vers adultes et 2) lors du lavage des embryons tardifs prélevés directement à partir d’une plaque NGM.
Remarque: la cellulose méthylique est utilisée pour empêcher les embryons de coller à la glissière du microscope. - Pour l’imagerie des embryons tardifs, prélever des embryons de C. elegans (de préférence à partir d’une plaque NGM avec des jeunes adultes présents) en utilisant un pic de cils et déplacer les embryons vers la cellulose de méthyle à 1% sur la glissière concave du microscope. Le pic de cils aide à réduire la force et réduit ainsi le stress ou les dommages aux embryons pendant la manipulation. La procédure pour faire un choix de cils est couverte par Hart20.
- Avec un deuxième pic de cils (dans la main opposée), Tapotez doucement les deux cils ensemble pour suspendre les embryons dans la cellulose méthylique.
- Facultatif: Si vous envisagez de lignage des cellules embryonnaires avec StarryNite, il faut monter des embryons de 1 cellule à 4 cellules. Pour ce faire, sélectionnez d’abord les jeunes adultes à partir d’une plaque NGM et déplacez-les dans la solution de cellulose M9-méthylique sur la glissière de microscope concave à l’aide d’un prélèvement de fil de platine.
- Facultatif: avec les pointes aiguisées des aiguilles hypodermiques (n ° 18G x 1 1/2), trancher l’animal transversalement au milieu du corps pour libérer des embryons de 1 cellule à 4 cellules.
3. la pipette à bouche: assemblage du tube de l’aspirateur avec une pipette microcapillaire
Remarque: nous utilisons un tube aspirateur avec une pipette microcapillaire tirée à la main insérée dans le joint en caoutchouc du tube. Cela nous permet de transférer des embryons de la lame de dissection à la surface enduite de poly-L-lysine dans la chambre d’imagerie remplie de tampons.
- Tirez manuellement la pipette microcapillaire sur une flamme ouverte pour créer deux moitiés avec des pointes étirées.
- Prendre une moitié de la pipette microcapillaire et insérer l’extrémité émoussée dans le joint en caoutchouc du tube de l’aspirateur (figure 1c). Mettre l’autre moitié de la pipette microcapillaire de côté pour une utilisation ultérieure (si nécessaire).
- Avec le tube d’aspiration assemblé muni d’une pipette microcapillaire, casser délicatement la pointe de la pipette microcapillaire et créer une ouverture qui s’adaptera à environ 1 – 2 deux embryons (à partir d’ici sur cet instrument est appelé «pipette à bouche»).
4. montage des embryons de C. elegans sur la lamelle enduite de poly-L-lysine
- Avec l’embouchure de l’aspirateur maintenu doucement entre les dents, pré-remplir la pipette microcapillaire avec 10 – 15 μL de tampon M9, puis aspirer doucement plusieurs embryons de la glissière concave dans le capillaire.
- Transférer les embryons dans la chambre d’imagerie en acier remplie de tampon M9, en positionnant la pointe capillaire de façon à ce que les embryons tombent dans le rectangle central de la Coverslip.
- En évitant de blesser les embryons, déplacez-les doucement avec un pic à cils ou utilisez la pipette pour positionner les embryons verticalement, pour orienter les embryons de sorte que l’axe long de l’embryon soit perpendiculaire à l’axe long de la lèvre (figure 1b encart , panneau inférieur).
Remarque: le positionnement de l’embryon dans cette orientation minimise le nombre de tranches à l’image, réduisant ainsi le dosage de la lumière et le temps de traitement des données tout en améliorant la vitesse d’acquisition. - Placer la chambre d’imagerie en acier dans le porte-échantillon au niveau du microscope (figure 1d).
5. montage, configuration logicielle et optimisation laser pour l’imagerie embryonnaire à l’aide du diSPIM
- Voir les instructions étape par étape sur la façon d’assembler l’ensemble du diSPIM à fibre optique à partir de pièces disponibles dans le commerce dans Kumar et al.10,11 et à http://www.dispim.org. Un protocole vidéo sur la façon d’assembler le diSPIM est également disponible sur le site Web de l’ASI (http://www.asiimaging.com).
Remarque: la configuration de l’instrument pour ce protocole est identique à Kumar et al.10,11 dispim, qui utilise des lentilles d’immersion 40x 0.8 na pour l’imagerie. La seule différence entre la configuration dans ce protocole et Kumar et al.10,11 est l’addition d’un miroir dichroïque (fractionnement à 560 nm) et de filtres passe-bande rouge et vert à l’intérieur d’un dispositif de fractionnement d’imagerie (modèle A12801-01) installé sur les deux bras d’imagerie du diSPIM. L’ajout de l’optique de fractionnement d’image permet la capture simultanée d’images de deux fluorophores distincts – excités par des lasers de 561 nm et 488 nm – en séparant les bandes d’émission sur deux moitiés de la même puce de caméra. - Après l’assemblage de l’instrument, vérifiez l’alignement optique du diSPIM avant l’imagerie.
Remarque: pour aligner correctement le diSPIM Voir https://youtu.be/qnOrg30NNuE, et pour les informations matérielles, http://dispim.org/hardware/objectives et http://www.asiimaging.com. - Utilisez la plate-forme open-source micro-Manager (https://micro-manager.org/)21, qui a été optimisé pour l’utilisation de microscopes de feuille de lumière pour l’imagerie cellulaire à haut débit22. Nous recommandons l’utilisation du plugin ASI diSPIM pour l’acquisition multi-positions, qui permet l’imagerie simultanée de jusqu’à 30 embryons comme décrit23.
- Avec le micro-Manager ouvert, réglez les intensités laser à ~ 179 μW (0,5) pour 488 nm et ~ 79 μW (0,25) pour 561 nm (figure 2a, rectangle rouge).
Remarque: il s’agit des paramètres recommandés pour l’imagerie à long terme des embryons de C. elegans utilisant des intervalles d’une minute. Au cours de l’imagerie à deux couleurs à long terme, le laser 561 nm est utilisé pour l’image noyaux (mCherry:: histone) jusqu’à ce que les embryons sont au stade de haricot, à quel point le laser 488 nm est ensuite allumer à l’image aussi les neurones étiquetés GFP. Ces conditions d’imagerie sont optimisées pour minimiser la phototoxicité et assurer la survie et l’éclosion des embryons tout en permettant une acquisition continue prolongée (12 – 14 heures) de données de neurodéveloppement et de linéarité. - Dans le micro-gestionnaire, choisissez les plugins de menu > contrôle de dispositif > ASI dispim pour ouvrir la fenêtre de Dispim d’ASI (figure 2b). Choisissez l’onglet acquisition . Dans la section paramètres d' enregistrement des données de cet onglet (rectangle vert), section paramètres de volume (carré bleu) et section paramètres de tranche (carré orange), assurez-vous que chaque paramètre est défini comme illustré à la figure 2B.
Remarque: notre logiciel d’analyse d’image CytoSHOW est adapté pour fonctionner avec d’autres formats de données de sortie facultatifs tels que les séries de fichiers OME-TIFF concaténées en vrac et les séries de fichiers TIFF-Stack créées après l’acquisition par l’utilisation d’une fonction d’exportation intégrée dans micro-Manager. En règle générale, le format de données de fichier OME-TIFF à pile point unique est utilisé car il permet l’affichage et le traitement en temps réel du volume d’image dès que les données brutes sont acquises.
6. paramètres d’autofocus optimisés pour l’imagerie à long terme des embryons de C. elegans
- Définissez les paramètres de l’autofocus micro-Manager sur les paramètres optimisés pour l’imagerie diSPIM de qualité de lignée à long terme des embryons de C. elegans . Dans la fenêtre ASI diSPIM, cliquez sur l’onglet autofocus (Figure 2c). Dans la section options d’autofocus générales (carré noir), spécifiez les paramètres exactement comme illustré. Notez que le canal autofocus (carré rouge) doit spécifier votre canal de fluorescence de canal nucléaire dans des expériences de linéarité.
Remarque: si le décalage maximum est supérieur à 5 μm, les images tendent à dériver hors de la mise au point. - Cliquez, Plugins > outils d’acquisition > superposition de motif.
- Dans la fenêtre superposition de modèle , cliquez sur afficher la grille.
- Dans la fenêtre ASI diSPIM , cliquez sur l’onglet de navigation .
- Cliquez sur les cases à cocher pour poutre et feuille de chemin A ou B, puis cliquez sur Live. L’acquisition d’images commence. Une fenêtre affichage en direct s’ouvre. Sélectionnez la région d’analyse du focus automatique de l’embryon en dessinant une boîte autour de l’embryon sur le canal sélectionné à partir de 6,1.
Remarque: nous capturons généralement 420 points de temps pour 10 embryons par session d’imagerie. Les données brutes par session d’image sont typiquement 1,7 to, alors que les données traitées par StarryNite et décongestiées sont de 1,4 to (voir les étapes 9 et 10). Nous vous recommandons d’utiliser un disque dur de grande capacité (18 to sur notre système actuel) pour l’acquisition d’images et des plateformes Cloud pour le stockage d’images. - Cliquez sur Démarrer l’acquisition dans l’ongletacquisition pour commencer la capture d’images multidimensionnelles à long terme (figure 2b).
7. ouverture des images RAW de micro-Manager dans CytoSHOW
- Téléchargez le bundle de logiciels à partir de http://dispimlineage.wormguides.org.
Remarque: le bundle de logiciels sera téléchargé en tant que fichier. zip et devra être extrait dans le «C:\» Répertoire avant utilisation. D’autres détails pour l’installation sont donnés à http://dispimlineage.wormguides.org/diSPIMlineaging_InstallationInstructions.htm. - Double-cliquez sur le fichier C:\cytoshowextrasforc\cytoshow_app.jnlp pour commencer à exécuter cytoshow.
- Choisissez le fichier de menu > nouveau > moniteur Dispim (micro-gestionnaire). Localisez le dossier de jeu de données racine où les dossiers de point d’acquisition ont été enregistrés. Sélectionnez n’importe quel dossier de point de temps et cliquez sur ouvrir. Les fenêtres de navigation multidimensionnelles (appelées Windows Monitor Dispim ) sont automatiquement ouvertes pour SPIMA et spimb (figure 3A).
Remarque: ces fenêtres surveillent le dossier de données racine pour les piles de point de temps brutes nouvellement enregistrées (dans le cas où un spécimen est encore en cours d’enregistrement). Après l’acquisition de chaque nouveau point de temps, chacune des fenêtres contrôlant les positions distinctes des bras et des échantillons SPIM sera actualisée pour afficher l’intégralité du jeu de données 4D multicanaux pour chaque embryon.
8. générer des images de projection Max avec CytoSHOW
Même avant la déconcentration, les données brutes peuvent être traitées rapidement pour évaluer les caractéristiques globales du spécimen.
- Cliquez sur le bouton Z-MIP sur le panneau latéral gauche de la fenêtre de l’image (figure 3A, rectangle rouge) pour effectuer des projections d’intensité maximale à travers le cours complet de profondeur et à temps plein d’une position donnée ou d’un bras SPIM. Une fenêtre d' hyperstack Z-projection apparaîtra.
- Dans la fenêtre de l' hyperstack Z-projection , définissez le type de projection sur intensité maximale. Spécifiez les canaux, les tranches et les trames de point temporel à traiter en fonction des préférences de l’utilisateur.
- Cliquez sur OK lorsque vous avez terminé.
- Sélectionnez l’emplacement du dossier pour enregistrer les sorties d' intensité max dans la fenêtre de dialogue du fichier, puis cliquez sur OK. Accordez un certain temps (15 à 20 minutes, en fonction de la taille du jeu de données et de la puissance de traitement de l’ordinateur) pour que CytoSHOW génère des images de projection.
9. analyse des lignées cellulaires dans les données volumétriques isotropes à haute résolution
- Facultatif: avec les données brutes ouvertes via le moniteur Dispim dans cytoshow, sélectionnez l' outil de sélection de polygone (figure 3A, flèches noires) et cliquez juste à l’extérieur des arêtes antérieures, postérieures, dorsale et ventrale de l’embryon (dans cet ordre précis) pour créer un motif «noeud papillon» sur l’embryon. Faites pour les deux vues (SPIM-A et SPIM-B, figure 3A).
NOTE: cette sélection spécifie la région elliptique d’intérêt (ROI) dans laquelle l’embryon est centré et enregistre l’axe antérieur-postérieur de l’embryon. Les repères de motif de noeud papillon Cytomontrent que l’utilisateur envisage de préciser une rotation précise des volumes finaux fusionnés isotropiquement dans une orientation qui est optimale pour les analyses d’alignement par StarryNite/AceTree. Dans les cas où l’alignement de StarryNite ne fait pas partie du plan expérimental, d’autres outils et formes de sélection peuvent être choisis pour définir le ROI pour le traitement d’image. - Si plusieurs embryons ont été simultanément imagés à l’aide de l’option d’acquisition multi-positions, ouvrez et exécutez l’étape 9,1 pour tous les embryons. Cela permettra l’exécution parallèle des étapes futures pour tous les embryons en une seule session. Fermez les fenêtres SIMA et SPIMB pour tous les embryons que vous ne souhaitez pas traiter.
- Cliquez sur le bouton dispim sur le panneau latéral gauche de la fenêtre du moniteur dispim (figure 3A, surligné en jaune). Cela révèle un sous-panel de contrôles spécifiques au traitement diSPIM.
- Alignez les canaux vert et rouge pour chaque bras SPIM. Étant donné que les répartiteurs de canaux d’émission sont utilisés pour capturer des images rouges et vertes distinctes simultanément sur la même caméra, il est important d’aligner visuellement l’enregistrement exact des pixels de ces deux champs d’image physiquement adjacents lorsqu’ils sont superposées. La réutilisation des mêmes ajustements d’alignement est généralement réalisable sur de nombreuses sessions d’imagerie consécutives, mais doit être vérifiée (comme dans les étapes 9.4.1 – 9.4.5).
- En commençant par le panneau SPIMA, sélectionnez le canal rouge en déplaçant la barre de défilement cm vers la gauche (figure 3A, flèche orange, panneau gauche).
- En utilisant les Ajusteurs x, y et z (figure 3A, carré orange), décaler le canal rouge pour qu’il corresponde au vert.
- Cliquez sur le bouton Dispim (figure 3A, surligné en jaune), pour fermer le sous-panneau et déclencher la propagation des mêmes décalages à toutes les autres fenêtres de position .
- Confirmez que l’alignement correct est propagé à d’autres trames et points de temps en déplaçant la barre de défilement «z» (figure 3A, flèche bleue, panneau de gauche) et/ou la barre de défilement «t» (figure 3A, flèche verte, panneau de gauche). Si l’acquisition multiposition a été effectuée et que plusieurs embryons ont été imagés (étape 5,3), l’alignement devrait également être propagé à ces embryons. Confirmez en examinant également les nombres pour les Ajusteurs x, y et z (figure 3A, carré orange, qui devrait être le même pour le panneau SPIMA de tous les embryons).
- Répétez les étapes 9.4.1 – 9.4.4 pour la fenêtre du moniteur SPIMB diSPIM (figure 3A, panneau de droite).
- Cliquez sur le bouton "Dispim", puis sur le bouton "fusible" (figure 3A, rectangle bleu) pour ouvrir une boîte de dialogue appelée "Deconvolve/FUSE Dispim Raw Data volumes" (figure 3b). Réglez les paramètres comme illustré à la figure 3b. Ces paramètres sont brièvement abordés dans les sous-étapes suivantes:
- Réglez l’enregistrement de la clé sur le canal 1 (488 nm laser) ou 2 (561 nm laser). Sélectionnez le canal avec un signal plus dense ou plus omniprésent. Pour les expériences d’alignement, sélectionnez toujours le canal utilisé pour l’image de la fluorescence de l’histone nucléaire omniprésente.
- Définissez l' orientation relative des volumes d’entrée sur + 1 ou-1. L’index d’orientation correct dépend d’un placement de caméra spécifique au diSPIM (figure 4).
Remarque: si incertain, testez chaque option en dupliquant un seul point de temps à partir des deux SPIM A et B Dispim moniteur fenêtre, suivant les étapes 9.1 – 2/9 et arbitrairement choisir un volume d’entrée relative orientation à tester. Des orientations incorrectes produirait des images floues avec des artefacts, tandis que les orientations correctes donneront des images claires. La valeur d’orientation relative des volumes en entrée qui donne l’image claire peut ensuite être réutilisée pour toutes les données futures de l’instrument dispim donné. - Indiquez si le volume fusionné doit être orienté de la même manière que le volume d’entrée A ou B (en fonction de la préférence de l’utilisateur).
- Sélectionnez "frais d’inscription pour chaque volume". Cette option contrôle la façon dont SpimFusion calcule les matrices d’enregistrement pour chaque paire de volumes à chaque point de temps. L’option "Fresh" permet à l’algorithme d’optimiser l’enregistrement de manière adaptative à chaque point de temps.
- Définissez le nombre d’itérations de déconvolution sur 10. Ce nombre tend à produire de façon fiable la haute résolution souhaitée de manière efficace dans le temps.
- Facultatif: si le ligage automatique est désiré (fortement recommandé), vérifiez l’alignement automatique de StarryNite des volumes fusionnés. Cette option lancera StarryNite automatiquement pour segmenter et suivre les cellules dans les volumes imagés produits par SpimFusion.
- Facultatif: pour une précision maximale dans l’alignement automatisé, il est préférable de repositionner les volumes d’embryons fusionnés isotropiquement dans l’orientation canonique «ADL» (Anterior [x-West], Dorsal [y-North], Left [z-near]). Sélectionnez l’option définir l’orientation de la sortie du volume dans l’aperçu pour indiquer ce choix. CytoSHOW répondra en traitant une paire initiale de volumes isotropiquement fusionnés, permettant à l’utilisateur d’observer attentivement et de spécifier les rotations nécessaires pour obtenir l’enregistrement ADL.
- Cliquez sur Oui une fois que tous les paramètres sont sélectionnés.
- Spécifiez le Répertoire de sortie dans lequel enregistrer les fichiers traités. Cliquez sur OK.
- Facultatif: si l’option définir l’orientation de la sortie de volume en aperçu a été sélectionnée, réglez la barre de défilement t (figure 3A, flèche verte, panneau de gauche) dans la fenêtre SPIM-A jusqu’au point de temps précoce où les cellules ABa et ABP ont atteint la métaphase. Réglez la barre de défilement t dans la fenêtre SPIM-B à l’étape de développement ultérieure de la virgule . Cela aidera à spécifier l’orientation ADL.
- Facultatif: cliquez sur OK lorsque vous êtes prêt. Si l’option de prévisualisation dans 9.5.7 ci-dessus a été sélectionnée, seuls deux volumes d’aperçu seront fusionnés isotropiquement pour les points de temps indiqués par les curseurs t des fenêtres d’image SPIM-A et SPIM-B. Ces deux points de temps d’aperçu peuvent être utilisés pour spécifier un réalignement précis des volumes d’embryons en sortie à l’orientation ADL, comme expliqué ci-dessous.
- Localisez le nouvel affichage 3DProjY_Decon-Fuse_.... fenêtre. Déplacez la barre de défilement t sur le point 2 de cette fenêtre d’aperçu. Déplacez le curseur Z jusqu’à ce que la vue directement vers le bas de l’axe long de l’embryon soit affichée.
- Déplacez la barre de défilement t de nouveau au point de temps 1 de la 3DProjY_Decon-Fuse_.... fenêtre. Choisissez l’outil de sélection de ligne et dessinez une sélection de ligne à partir de la cellule EMS (noyau rond le plus ventral) à travers le plan des plaques de métaphase AB-Cell.
- Cliquez sur le bouton d’aperçu orange diSPIM sur le 3DProjY_Decon-Fuse_.... fenêtre. Les ajustements fins à l’orientation du volume imagé prévisualisé seront sauvegardés pour être utilisés dans le traitement du jeu de données complet.
- Facultatif: la boîte de dialogue Deconvolve/FUSE diSPIM Raw Data volumes réapparaîtra, tout comme à l’étape 9,5 ci-dessus. Cliquez sur Oui sans choisir l’option définir l’orientation de la sortie du volume dans l’aperçu . Spécifiez le dossier de sortie pour l’exécution complète du traitement des données.
- Définissez les barres de défilement t (figure 3A, flèche verte, panneau de gauche) des fenêtres du moniteur dispim sur le point de départ (SPIMA) et le point de terminaison (spimb) de l’étendue complète des images à traiter. Cliquez ensuite sur OK.
- Au fur et à mesure que SpimFusion progresse, CytoSHOW ouvre et actualise une fenêtre multidimensionnelle montrant le volume de fusion isotrope tranché-4D pour chaque embryon, ainsi que deux fenêtres avec des projections en rotation-4D Max-intensité du volume isotrope. Pendant ce temps, ne pas perturber ou fermer une fenêtre cytoshow jusqu’à ce que la fusion isotrope et le suivi de la lignée soient terminées.
- Facultatif: Notez qu’une fois l’écran de démarrage StarryNite apparu et plus tard disparu, le pipeline complet de traitement des données a été terminé. Cette fenêtre ne doit pas être fermée pendant le traitement ou StarryNite sera interrompue.
10. ouverture de la série de traces de la lignée StarryNite dans AceTree (facultatif)
- Ouvrez la version personnalisée de "AceTree_16BitCompat. jar" fournie.
- Choisissez le menu le fichier > ouvrir configurer le fichier. Localisez votre Répertoire de sortie précédemment indiqué à cytoshow. Ouvrez le sous-dossier Decon_Fuse_... _ POS [n] pour l’embryon [n]. Sélectionnez aaa_edited. xml et Open.
- Utilisez le menu d’AceTree edit > Outils d’édition pour ouvrir la piste d’édition et ajuster ou supprimer des fenêtres de cellules.
- Cliquez sur le cercle demi-ombragé figure 5b, carré rouge pour ajuster les intensités rouge et verte.
- Procédez à la visualisation et à l’édition des lignées comme décrit précédemment5,6,8 (les manuscrits sont également inclus dans notre Bundle de téléchargement).
Representative Results
Nous avons d’abord validé la viabilité des embryons imagés en utilisant les paramètres du protocole pour l’acquisition de diSPIM (sections 1-6). Dix embryons ont été simultanément imagés à 20 ° c, un volume/embryon/minute, du stade 2 cellules au stade 2 fois (7,5 heures, 451 volumes/embryons). Pour surveiller les divisions cellulaires tout au long de l’embryogenèse, nous avons utilisé la souche BV514, qui façon ubiquitaire exprime le mCherry::histone reporter construit à partir de la matrice intégrée de transgène ujIs11324. La figure 6 montre une chronologie de cette première moitié du développement embryonnaire pour l’un des embryons imagés. Chaque image représente une projection d’intensité maximale à vue unique (produite par les étapes 7-8) de l’embryon imagé. Nous avons constaté que les protocoles optimisés n’induisent pas de phototoxicité détectable pour les embryons, tel qu’évalué par la synchronisation des divisions cellulaires (non illustrées), le moment de l’éclosion et le moment lié aux jalons du développement (figure 6 et références1 , le 25 , 26).
Nous avons ensuite appliqué le protocole pour analyser la dynamique de l’excroissance des neurones simples dans les embryons en développement. Nous avons imagé DCR7692 (olaex4655), une souche de nématodes transgéniques qui exprime GFP sur le promoteur du neuropeptide FLP-19 dans un sous-ensemble de cellules non identifiées (DACR2819, PFLP-19 (3.6 KB):: Syn21:: GFP:: CAAX::p 10 3 'UTR) . Suivant les étapes du protocole décrit ici, nous avons déterminé que les cellules non identifiées correspondent aux neurones moteurs RMDDL et RMDDR, à la cellule du canal excrétoire, et à deux cellules musculaires (figure 7). Nous avons ensuite examiné et quantifié la dynamique de l’excroissance des neurones rmddl et rmddr. Nous avons observé que les neurones RMDDL et RMDDR sont en forme oblique dès 360 minutes après la fertilisation, avec l’axe cellulaire plus long représentant l’axe suivant pour l’excroissance de la neurites (figure 7 et film S1). En utilisant le plugin "simple neurites Tracing" aux Fidji et en l’appliquant aux reconstructions 3D de volumes isotropiquement fusionnés, nous avons ensuite quantifié l’excroissance stéréotypée des neurites RMDDL et RMDDR pour six embryons. Nous avons déterminé que la dynamique de l’excroissance était stéréotypée pour le rmddl et le rmddr à travers les embryons (ci-après appelés rmdds). À partir de 385-410 minutes après la fertilisation, les neurites des RMDD ont prolongé 6,0 ± 0,5 μm (moyenne ± SEM; n = 12 neurites) antérieur des corps cellulaires (figure 7B, C, I). À partir de 415-445 minutes après la fertilisation, les deux neurites s’étendent dorsalement dans et autour de l’anneau nerveux présumé (astérisque dans la figure 7D). En moyenne, chaque neurites RMDD prolongée 11,0 ± 0,6 μm (moyenne ± SEM; n = 12 neurites) du corps cellulaire avant de rencontrer de façon synchrone son homologue contralatéral au sommet de l’anneau (figure 7i). Il est important de noter que nos résultats représentatifs démontrent que nous sommes en mesure d’examiner, de comparer et de quantifier les caractéristiques du développement neuronale pour les cellules individuelles identifiables en utilisant notre protocole intégré (figure 7 et figure 8).
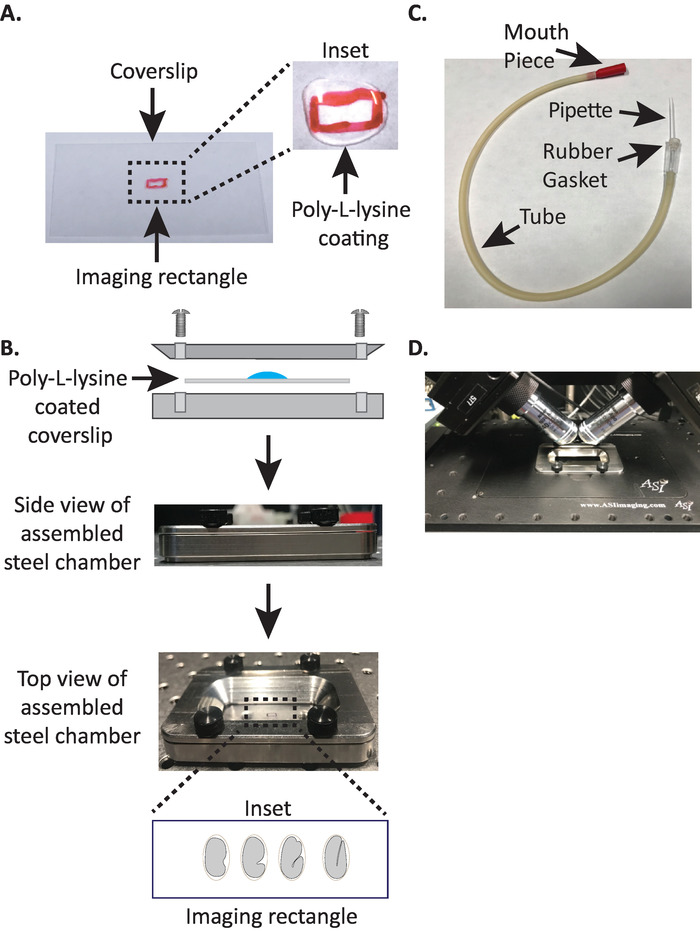
Figure 1: configuration du montage de l’échantillon diSPIM. (A) préparation de lamelle avec poly-L-lysine. Dans l’encart, 10 μl de poly-L lysine a été utilisé pour enduire la lamelle pendant 5 minutes. La poly-L-lysine permet à la coquille d’oeuf d’embryon de coller fermement à la lamelle dans le rectangle. (B) schéma de la chambre d’imagerie en acier et de la chambre Assemblée. Dans l’encart, les embryons représentatifs sont orientés avec l’axe antérieur-postérieur perpendiculaire à l’axe long de la Coverslip. C) tube d’aspiration assemblé avec pipette microcapillaire. (D) chambre d’imagerie en acier montée dans le porte-échantillon selon les objectifs du dispim 40x. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 2: configuration de l’imagerie diSPIM à long terme dans micro-Manager. (A) paramètres recommandés de puissance du laser dispim (rectangle rouge) optimisés pour une imagerie prolongée tout en réduisant la phototoxicité (évaluée par un taux d’éclosion plus élevé d’embryons de C. elegans ). Réglez le laser 561 nm à 79 μW (0,25) et 488 nm au laser à 179 μW (0,5). Notez que l’étalonnage exact des paramètres du logiciel à la puissance laser varie entre les installations de diSPIM. Il est recommandé que les utilisateurs mesurent et étalonnent la puissance du laser afin d’atteindre 79 μW (561 nm) et 179 μW (488 nm) de puissance laser. (B) paramètres de dispim pour l’enregistrement des données (rectangle vert), les paramètres de volume (carré bleu) et les paramètres de tranche (carré orange). (C) paramètres autofocus du dispim pour l’imagerie à long terme de l’embryogenèse C. elegans (voir les étapes 6.1 à 6.6). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 3: visualisation de l’image et configuration du traitement des données à l’aide de CytoSHOW. (A) images brutes de dispim ouvertes par cytoshow. CytoSHOW est capable d’ouvrir les images capturées par les deux chemins de caméra (SPIM A et B). Ces images brutes sont ouvertes dans les fenêtres multidimensionnelles appelées moniteur Dispim. Dans le moniteur du Dispim, un «motif de noeud papillon» est généré pour sélectionner les arêtes antérieures, postérieures, dorsale et ventrale de l’embryon (Voir l’étape 9,1). Les sélections de noeuds papillon indiquent l’orientation de l’embryon pour la déconvolution et le traçage de l’alignement assisté par StarryNite. (B) paramètres optimisés utilisés pour générer des images isotropes. Dans le Deconvolved lors de l’acquisition de la fenêtre, définissez les paramètres spécifiés dans les étapes 9.5.1-9.5.8. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 4: configuration de la caméra diSPIM. (A) photographie des emplacements et des orientations de la caméra dispim. (B) représentation des rotations de + 90 degrés de spim A pour faire correspondre les images de spim B recueillies. (C) volumes d’entrée par rapport à l’indice d’orientation + 1 en fonction de la configuration de la caméra de notre dispim (voir étape Nous tournons SPIM une image (s) + 90 degrés autour de l’axe Y avant l’enregistrement pour correspondre à SPIM B image (s). Barres d’échelle = 10 μM. les images sont des projections représentatives à une seule vue, à une intensité maximale et des images de déconvolution de l’embryon 1,5 avec des noyaux marqués (561-nm, rouge) et des neurones (488-nm, vert). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 5: curation et modification de la lignée embryonnaire de C. elegans dans acetree. (A) nous utilisons l’acétée pour éditer les traces de la lignée de starrynite (voir références5,6,8; les manuscrits sont également inclus dans notre Bundle de téléchargement). AceTree affichera C. elegans des noms systématiques pour chaque noyau (rectangle vert) à la fin des étapes 10.1 à 10.2. Cette fenêtre (A) fournit des informations (rectangle noir) sur chaque cellule dans la trace de lignage (ABa, surligné en bleu) qui aident à guider les utilisateurs lors du suivi et de la modification des traces de lignage. Il est recommandé aux utilisateurs de vérifier et de comparer les cellules lignées et leurs positions à la lignée de cellules embryonnaires C. elegans précédemment rapportée par Sulston et al.1 en outre, si les utilisateurs sont intéressés à localiser des cellules spécifiques dans le série de données auscultatoire (voir ci-dessous, B) Entrez le nom systématique de C. elegans dans la barre de recherche (rectangle orange). (B) la série de données décongestiues de l’utilisateur s’ouvre également automatiquement à la fin des étapes 10.1 à 10.2. Montré ici une image isotropiquement fusionnée d’un embryon de stade à quatre cellules avec des noyaux marqués en rouge. Pendant le suivi d’un nucléi, les utilisateurs doivent modifier l’intensité de l’image (carré rouge) et naviguer dans le temps et z en utilisant les touches fléchées sur leur clavier (Time-gauche/droite, z-haut/bas). (C) dessin animé 3D du point de temps en (B) avec certaines fonctionnalités (rectangle violet) qui permet la visualisation 3D rotative. Pour une vue d’ensemble de l’acetree et de ses fonctionnalités 3D, voir références5,6,8. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 6: dynamique de développement chronométré des embryons de C. elegans sur le dispim. Panneau supérieur, images diSPIM montrant la première moitié du développement embryonnaire pour l’un des embryons imagés (souche BV514 ujIs11324). Les embryons ont été imagés continuellement, chaque minute pendant 7,5 heures (à 20 ° c). Les deux premières images du panneau supérieur représentent des embryons à 4 et 8 cellules avec des noyaux (rouges) et des positions de corps polaires (sphères rouges denses, marquées d’astérisques bleus). Chaque image représente une projection à une seule vue d’intensité maximale de l’embryon imagé. Barres d’échelle = 10 μM. La chronologie (barre horizontale) représente les minutes après la fertilisation (m.p.f.) du développement des embryons de C. elegans . Nous avons validé que les paramètres de notre protocole pour l’acquisition de diSPIM n’induisent pas de phototoxicité détectable pour les embryons imagés évalués par la viabilité, le moment des divisions cellulaires, le moment de l’éclosion et le moment des jalons du développement (voir références le premier , le 25 , 26). nous notons que le calendrier des étapes de développement a été reproductible pour les embryons avec nos paramètres d’imagerie (SEM ± 8,174 minutes pour les séances d’imagerie de 6,4 heures de long; n = 10). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

La figure 7. Identification cellulaire et caractérisation cellulaire unique de la dynamique de l’excroissance neurites dans le développement d’embryons de C. elegans . Imagerie à deux couleurs d’une souche faite en traversant BV514 ujIs11324 (pour l’alignement) et DCR7692 (olaex4655), une souche de nématodes transgéniques qui exprime le GFP hors du promoteur du neuropeptide FLP-19 dans un sous-ensemble de cellules non identifiées. (A-H) Suivant les étapes du protocole décrit ici, nous avons déterminé que les cellules non identifiées correspondent aux neurones moteurs RMDDL et RMDDR (flèches jaunes), à la cellule du canal excrétoire (flèches bleues), et à deux cellules musculaires (flèches blanches). (I) quantification de la dynamique de l’excroissance des neurones rmddl et rmddr en utilisant le plugin fidjien "traçage simple des neurites" et en l’appliquant aux reconstructions 3D de volumes fusionnés isotropiquement. Notez comment le rmddl et le rmddr montrent la dynamique de l’excroissance stéréotypique, chacune s’étendant de façon synchrone pour une longueur totale de 11,0 ± 0,6 μm (moyenne ± SEM; n = 12 neurites) et rencontre à l’apex dorsal de l’anneau nerveux (voir aussi le film S1). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 8: examen des images du diSPIM isotrope des morphologies neuronales chez les embryons de C. elegans . Visualisation isotrope des neurones AVHL et AVHR (flèches jaunes). En utilisant le diSPIM, les morphologies neuronales peuvent être capturées en produisant des images en quatre dimensions (4D) avec une résolution spatiale isotrope d’environ 330 nm. Le diSPIM permet aux utilisateurs de faire pivoter virtuellement les volumes d’image avec une résolution identique dans toutes les directions. Les images en A-D sont des projections d’intensité maximale du même volume d’images de dispim fusionnés isotropiquement à partir de rotations distinctes autour de l’axe long de l’embryon. Barres d’échelle = 5 μm. s’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
Film supplémentaire S1: C. elegans embryon développant de 280 à 434 minutes après la fertilisation. Film isotrope de la souche DCR7692 (olaex4655) exprimant UJIS113 façon ubiquitaire avec DACR2819 des neurites RMDD à étiquetage faible (figure 7A-d, flèches jaunes). DACR2819 étiquette également deux cellules musculaires (figure 7A-d, flèches blanches) et une cellule de canal excrétoire (figure 7A-d, flèche bleue) pendant le développement embryonnaire (figure 7A-d). Barres d’échelle = 10 μM. veuillez cliquer ici pour télécharger ce fichier.
Discussion
C. elegans se distingue comme le seul organisme avec les positions finales et la connectivité de chaque neurone adulte connu27. Cependant, la dynamique de développement conduisant à l’Organisation des circuits de travail et des réseaux qui composent le C. elegans connectome reste inconnue. Sur la base des occasions qui émergent des progrès en microscopie photonique, nous pouvons maintenant capturer et analyser les positions cellulaires, la morphogenèse et la neurogenèse tout au long du développement embryonnaire de C. elegans .
La procédure que nous avons décrite et que nous utilisons couramment dans le laboratoire donne des images 4D-isotropes de neurones étiquetés et de noyaux pour l’alignement cellulaire chez des embryons de C. elegans . Plus important encore, nous avons optimisé les conditions d’imagerie à long terme avec le diSPIM et les capacités de linéarité semi-automatisée couplées avec des images haute résolution pour améliorer la vitesse et la précision de l’analyse de l’embryogenèse C. elegans . Ce protocole intégré permettra aux utilisateurs de visualiser et d’identifier les cellules et de quantifier les caractéristiques tridimensionnelles telles que la migration des neurites et la fasciculation des axons au début des contractions précoces. Cette procédure peut être facilement adaptée à n’importe quelle installation avec un système ASI diSPIM, et nous recommandons ce système spécifiquement pour ce protocole. D’autres formulations de SPIM offertes commercialement peuvent différer de la configuration ASI dans la chambre d’échantillonnage et les propriétés optiques. Cependant, les données exportées à partir d’autres plateformes peuvent également être mises à travers notre pipeline de données. Par conséquent, l’évaluation de leur valeur dans l’alignement, un test exigeant de la qualité de l’image et la stabilité de l’instrument, est faisable. Même si nous utilisons activement le diSPIM pour faire régulièrement l’image d’autres spécimens (comme les embryons de drosophile et de zébrafish), l’analyse détaillée et exhaustive des embryons est encore actuellement limitée aux espèces de nématodes. Pour les échantillons plus grands ou épais, nous optons pour utiliser des approches de balayage de scène, qui scannent les échantillons à travers une feuille de lumière stationnaire. Kumar et coll. ont déjà démontré cette sectionnement améliorée de diSPIM pour produire des images de haute qualité à partir d’échantillons épais sans modifications supplémentaires au diSPIM10.
Les étapes critiques dans le protocole incluent le montage des embryons C. elegans sur la Coverslip revêtue de poly-L-lysine, l’acquisition de données et le traitement des données. La récolte et le montage des embryons de C. elegans sur la lamelle de verre peuvent être difficiles pour les utilisateurs inexpérimentés, mais ici nous fournissons un protocole détaillé des étapes clés pour faciliter l’apprentissage. Si l’imagerie à long terme est souhaitée, nous obtenons les meilleurs résultats en récoltant des embryons à quatre cellules ou plus tôt à partir de 8-10 jeunes adultes28. Notez que les adultes âgés sont moins souhaitables pour récolter des embryons de stade précoce parce qu’ils ont tendance à contenir des embryons plus âgés dans l’utérus et des oeufs non fertilisés. En ce qui concerne les embryons de montage, des problèmes tels que le blocage dans l’aspirateur assemblé (pipette à bouche) ou un trop grand d’une ouverture dans la pipette microcapillaire peuvent empêcher le montage et l’orientation appropriés des embryons. Pour se préparer à une imagerie optimale, nous effectuons des tests de pré-acquisition sur les embryons précoces et tardifs de pré-contractions pour vérifier la performance des feuilles lumineuses, des caméras, des objectifs et de l’autofocus. Nous obtenons de meilleurs résultats lorsque toutes ces opérations sont testées et donnent des images de haute qualité lors de nos tests de pré-acquisition. Ceci est particulièrement pertinent pour générer des images avec une résolution spatiale isotrope, pour lesquelles les images brutes acquises à partir des deux vues (objectifs) doivent être de haute qualité. Après l’acquisition, les volumes acquis pour chaque vue sont traités pour produire des images isotropes. Il est important d’utiliser une carte d’unité de traitement graphique (GPU) appropriée comme décrit dans ce protocole (voir ci-dessous). Cela améliore la vitesse de traitement à laquelle les images fusionnées isotropiquement sont générées, raccourcissant le temps d’analyse des données. Il est également impératif que les utilisateurs exécutent la dernière version de CytoSHOW et utilisent les paramètres fournis avec notre Bundle de téléchargement pour StarryNite auto-lineaging. Si les utilisateurs sont intéressés à utiliser l’auto-ligage pour d’autres échantillons (par exemple, zebrafish, Drosophila etc.), l’optimisation supplémentaire aux paramètres utilisés dans starrynite sera nécessaire (voir références3,4).
Bien que notre protocole intégré fournisse des images et des résultats d’alignement dans l’embryon pré-twdémangeant, les utilisateurs doivent être conscients que l’alignement automatisé dans l’embryon post-twdémangeaisons n’est actuellement pas réalisable: les positions nucléaires changent de l’ordre des secondes dans le embryonnaire post-contractions, trop rapidement pour permettre le suivi de la lignée. Cependant, le dispim a en effet démontré une capacité prometteuse de capturer des événements neurodéveloppementaux et de suivre certaines positions cellulaires dans les stades post-contractions de l’embryogenèse23,29. Si les utilisateurs sont intéressés à examiner l’embryon post-twdémangeant, le diSPIM fournit la vitesse pour obtenir des instantanés volumétriques et suivre des événements neurodéveloppementaux fins, tels que l’excroissance de neurites, dans les embryons en mouvement rapide.
Ce protocole sera fondamental pour l’achèvement de la cellule par cellule de l’Atlas30de wormguides, car il fournira une approche intégrée avec des images isotropes à haute résolution pour identifier et capturer les morphologies 3D des neurones étiquetés pendant 430 premières minutes d’embryogenèse. En l’État, le prototype de l’Atlas WormGUIDES fournit des positions nucléaires de cellules dans l’embryon en développement et vise à capturer la dynamique du développement d’un sous-ensemble de neurones embryonnaires. Ce protocole sera une clé pour l’intégration de neurones en développement supplémentaires dans l’Atlas de WormGUIDES30.
Notre protocole intégré simplifiera également l’exploration de nouveaux profils d’expression génique dans l’embryon de C. elegans . Chez les C. eleganstransgéniques, de nombreux promoteurs spécifiques à la cellule contrôlent spatialement et temporequement l’expression transgénique. Alors que les patrons d’expression de la plupart des gènes ont été largement caractérisés dans l’animal adulte31,32,33,34, presque tous n’ont pas encore été caractérisés dans les pays en développement (en particulier embryonnaire tardif). Le C. elegans promoterome a été une ressource utile pour la communauté de ver pour conduire l’expression transgénique spécifique à une cellule, ainsi que de déterminer si la fonction génique est cellulaire-autonome ou non-autonome. Capturer des schémas isotropes de haute résolution et d’expression dynamique des gènes, et identifier précisément les cellules exprimant par le biais d’un ligage sera utile à beaucoup dans la communauté scientifique.
L’embryogenèse comprend deux processus majeurs entrelacés, la différenciation cellulaire et la morphogenèse tissulaire. On sait beaucoup sur les mécanismes et les molécules qui définissent des types distincts de cellules pendant le développement de C. elegans. Cependant, on sait peu de chose sur les mécanismes importants pour la migration cellulaire, l’adhérence des cellules et la forme cellulaire dans l’embryon de C. elegans . Avec la lignée de cellules invariantes C. elegans connue, notre protocole nous permet de discerner facilement la microanatomie 3D cataloguée de l’embryon au cours de la morphogenèse à de nouveaux niveaux de détail: p. ex. fascicule d’axones, synaptogenèse et activité neuronale. Ardiel et coll. ont déjà démontré la puissance du diSPIM pour capturer les transitoires calciques au niveau d’un seul neurones dans les embryons de C. elegans 23. De nombreux autres aspects de la physiologie du développement sont mûrs pour l’enquête par ces méthodes.
Enfin, ce protocole est largement automatisé et réduit systématiquement le temps nécessaire pour générer des images de déconvolution et effectuer des lignées cellulaires via StarryNite et Acetree. Les stratégies logicielles utilisées dans ce protocole peuvent être appliquées à de nombreuses questions de biologie loin des domaines très spécifiques dans lesquels nous les avons démontrés ici.
Détails sur la compatibilité logicielle et l’accès au téléchargement
Des informations sur le micro-gestionnaire et les plugins pour l’imagerie diSPIM sont disponibles sur http://dispim.org/software/micro-manager et https://micro-manager.org/wiki/ASIdiSPIM_Plugin.
Le pipeline de traitement des données nécessite actuellement un système d’exploitation Windows. Nous avons regroupé un seul fichier d’archive pour simplifier l’installation de tous les programmes de traitement de données et fichiers de support requis. Il est disponible au téléchargement à http://dispimlineage.wormguides.org.
CytoSHOW (http://run.cytoshow.org/) est basé sur la plate-forme d’analyse d’image Open source largement utilisée et ImageJ (v1). Java doit être installé et mis à jour sur l’ordinateur pour utiliser CytoSHOW, et les mises à jour de CytoSHOW sont déployées automatiquement via Java Web Start. De nombreuses fonctions de CytoSHOW basées sur ImageJ sont décrites et illustrées à https://imagej.nih.gov/ij/docs/examples/index.html. CytoSHOW a été personnalisé pour afficher des données brutes multidimensionnelles à partir de l’ASI diSPIM, ainsi que d’autres logiciels d’imagerie qui crée la sortie TIFF. En principe, d’autres systèmes d’imagerie SPIM à vues multiples pourraient être soutenus par des modifications mineures de CytoSHOW pour permettre que ce protocole soit réalisé sur différents systèmes de microscope.
SpimFusion a été écrit en CUDA/C++ à l’aide de Visual Studio 2013 avec CUDA Toolkit v 7.5. L’exécution de SpimFusion nécessite un matériel informatique spécifique: une carte graphique NVIDIA (GPU) avec une capacité de calcul CUDA 1,0 ou supérieure et un minimum de 2 Go de mémoire de carte graphique. Au moment de la publication de notre protocole, SpimFusion n’est pas publié (min Guo et Hari Shroff) mais disponible dans les Archives du bundle de logiciels mentionnées ci-dessus.
Une version spécialement conçue pilotée par la ligne de commande de StarryNite exige que le runtime MATLAB compiler librement disponible soit installé, mais ne nécessite pas une licence pour le logiciel commercial MATLAB. Le MATLAB compiler Runtime est inclus dans l’archive de Bundle de logiciels mentionnée ci-dessus. Le code de StarryNite utilisé dans ce protocole est essentiellement inchangé par rapport à celui utilisé pour les images confocales6. Cependant, plusieurs questions opérationnelles dans la création d’images d’entrée pour le traitement de StarryNite et la gestion des résultats de StarryNite ont été abordées ici par des méthodes dans CytoSHOW qui permettent un pipeline de traitement de données continu pour le diSPIM isotrope fusionné Volumes. Ces modifications sont automatisées par le code CytoSHOW qui gère ces étapes de pré-et post-traitement. CytoSHOW modifie également un ensemble de paramètres StarryNite de modèle spécifique au diSPIM pré-optimisé pour ajuster automatiquement l’algorithme de segmentation à l’intensité de fluorescence des noyaux dans les données imagées. Les paramètres uniques utilisés par StarryNite sur chaque ensemble de données diSPIM sont ensuite enregistrés dans un fichier avec l’image de sortie et les données de linéarité.
Une version personnalisée d’AceTree qui fonctionne avec des images 16 bits et maintient la compatibilité avec le rendu Java3D est le mieux adapté à ce protocole. Il est également inclus dans les Archives de Bundle de logiciels mentionnées ci-dessus.
Disclosures
Les auteurs n’ont rien à divulguer.
Acknowledgments
Nous remercions John Murray pour la souche intégrée, ujIs113, pour générer la souche de ligage BV514; Brandon Harvey (NIBIB) pour l’aider à tester le protocole; Jon Daniels et Gary Rondeau (Applied Scientific Instrumentation) pour l’aide au micro-Manager et à l’instrument diSPIM; et Andrew York et Hank Eden pour leurs commentaires critiques sur le système diSPIM. Nous remercions également le programme du centre de recherche pour les institutions minoritaires et l’Instituto de Neurobiología Jose del Castillo (Universidad de Puerto Rico) pour avoir fourni une plate-forme de réunion et de remue-méninges. Une grande partie de ce travail a été menée au laboratoire de biologie marine de Woods Hole à travers le programme Whitman. Ce travail a été appuyé par les programmes de recherche intra-muros de l’Institut national d’imagerie biomédicale et de bioingénierie de la NIH et par la subvention du NIH no. U01-HD075602 et no. OD016474. Mark W. Moyle a été soutenu par NS098616 et Leighton H. Duncan a été appuyé par un supplément de diversité à OD016474.
Materials
| Name | Company | Catalog Number | Comments |
| Steps 1-4 | |||
| Concavity slides | ThermoFisher Scientific | 1519006 | 5-18mm diameter, 0.6-0.8mm deep, 1.2-1.5mm |
| Dissecting microscope with 10×–50× zoom range | Motic | SMZ-171 | |
| E. coli (OP50) | Caenorhabditis Genetics Center (CGC) | OP50 | |
| Glass coverslips, no. 1.5, 24 × 50 mm | VWR International | 48393-241 | |
| M9 Buffer | Stiernagle, T. Maintenance of C. elegans. WormBook. 1-11, doi:10.1895/wormbook.1.101.1, (2006). | ||
| Methyl cellulose | Sigma-Aldrich | H7509-25G | |
| Microcapillary pipette aspirator tube | Sigma-Aldrich | A5177 | |
| Microcapillary pipettes, 0.4-mm i.d | Drummond Scientific | 1-000-800 | |
| Needle, no. 18G x 1 ½ (1.2mm x 40mm) | BD Precision Glide | 305196 | |
| NGM plates | prepared as described by Brenner (1974) | ||
| O-ring for imaging chamber | O-Rings West | M1.5X40 | |
| Pasteur pipette | Corning/Sigma-Aldrich | CLS7095D5X | |
| Platinum wire, 0.5-mm diameter | Sigma-Aldrich | 267201 | |
| Poly-L-lysine | Sigma-Aldrich | P1524 | |
| Stainless steel rectangular chamber (76.0 mm x 50.5 mm) | Applied Scientifics Instrumentations (ASI) | I2450 | |
| Worm Eyelash Pick | Hart, A. C. Behavior. WormBook. (2006). | ||
| Worm Pick | Stiernagle, T. Maintenance of C. elegans. WormBook. 1-11, doi:10.1895/wormbook.1.101.1, (2006). | ||
| Name | Company | Catalog Number | Comments |
| Steps 5-6 | |||
| 488 nm long-pass filter | Semrock | LP02-488 RU-2 | |
| 561-nm notch filter | Semrock | NF03-561E-25 | |
| BLP02-561R-25, quantity 2 | Semrock | 561 nm EdgeBasic best-value long-pass edge filter | |
| Control software for bottom camera | Jenoptik | ProgRes CapturePro | |
| diSIPM assembly video | Applied Scientifics Instrumentations (ASI) | https://youtu.be/TAgbr6IrTqw ; http://www.asiimaging.com | |
| diSPIM alignment video | Applied Scientifics Instrumentations (ASI) | https://youtu.be/qnOrg30NNuE | |
| diSPIM imaging PC | Intel | Intel Xeon CPU E5-2630 2.6GHz, 12 cores in total, 64 GB memory, Windows 7 | |
| FF01-525/45-25, quantity 2 | Semrock | 525/45 nm BrightLine single-band bandpass filter | |
| FF555-DI03-25X36, quantity 2 | Semrock | 555 nm edge BrightLine single-edge dichroic beamsplitter | |
| Imaging PC Graphics Card | NVIDIDA | NVIDIA GeForce GTX 1080 Ti graphics cards | |
| Kumar et al diSPIM Setup | Applied Scientifics Instrumentations (ASI) | Instrument setup for this protocol is identical to Kumar et al 10,11 diSPIM, which makes use of 40x 0.8NA water immersion lenses for imaging. (See steps 5.1 and note) | |
| Micro Manager | Micro-Manager | https://micro-manager.org/ | |
| Modifications to Kumar et al diSPIM Setup (see below) | |||
| Optical table with isolators, 4 feet × 6 feet × 12 inches | TMC | 784-651-02DR and 14-416-34 | |
| Name | Company | Catalog Number | Comments |
| Steps 7-10 | |||
| Analysis PC | Intel | Intel Core i7-8700K CPU 3.70GHz, 6 cores in total, 64 GB memory, Windows 10 | |
| Analysis PC Graphics Card | NVIDIDA | NVIDIA GeForce GTX 1080 Ti graphics cards | |
| Installation instructions | Software bundle | http://dispimlineage.wormguides.org/diSPIMlineaging_InstallationInstructions.htm | |
| Software bundle | Software bundle | http://dispimlineage.wormguides.org |
References
- Sulston, J. E., Schierenberg, E., White, J. G., Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology. 100 (1), 64-119 (1983).
- Bao, Z., et al. Automated cell lineage tracing in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 103 (8), 2707-2712 (2006).
- Santella, A., Du, Z., Bao, Z. A semi-local neighborhood-based framework for probabilistic cell lineage tracing. BMC Bioinformatics. 15, 217 (2014).
- Santella, A., Du, Z., Nowotschin, S., Hadjantonakis, A. K., Bao, Z. A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics. 11, 580 (2010).
- Boyle, T. J., Bao, Z., Murray, J. I., Araya, C. L., Waterston, R. H. AceTree: a tool for visual analysis of Caenorhabditis elegans embryogenesis. BMC Bioinformatics. 7, 275 (2006).
- Katzman, B., Tang, D., Santella, A., Bao, Z. AceTree: a major update and case study in the long term maintenance of open-source scientific software. BMC Bioinformatics. 19 (1), 121 (2018).
- Murray, J. I., et al. Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nature Methods. 5 (8), 703-709 (2008).
- Murray, J. I., Bao, Z., Boyle, T. J., Waterston, R. H. The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nature Protocols. 1 (3), 1468-1476 (2006).
- Wu, Y., et al. Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nature Biotechnology. 31 (11), 1032-1038 (2013).
- Kumar, A., et al. Using Stage- and Slit-Scanning to Improve Contrast and Optical Sectioning in Dual-View Inverted Light Sheet Microscopy (diSPIM). The Biological Bulletin. 231 (1), 26-39 (2016).
- Kumar, A., et al. Dual-view plane illumination microscopy for rapid and spatially isotropic imaging. Nature Protocols. 9 (11), 2555-2573 (2014).
- Wu, Y., Christensen, R., Colon-Ramos, D., Shroff, H. Advanced optical imaging techniques for neurodevelopment. Current Opinion in Neurobiology. 23 (6), 1090-1097 (2013).
- Wu, Y., et al. Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 108 (43), 17708-17713 (2011).
- Huisken, J., Stainier, D. Y. Selective plane illumination microscopy techniques in developmental biology. Development. 136 (12), 1963-1975 (2009).
- Stelzer, E. H. Light-sheet fluorescence microscopy for quantitative biology. Nature Methods. 12 (1), 23-26 (2015).
- Winter, P. W., Shroff, H. Faster fluorescence microscopy: advances in high speed biological imaging. Current Opinion in Chemical Biology. 20, 46-53 (2014).
- Lucy, L. B. An iterative technique for the rectification of observed distributions. Astronomical Journal. 76 (6), 745-754 (1974).
- Richardson, W. H. Bayesian-Based Iterative Method of Image Restoration. JOSA. 62 (1), 55-59 (1972).
- Stiernagle, T. Maintenance of C. elegans. WormBook. , 1-11 (2006).
- Hart, A. C. Behavior. WormBook. , (2006).
- Edelstein, A., Amodaj, N., Hoover, K., Vale, R., Stuurman, N. Computer control of microscopes using microManager. Current Protocols in Molecular Biology. Chapter 14, Unit14 20 (2010).
- Gualda, E. J., et al. SPIM-fluid: open source light-sheet based platform for high-throughput imaging. Biomedical Optics Express. 6 (11), 4447-4456 (2015).
- Ardiel, E. L., et al. Visualizing Calcium Flux in Freely Moving Nematode Embryos. Biophysical Journal. 112 (9), 1975-1983 (2017).
- Walton, T., et al. The Bicoid class homeodomain factors ceh-36/OTX and unc-30/PITX cooperate in C. elegans embryonic progenitor cells to regulate robust development. PLoS Genetics. 11 (3), e1005003 (2015).
- Altun, Z. F. WormAtlas. , (2002).
- Wood, W. B. Embryology: In the nematode C. elegans. Cold Spring Harbor Laboratory Press. Chapter 8, 215-241 (1988).
- White, J. G., Southgate, E., Thomson, J. N., Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical Transactions of the Royal Society B: Biological Sciences. 314 (1165), (1986).
- Bao, Z., Murray, J. I. Mounting Caenorhabditis elegans embryos for live imaging of embryogenesis. Cold Spring Harb Protoc. (9), (2011).
- Christensen, R. P., et al. Untwisting the Caenorhabditis elegans embryo. eLife. 4, (2015).
- Santella, A., et al. WormGUIDES: an interactive single cell developmental atlas and tool for collaborative multidimensional data exploration. BMC Bioinformatics. 16, 189 (2015).
- Dupuy, D., et al. A first version of the Caenorhabditis elegans Promoterome. Genome Research. 14 (10B), 2169-2175 (2004).
- Reece-Hoyes, J. S., et al. Insight into transcription factor gene duplication from Caenorhabditis elegans Promoterome-driven expression patterns. BMC Genomics. 8 (27), (2007).
- WormBase. , Available from: https://www.wormbase.org (2019).
- Lee, R. Y. N., et al. WormBase 2017: molting into a new stage. Nucleic Acids Research. 46 (D1), D869-D874 (2018).
