

Summary
Aqui, nós apresentamos uma aproximação combinatória usando a microscopia de alta resolução, as ferramentas computacionais, e a rotulagem da único-pilha em embriões vivos de C. elegans para compreender a única dinâmica da pilha durante o neurodevelopment.
Abstract
Caenorhabditis elegans (C. elegans) destaca-se como o único organismo em que o desafio de compreender as origens celulares de um sistema nervoso inteiro pode ser observado, com resolução de células únicas, in vivo. Aqui, apresentamos um protocolo integrado para o exame do neurodesenvolvimento em embriões de C. elegans . Nosso protocolo combina a imagem latente, o lineaging e o traçado neuroanatômico de únicas pilhas em embriões tornando-se. Nós alcançamos a imagem latente a longo prazo, Four-dimensional (4D) de embriões vivos de C. elegans com a definição espacial quase isotrópico com o uso da microscopia de iluminação de plano seletivo invertida da Duplo-Vista (dispim). Núcleos e estruturas neuronais nos embriões de nematódeos são imaged e fundidos isotropicamente para produzir imagens com resolução de ~ 330 nm em todas as três dimensões. Esses conjuntos de dados 4D de alta resolução minuto a minuto são então analisados para correlacionar identidades definitivas de linhagem celular com expressão gênica e dinâmica morfológica em níveis de detalhe de células únicas e subcelulares. Nosso protocolo está estruturado para possibilitar a implementação modular de cada uma das etapas descritas e aprimorar estudos sobre embriogênese, expressão gênica ou neurodesenvolvimento.
Introduction
C. elegans destaca-se como o único organismo em que cada célula do embrião pode ser observada em todo o neurodesenvolvimento. Com toda a linhagem celular conhecida e invariante1, e com o desenvolvimento de novas ferramentas que permitem a rotulagem e a imagem contínua de células individuais em embriões, os biólogos podem agora começar a examinar diferentes etapas no desenvolvimento do nematoide nervoso sistema de todos os ângulos-células de nascimento; migração e diferenciação; formação de neurite, crescimento direcionado e fasciculação; formação de sinapse; e sintonia de circuitos funcionais. A captura de dinâmicas de crescimento neuronal no embrião C. elegans , combinando repórteres e microscopia de fluorescência de forma estavelmente expressa, é valiosa para a comunidade científica.
Estudos de desenvolvimento em C. elegans costumam alavancar os mapas invariantes da linhagem celular e do destino celular desta espécie para aumentar a compreensão contextual no nível de célula única dentro do organismo intacto1. Auto-análise de lineaging-usando starrynite2,3,4 e acetree5,6, 7,8 software-benefícios de alto contraste, alta resolução imagens de núcleos fluorescentes. Para trabalhar otimamente, StarryNite/AceTree também depende da orientação restrita previsível de embriões imaged durante o desenvolvimento. A microscopia confocal, feita em embriões de C. elegans comprimida entre dois COVERSLIP, foi o método padrão da microscopia da auto-lineaging por mais de uma década porque fornece o elevado contraste/alta resolução e um restrito previsível orientação do embrião7,8. Nós descrevemos previamente a construção e o uso de um microscópio plano seletivo invertido luz-folha-baseado novela da iluminação da Duplo-Vista (dispim) para a imagem latente viva da amostra tal como o C. elegans embriogênese9,10 , 11 anos de , 12 anos de , 13. a microscopia da luz-folha, geralmente, fornece a baixa fototoxicidade, a alta velocidade, e a imagem latente a longo prazo de espécimes 3D vivos14,15. O método diSPIM, especificamente, produz imagens de quatro dimensões (4D) com resolução espacial quase isotrópica de aproximadamente 330 nm9.
Comparado com a microscopia confocal, dispim oferece um sinal-à-ruído e uma velocidade mais elevados, uma definição espacial mais isotrópico, e é mais apropriado para a imagem latente in vivo de longo prazo16. Por isso, trabalhamos para adaptar os dados de diSPIM para entrada em StarryNite/AceTree e investigamos se isso melhoraria as análises de lineaging. Um obstáculo principal é que os espécimes de diSPIM não podem facilmente ser restringidos pela casca de ovo-compressão para adotar orientações esperadas para StarryNite/AceTree. A orientação aleatória das posições das células no volume que está sendo analisado degrada a precisão da análise de lineaging automático.
Por isso, empregamos o CytoSHOW, uma interface de usuário guiada por visualizador, que permite aos usuários selecionar a orientação 3D precisa de embriões durante o pré-processamento de imagens diSPIM, produzindo dados de imagem que são otimizados para a qualidade e com reconhecimento de contexto para entrada em StarryNite /AceTree. Após a seleção do usuário de embriões imaged, o CytoSHOW orquestra um pipeline automatizado de processamento de dados. As imagens cortadas e de fundo subtraída de embriões são salvas em arquivos de pilha TIFF para cada posição, ponto de tempo e exibição. O cytoshow, então, iterativamente chama o programa spimfusion para coinscrever-se e deconvolve conjuntamente as duas visualizações pré-processadas, usando o algoritmo Richardson-Lucy17,18 para produzir imagens volumétricas de alta resolução isotrópicas. Um conjunto dispim-specific dos parâmetros foi aperfeiçoado para que o starrynite governe seu comportamento durante a imagem-segmentação e o núcleo-seguimento em imagens isotropicamente fundidas. Imagens fundidas e resultados de lineaging são então editados usando AceTree, que permite aos usuários identificar e corrigir quaisquer erros no rastreamento de linhagem automática gerado pelo StarryNite. Acetree também pode apresentar linhagem-árvore e 3D modelado renderizações de núcleos rastreados no embrião. Nós achamos que a velocidade e a exatidão da auto-lineaging são realçadas marcada usando imagens isotropicamente fundidas, quando comparadas às imagens cruas de uma ou outra câmera de spim. Nosso protocolo, enquanto otimizado para o C. elegans aplicação descrita aqui, poderia ser geralmente adaptado para auto-lineaging de dispim dados produzidos para outras espécies ou espécimes. Se este for o uso pretendido do protocolo, por favor, note que a afinação adicional dos parâmetros starrynite provavelmente será necessária para novos espécimes, como descrito3,4.
A implementação bem-sucedida deste protocolo resulta em imagens com resolução 4D-isotrópica e permite que os biólogos rastreem linhagens celulares, identificando e analisando simultaneamente os neurônios no embrião C. elegans em desenvolvimento. Além disso, ao mesclar vários algoritmos de pós-processamento-com aceleração de hardware sendo o mais demorado destes-agora podemos analisar os detalhes subcelulares finos e as linhagens celulares e células-Fates de embriões vivos em tempo essencialmente real. Este novo protocolo permite a manipulação precisa, informada e a observação do comportamento celular durante estudos probatório de diferenciação e morfogênese in vivo. Neste manuscrito, apresentamos uma explicação detalhada dos protocolos melhorados que desenvolvemos para lineaging e rastreamento de células no desenvolvimento de embriões de C. elegans , para aprimorar estudos de embriogênese, expressão gênica ou neurodesenvolvimento.
Protocol
1. montando a câmara da imagem latente de aço de dispim com o lamela revestido poli-L-lysine
Nota: os passos abaixo são todos necessários para otimizar e automatizar a análise de linhagem de embriões de nematódeos por StarryNite/acetree. Várias opções (indicadas por como tal) podem ser omitidas para experimentos que exigem o rastreamento de linhagens de células C. elegans .
- Desenhe um pequeno retângulo (2mm x 5mm) no centro de uma lamínula retangular limpa (n º 1,5, 24 mm x 50 mm) com uma Sharpie (ou caneta semelhante).
- Vire a lamínula sobre o lado não marcado e coloque 10 μL de poli-L-lisina (gato. não. P1524) sobre o retângulo marcado.
Nota: faça uma concentração de trabalho de 1 mg/mL de poli-L-lisina dissolvida em água filtrada (ou equivalente). Para armazenamento a longo prazo, prepare alíquotas de 5 a 10 mL e armazene a − 20 ° c. Uma vez descongelados, as alíquotas podem ser armazenadas à temperatura ambiente (23 ° c) durante 3 – 4 semanas. - Deixe a poli-L-lisina revestir a lamínula durante 5 minutos (Figura 1a). Poly-L-lysine é usado para o revestimento de tampa de vidro desliza onde os embriões serão montados, permitindo que a casca de ovo do embrião para furar firmemente a lamínula, mesmo quando os objetivos são imersos no meio de imagem.
- Coloque a lamínula revestida de poli-L-lisina na metade inferior da câmara de imagem de aço.
- Coloque a metade superior da câmara de imagem de aço na metade inferior com a lamínula e aperte com os quatro parafusos associados à câmara. Verifique visualmente a partir do lado para se certificar de que a metade superior é uniformemente sentado na metade inferior (Figura 1b).
- Encha a câmara com 7 – 8 ml de tampão M919, um tampão isotónico que ajude a impedir que os embriões do estágio adiantado sucumbir à pressão osmótica anormal. 1-Cell, e mesmo os embriões de 2 células são osmoticamente sensíveis e podem se desenvolver anormalmente se não neste tampão isotônico. Os embriões também tendem a prender no estágio 3 vezes se M9 é substituído com água.
2. preparando amostras de embrião C. elegans para montagem
Nota: aproximadamente 18 horas antes da imagem, cinco jovens (1 dia desde o Molt final) adultos e dez larval fase 4 (L4) C. elegans são escolhidos para um meio de crescimento nematódeo (ngm) placa de agar semeada com E. coli Strain OP50. As picaretas do fio da platina são usadas para mover larvas e os elegans adultos novos de C. sem prejudicar o animal19.
- Prepare um 1% metil celulose (gato. não. H7509-25G) solução em buffer M9.
Nota: a celulose metílica deve ser agitada em M9 quente até ser dissolvida. Uma vez preparada, esta solução pode ser armazenada à temperatura ambiente. - Adicionar 500 μL de solução de celulose em M9 de 1% de metilo na depressão de uma lâmina de microscópio côncavo. Este tampão viscoso será usado em duas ocasiões: 1) ao colher embriões adiantados pela dissecção de sem-fins adultos e 2) ao lavar os embriões do tarde-estágio escolhidos diretamente de uma placa de NGM.
Nota: a celulose metílica é utilizada para evitar que os embriões aderem à lâmina do microscópio. - Para os embriões da fase tardia da imagem latente, a picareta colocou embriões de C. elegans (preferivelmente de uma placa de ngm com os adultos novos atuais) usando uma picareta da pestana, e mova os embriões à celulose 1% methyl na corrediça côncava do microscópio. A picareta da pestana ajuda a reduzir a força e, assim, minimizar o stress ou danos aos embriões durante o manuseio. O procedimento para fazer uma picareta da pestana é coberto por Hart20.
- Com uma segunda picareta da pestana (na mão oposta), bata delicadamente ambas as pestanas junto para suspender os embriões na celulose de metilo.
- Opcional: se planear às pilhas embrionário da linhagem com starrynite, um deve montar 1 pilha aos embriões de 4 pilhas. Para fazer isso, primeiro selecione jovens adultos de uma placa NGM e movê-los para a solução de celulose M9-metil na corrediça côncava microscópio usando uma picareta de fio de platina.
- Opcional: com as pontas afiadas de agulhas hipodérmica (no. 18g x 1 1/2), fatie o animal transversalmente no corpo meados de liberar 1 pilha aos embriões de 4 células.
3. a pipeta da boca: montando o tubo do aspirador com pipeta microcapilar
Nota: utilizamos um tubo aspirador com uma pipeta microcapilar puxada à mão inserida na junta de borracha do tubo. Isto permite-nos transferir embriões da corrediça da dissecção à superfície poli-L-lysine-revestida na câmara tampão-enchida da imagem latente.
- Puxe manualmente a pipeta microcapilar sobre uma chama aberta para criar duas metades com pontas esticadas.
- Tome uma metade da pipeta microcapilar e insira a extremidade sem corte na junta de borracha do tubo aspirador (Figura 1C). Defina a outra metade da pipeta microcapilar de lado para uso mais tarde (se necessário).
- Com o tubo aspirador montado equipado com pipeta microcapilar, quebre suavemente a ponta da pipeta microcapilar e crie uma abertura que caiba cerca de 1 – 2 dois embriões (a partir daqui neste instrumento é chamado de "pipeta bucal").
4. montagem C. elegans embriões em poli-L-lisina revestido lamela
- Com o bocal aspirador mantido suavemente entre os dentes, pré-encha a pipeta microcapilar com 10 – 15 μL de tampão M9 e, em seguida, chupe suavemente vários embriões da corrediça côncava no capilar.
- Transfira os embriões para a câmara de imagem de aço preenchida com o tampão M9, posicionando a ponta capilar para que os embriões caiam no retângulo central da lamínula.
- Evitando ferimentos aos embriões, mova-os suavemente com uma pestana pick ou use a pipeta da boca para posicionar os embriões verticalmente, para orientar os embriões de modo que o eixo longo do embrião seja perpendicular ao eixo longo da lamínula (Figura 1b Inset , painel inferior).
Nota: posicionar o embrião nesta orientação minimiza o número de fatias à imagem, reduzindo assim a dosagem clara e o tempo de processamento de dados ao melhorar a velocidade da aquisição. - Coloque a câmara de imagem de aço no suporte da amostra no estágio do microscópio (Figura 1D).
5. montagem, configuração de software e otimização de laser para a imagem embrionária usando o diSPIM
- Veja instruções passo a passo sobre como montar todo o diSPIM acoplado a fibra de peças comercialmente disponíveis em Kumar et al.10,11 e em http://www.dispim.org. Um vídeo-protocolo de como montar o diSPIM está igualmente disponível no Web site de ASI (http://www.asiimaging.com).
Nota: a configuração do instrumento para este protocolo é idêntica a Kumar et al.10,11 dispim, que faz uso de 40x 0.8 na água lentes de imersão para imagem. A única diferença entre a configuração neste protocolo e Kumar et al.10,11 é a adição de um espelho dicróico (dividindo a 560 nm) e filtros de bandpass vermelho e verde dentro de um dispositivo de divisão de imagem (modelo A12801-01) instalado no ambos diSPIM braços de imagem. A adição de óptica de divisão de imagem permite a captura simultânea de imagens de dois fluoróforos distintos – animado por 561 nm e lasers de 488 nm – separando as bandas de emissão em duas metades do mesmo chip de câmera. - Após o conjunto do instrumento, verifique o alinhamento óptico do diSPIM antes da imagem latente.
Observação: para alinhar corretamente o diSPIM consulte https://youtu.be/qnOrg30NNuE e para informações de hardware, http://dispim.org/hardware/objectives e http://www.asiimaging.com. - Use a plataforma de código aberto micro-Manager (https://micro-manager.org/)21, que foi otimizado para operar microscópios de folha de luz para imagens celulares de alta produtividade22. Recomendamos o uso do plugin ASI diSPIM para aquisição de várias posições, que permite imagens simultâneas de até 30 embriões, conforme descrito23.
- Com o micro-gerente aberto, ajuste intensidades do laser a ~ 179 μW (0,5) para 488 nanômetro e ~ 79 μW (0,25) para 561 nanômetro (Figura 2a, retângulo vermelho).
Nota: Estas são as definições recomendadas para imagens a longo prazo de embriões C. elegans utilizando intervalos de 1 minuto. Durante a imagem latente a longo prazo da duplo-cor, o laser de 561 nanômetro é usado aos núcleos da imagem (Mcherry:: histone) até que os embriões estejam no estágio do feijão, em que ponto o laser do nm 488 é então gira sobre para igualmente imagem os neurônios GFP-etiquetados. Estas condições de imagem são otimizadas para minimizar a fototoxicidade e garantir a sobrevivência e eclosão dos embriões, permitindo a aquisição contínua prolongada (12 – 14 horas) de dados de neurodesenvolvimento e lineaging. - No micro-Manager, escolha menu Plugins ≫ controle de dispositivo > ASI DISPIM para abrir o ASI dispim Window (Figura 2b). Escolha a guia aquisição . Na seção Configurações de salvamento de dados desta guia (retângulo verde), seção configurações de volume (quadrado azul) e seção configurações de fatia (quadrado laranja), assegure-se de que cada parâmetro seja definido como mostrado na Figura 2b.
Nota: nosso software de análise de imagem CytoSHOW é adaptado para trabalhar com outros formatos de dados de saída opcionais, como a série de arquivos OME-TIFF em massa concatenada e a série de arquivos TIFF-Stack criadas após a aquisição através do uso de uma função de exportação incorporada ao micro-Manager. Normalmente, o formato de dados de arquivo do OME-TIFF de pilha de ponto único é usado porque permite a visualização em tempo real e o processamento do volume da imagem assim que os dados brutos são adquiridos.
6. parâmetros otimizados do autofocus para a imagem latente a longo prazo de embriões de C. elegans
- Defina os parâmetros de autofoco micro-Manager para as configurações otimizadas para a qualidade de linhagem de longo prazo diSPIM-Imaging de embriões C. elegans . Na janela do ASI diSPIM, clique na guia autofocus (Figura 2C). Na seção geral de opções de autofoco (quadrado preto), especifique os parâmetros precisamente como mostrado. Observe que o canal de autofoco (quadrado vermelho) deve especificar seu canal de fluorescência de canal nuclear em experimentos de lineaging.
Nota: se o deslocamento máximo for maior que 5 μm, as imagens tendem a deriva fora de foco. - Clique, Plugins > ferramentas de aquisição > padrão Overlay.
- Na janela sobreposição de padrão , clique em Mostrar grade.
- Na janela do ASI diSPIM , clique na guia navegação .
- Clique em caixas de seleção para feixe e folha de caminho A ou B, em seguida, clique em Live. A aquisição da imagem começa. Uma janela do Live View é aberta. Selecione a região de análise de foco automático do embrião desenhando uma caixa ao redor do embrião no canal selecionado de 6,1.
Nota: normalmente capturamos 420 pontos de tempo para 10 embriões por sessão de imagem. A sessão de dados brutos por imagem normalmente é de 1,7 TB, enquanto os dados processados e de StarryNite são 1,4 TB (consulte as etapas 9 e 10). Recomendamos o uso de HDD de grande capacidade (18TB no nosso sistema atual) para aquisição de imagens e plataformas de nuvem para armazenamento de imagens. - Clique em Iniciar aquisição na guia "aquisição para iniciar a captura de imagens multidimensionais de longo prazo (Figura 2b).
7. abrindo imagens micro-Manager RAW no CytoSHOW
- Baixe o pacote de software de http://dispimlineage.wormguides.org.
Nota: o pacote de software será transferido como um ficheiro. zip e terá de ser extraído para o "C:\" diretório antes de usar. Mais detalhes para a instalação são dadas em http://dispimlineage.wormguides.org/diSPIMlineaging_InstallationInstructions.htm. - Clique duas vezes no arquivo C:\cytoshowextrasforc\cytoshow_app.jnlp para começar a executar o cytoshow.
- Escolha o arquivo de menu > novo > Monitor Dispim (micro-Manager). Localize a pasta de conjunto de dados raiz onde as pastas de ponto de tempo de aquisição foram salvas. Selecione qualquer pasta de ponto de tempo e clique em abrir. As janelas de navegação multidimensionais (chamadas de Monitor Dispim Windows) são abertas automaticamente para spima e SPIMB (Figura 3a).
Nota: estas janelas irão monitorizar a pasta de dados raiz para pilhas de ponto de temporal RAW recém-guardadas (no caso de uma amostra ainda está a ser registada). Após cada novo temporal ser adquirido, cada uma das janelas que monitoram os braços spim distintos e as posições de amostra serão atualizadas para exibir todo o conjunto de dados 4D multicanal para cada embrião.
8. gerando imagens de projeção Max com CytoSHOW
Mesmo antes da deconvolução, os dados brutos podem ser processados rapidamente para avaliar as características globais do espécime.
- Clique no botão Z-MIP no painel do lado esquerdo da janela de imagem (Figura 3a, retângulo vermelho) para fazer projeções de intensidade máxima através da profundidade total e do curso em tempo integral de uma determinada posição ou braço spim. Aparecerá uma janela de hiperpilha de projeção Z .
- Na janela de hiperpilha de projeção Z , defina o tipo de projeção como intensidade máxima. Especifique quais canais, fatias e quadros de ponto de tempo para processar com base na preferência do usuário.
- Clique em OK quando concluído.
- Selecione o local da pasta para salvar as saídas de intensidade máxima da janela de diálogo do arquivo e, em seguida, clique em OK. Permita algum tempo (15 – 20 minutos, dependendo do tamanho do conjunto de dados e do poder de processamento do computador) para que o CytoSHOW gere imagens de projeção.
9. analisando linhagens celulares em dados volumétricos de alta resolução isotrópicas
- Opcional: com os dados brutos abertos através do Monitor Dispim no cytoshow, selecione a ferramenta de seleção do polígono (Figura 3a, setas pretas) e clique apenas fora das bordas anterior, posterior, dorsal e ventral do embrião (nessa ordem exata) para gerar um padrão de "bowtie" sobre o embrião. Faça para ambas as visualizações (SPIM-A e SPIM-B, Figura 3a).
Nota: esta seleção especifica a região elíptica de interesse (ROI) na qual o embrião é centrado e registra o eixo anterior-posterior do embrião. O teste padrão do bowtie mostra o cytoshow que o usuário planeia especificar mais uma rotação precisa dos volumes isotropicamente fundidos finais em uma orientação que seja óptima para análises de lineaging pelo starrynite/acetree. Nos casos em que a lineaging de StarryNite não faz parte do plano experimental, outras ferramentas de seleção e formas podem ser escolhidas para definir o ROI para processamento de imagem. - Se vários embriões foram simultaneamente imaged usando a opção de aquisição de várias posições, abra e execute o passo 9,1 para todos os embriões. Isso permitirá a execução paralela de etapas futuras para todos os embriões em uma sessão. Feche as janelas SIMA e SPIMB para quaisquer embriões que não deseje processar.
- Clique no botão Dispim no painel do lado esquerdo da janela do monitor dispim (Figura 3a, destacada em amarelo). Isso revela um Subpainel de controles específicos para o processamento de diSPIM.
- Alinhe os canais verdes e vermelhos para cada braço SPIM. Como os divisores de canal de emissão são usados para capturar imagens vermelhas e verdes distintas simultaneamente na mesma câmera, é importante alinhar visualmente o registro exato de pixel desses dois campos de imagem fisicamente adjacentes quando eles são sobrepostos. A reutilização dos mesmos ajustes de alinhamento é normalmente viável em muitas sessões de imagens consecutivas, mas deve ser verificada (como nas etapas 9.4.1 – 9.4.5).
- Começando com o painel SPIMA, selecione o canal vermelho movendo a barra de rolagem cm para a esquerda (Figura 3a, seta laranja, painel esquerdo).
- Usando os x-, y-, e z-ajustadores (Figura 3a, quadrado alaranjado), desloc o canal vermelho para combinar o verde.
- Clique no botão Dispim (Figura 3a, realçado em amarelo), para fechar o Subpainel e acionar a propagação dos mesmos turnos para todas as outras janelas de posição .
- Confirme se o alinhamento correto é propagado para outros quadros e pontos de tempo movendo a barra de rolagem "z" (Figura 3a, seta azul, painel esquerdo) e/ou barra de rolagem "t" (Figura 3a, seta verde, painel esquerdo). Se a aquisição de várias posições foi realizada e vários embriões foram fotografados (etapa 5,3), o alinhamento também deve ter se propagado para esses embriões. Confirme examinando também os números para x-, y-, e z-ajustadores (Figura 3a, quadrado alaranjado, que deve ser o mesmo para o painel de spima de todos os embriões).
- Repita as etapas 9.4.1 – 9.4.4 para a janela do monitor diSPIM do SPIMB (Figura 3a, painel direito).
- Clique no botão "Dispim" e, em seguida, no botão "Fuse" (Figura 3a, retângulo azul) para abrir uma caixa de diálogo denominada "Deconvolve/Fuse Dispim Raw Data volumes" (Figura 3B). Defina os parâmetros como mostrado na Figura 3B. Esses parâmetros são abordados brevemente nas seguintes subetapas:
- Definir registro de chave no canal 1 (488 nm laser) ou 2 (561 nm laser). Selecione o canal com sinal mais denso ou onipresente. Para experimentos de lineaging sempre selecione o canal usado para a imagem da fluorescência histona nuclear onipresente.
- Defina a orientação relativa dos volumes de entrada para + 1 ou-1. O índice de orientação correto depende de posicionamentos de câmera específicos do diSPIM (Figura 4).
Nota: se incerto, teste cada opção duplicando um único ponto de tempo da janelade Monitor dispim spim a e B, seguindo os passos 9.1 – 9.12 e arbitrariamente escolhendo uma orientação relativa de volumes de entrada para testar. As orientações incorretas produzirão imagens borradas com artefatos, enquanto as orientações corretas produzirão imagens claras. O valor de orientação relativa de volumes de entrada que produz a imagem nítida, em seguida, pode ser reutilizado para todos os dados futuros do instrumento dispim determinado. - Escolha se o volume fundido deve ser orientado da mesma forma que o volume de entrada a ou B (com base na preferência do usuário).
- Selecione "registro fresco para cada volume". Essa opção controla como o SpimFusion calcula as matrizes de registro para cada par de volumes em cada ponto de tempo. A opção "Fresh" permite que o algoritmo Otimize o registro de forma adaptativamente em cada ponto temporal.
- Defina o número de iterações de deconvolução como 10. Esse número tende a produzir de forma confiável a alta resolução desejada de maneira eficiente em termos de tempo.
- Opcional: se a auto-lineaging é desejada (altamente recomendável), verifique auto-lançamento StarryNite lineaging de volumes fundidos. Essa opção lançará StarryNite automaticamente para segmentar e rastrear células nos volumes de imagem produzidos pelo SpimFusion.
- Opcional: para maior precisão na lineaging automatizada, é melhor reposicionar os volumes de embriões isotropicamente fundidos na orientação canônica "ADL" (anterior [x-West], Dorsal [y-North], Left [z-Near]). Selecione a opção definir orientação de saída de volume na visualização para indicar essa escolha. O CytoSHOW responderá processando um par inicial de volumes isotropicamente fundidos, permitindo que o usuário observe de perto e especifique as rotações necessárias para alcançar o registro da ADL.
- Clique em Sim uma vez que todos os parâmetros são selecionados.
- Especifique o diretório de saída no qual salvar os arquivos processados. Clique em OK.
- Opcional: se a opção definir a orientação de saída de volume na visualização for selecionada, defina a barra de rolagem t (Figura 3a, seta verde, painel esquerdo) na janela spim-a para o ponto de tempo inicial em que as células aba e ABP atingiram a metafase. Defina a barra de rolagem t na janela spim-B para o estágio de vírgula posterior do desenvolvimento. Isso ajudará a especificar a orientação da ADL.
- Opcional: clique em OK quando estiver pronto. Se a opção de pré-visualização em 9.5.7 acima foi selecionada, apenas dois volumes de visualização serão isotropicamente fundidos para os temporais indicados pelos t-sliders das janelas de imagem spim-a e spim-B. Esses dois temporais de visualização podem ser usados para especificar o realinhamento preciso dos volumes de embriões de saída para a orientação do ADL, conforme explicado abaixo.
- Localize o recém-exibido 3DProjY_Decon-Fuse_.... Janela. Mova a barra de rolagem t para o ponto de tempo 2 desta janela de visualização. Mova o controle deslizante Z até que a vista diretamente para baixo do eixo longo do embrião seja mostrada.
- Mova a barra de rolagem t de volta para o ponto de tempo 1 do 3DProjY_Decon-Fuse_.... Janela. Escolha a ferramenta de seleção de linha e desenhe uma seleção de linha da célula do EMS (núcleo ventral-mais redondo) através do plano das placas da metafase da AB-pilha.
- Clique no botão laranja diSPIM Preview no 3DProjY_Decon-Fuse_.... Janela. Os ajustes finos para a orientação do volume imaged visualizado serão salvos para uso no processamento do conjunto de dados completo.
- Opcional: a caixa de diálogo Deconvolve/Fuse Dispim Raw Data volumes irá reaparecer, assim como na etapa 9,5 acima. Clique em Sim sem escolher a orientação de saída de volume definir na opção de visualização . Especifique a pasta de saída para a execução completa de processamento de dados.
- Defina as barras de rolagem t (Figura 3a, seta verde, painel esquerdo) das janelas do monitor dispim para o temporal inicial (spima) e terminando o temporal (spimb) do período completo de imagens para processar. Em seguida, clique em OK.
- Como o SpimFusion progride, o CytoSHOW abre e atualiza uma janela multidimensional que mostra o volume fundido isotrópico cortado em 4D para cada embrião, bem como duas janelas com projeções de intensidade máxima rotativa-4D do volume isotrópico. Durante este tempo, não interrompa nem feche nenhuma janela do cytoshow até que a fusão isotrópico e o seguimento da linhagem estejam completos.
- Opcional: Observe que, depois que a tela inicial StarryNite aparecer e mais tarde desaparecer, o pipeline de processamento de dados completo foi concluído. Esta janela não deve ser fechada durante o processamento ou StarryNite será interrompida.
10. abrindo série de traços de linhagem StarryNite em AceTree (opcional)
- Abra a versão personalizada do "AceTree_16BitCompat. jar" fornecida.
- Escolha o menu arquivo > abrir configurar arquivo. Localize o diretório de saída indicado anteriormente para o cytoshow. Abra a subpasta Decon_Fuse_... _ pos [n] para embrião [n]. Selecione aaa_edited. xml e abrir.
- Use o menu AceTree edit > Editar ferramentas para abrir a faixa de edição e ajustar ou excluir janelas de células.
- Clique no círculo meia-sombreado Figura 5b, quadrado vermelho para ajustar as intensidades vermelho e verde.
- Prossiga com a visualização e edição de linhagem conforme descrito anteriormente5,6,8 (manuscritos também estão incluídos em nosso pacote de download).
Representative Results
Primeiramente validamos a viabilidade de embriões imaged utilizando os parâmetros do protocolo para aquisição de diSPIM (seções 1-6). Dez embriões foram simultaneamente imaged a 20 ° c, um volume/embrião/minuto, do estágio de 2 células para o estágio 2 vezes (7,5 horas, 451 volumes/embrião). Para monitorar as divisões celulares ao longo da embriogênese, utilizou-se a cepa BV514, que expressa de forma onipresente as construções de Mcherry::histone reporter a partir da matriz transgênica integrada ujIs11324. A Figura 6 mostra uma linha temporal desta primeira metade do desenvolvimento embrionário para um dos embriões imaged. Cada imagem representa uma projeção de intensidade máxima de visualização única (produzida pelas etapas 7-8) do embrião imaged. Verificou-se que os protocolos otimizados não induzem qualquer fototoxicidade detectável aos embriões, conforme avaliado pelo tempo de divisões celulares (não mostradas), época de eclosão e cronometragem relacionada a Marcos de desenvolvimento (Figura 6 e referências1 , 25 anos de , 26).
Em seguida, aplicou-se o protocolo para analisar a dinâmica de conseqüência de neurônios únicos no desenvolvimento de embriões. Nós imaged DCR7692 (olaex4655), uma cepa transgênica do nematoide que expresse o GFP fora do neuropeptídeo FLP-19 promotor em um subconjunto de pilhas não identificadas (DACR2819, PFLP-19 (3.6 KB):: Syn21:: GFP:: CAAX::p 10 3 ' UTR) . Seguindo as etapas do protocolo delineado aqui, determinou-se que as células não identificadas correspondem aos neurônios motores RMDDL e RMDDR, à célula excretora do canal e a duas células musculares (Figura 7). Nós examinamos então e quantificamos a dinâmica do conseqüência dos neurônios de rmddl e de rmddr. Observamos que os neurônios RMDDL e RMDDR são obliquamente moldados em 360 minutos após a fertilização, com o eixo celular mais longo representando o eixo subseqüente para o crescimento do neurite (Figura 7 e filme S1). Usando o "simples neurite rastreamento" plugin em FIJI e aplicá-lo a reconstruções 3D de volumes isotropicamente fundidos, então quantificamos o crescimento estereotipado dos neuritos RMDDL e RMDDR para seis embriões. Determinamos que a dinâmica de crescimento foi estereotipada para RMDDL e RMDDR através de embriões (aqui denominados RMDDs). A partir de 385-410 minutos após a fertilização, os neuritos de RMDDs estendidas 6,0 ± 0,5 μm (média ± SEM; n = 12 neuritos) anterior dos corpos celulares (Figura 7B, C, I). A partir de 415-445 minutos após a fertilização, ambos os neuritos estendem dorsalmente e em torno do anel do nervo presuntivo (asterisco na Figura 7D). Em média, cada neurite de RMDD estendeu 11,0 ± 0,6 μm (média ± SEM; n = 12 neuritos) a partir do corpo celular antes de sincronizar de forma síncrona sua contraparte contralateral no ápice do anel (Figura 7i). É importante ressaltar que nossos resultados representativos demonstram que somos capazes de examinar, comparar e quantificar as características de desenvolvimento neuronal para células de identificação única usando nosso protocolo integrado (Figura 7 e Figura 8).
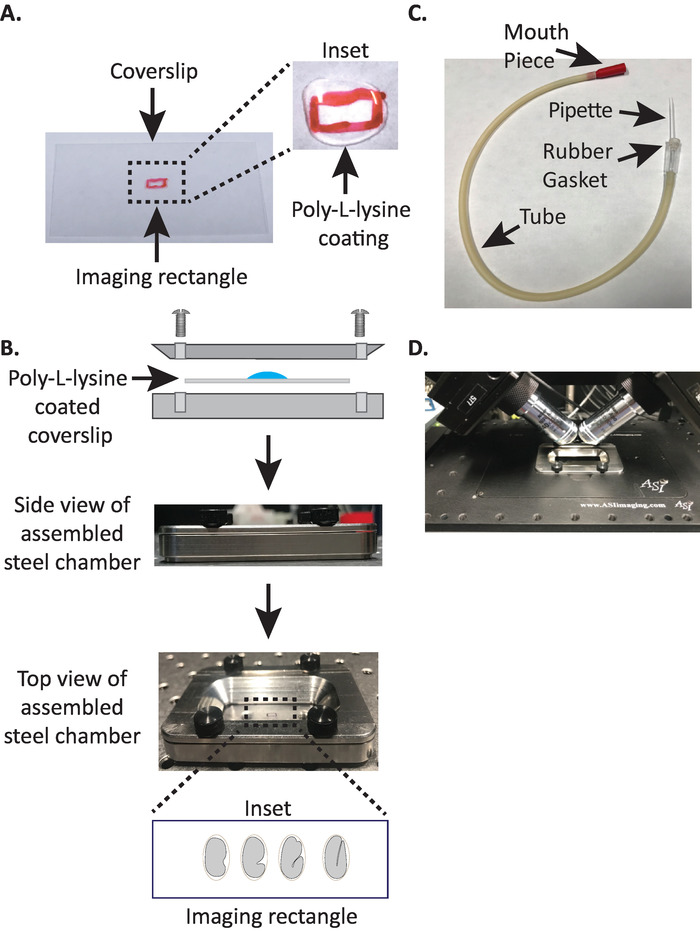
Figura 1: configuração de montagem de amostra diSPIM. (A) preparação de lamínula com poli-L-lisina. No Inset, 10 μL de poli-L lisina foi usado para revestir a lamínula por 5 minutos. Poli-L-lisina permite que a casca de ovo do embrião para furar firmemente à lamínula no retângulo. (B) esquemático da câmara de imagem de aço e da câmara montada. No Inset, os embriões representativos são orientados com o eixo anterior-posterior perpendicular ao eixo longo na lamínula. (C) tubo aspirador montado com pipeta microcapilar. (D) câmara de imagem de aço montada no suporte da amostra objetivos de dispim 40x. Por favor clique aqui para ver uma versão maior desta figura.

Figura 2: configuração de imagens diSPIM de longo prazo no micro-Manager. (A) parâmetros recomendados do poder do laser de dispim (retângulo vermelho) aperfeiçoados para a imagem latente prolongada ao reduzir o fototoxicidade (como avaliado pela taxa de eclosão mais elevada de embriões de C. elegans ). Definir 561 nm laser para 79 μW (0,25) e 488 nm laser para 179 μW (0,5). Observe que a calibração exata das configurações de software para a potência do laser varia entre as instalações diSPIM. Recomenda-se que os usuários medem e calibram o poder do laser a fim conseguir o poder do laser de 79 μW (561 nanômetro) e de 179 μW (488 nanômetro). (B) dispim parâmetros para economia de dados (retângulo verde), configurações de volume (quadrado azul) e configurações de fatia (quadrado laranja). (C) dispim autofocus parâmetros para imagens a longo prazo de C. elegans embriogênese (ver passos 6.1-6.6). Por favor clique aqui para ver uma versão maior desta figura.

Figura 3: visualização de imagem e configuração de processamento de dados usando o CytoSHOW. (A) RAW dispim imagens abertas pelo cytoshow. O CytoSHOW é capaz de abrir imagens capturadas por ambos os caminhos da câmera (SPIM A e B). Essas imagens RAW são abertas em janelas multidimensionais chamado Monitor Dispim. No Monitor Dispim, um "padrão de bowtie" é gerado para selecionar as bordas anterior, posterior, dorsal e ventral do embrião (ver etapa 9,1). As seleções Bow-tie indicam a orientação do embrião para a deconvolução e o traçado de lineaging assistido por StarryNite. (B) parâmetros otimizados usados para gerar imagens isotrópicas. Na janela Deconvolved ao adquirir , defina os parâmetros especificados nas etapas 9.5.1-9.5.8. Por favor clique aqui para ver uma versão maior desta figura.

Figura 4: configuração da câmera diSPIM. (A) fotografia de posicionamentos e orientações da câmera dispim. (B) representação de + 90-graus rotações de spim a para coincidir com as imagens spim B coletadas. (C) volumes de entrada em relação ao índice de orientação + 1 com base na configuração da câmera do dispim (consulte a etapa 9.5.2). Nós giramos SPIM uma imagem (s) + 90 graus em torno de Y-axis antes do registro para combinar a imagem de SPIM B (s). Barras de escala = 10 μm. as imagens são representativas de visualização única, projeções de máxima intensidade e imagens de desvolução de embriões de 1,5 vezes com núcleos rotulados (561-nm, vermelho) e neurônios (488-nm, verde). Por favor clique aqui para ver uma versão maior desta figura.

Figura 5: Curação e edição C. elegans linhagem embrionária em acetree. (A) usamos acetree para editar traços de linhagem de starrynite (ver referências5,6,8; manuscritos também estão incluídos em nosso pacote de download). AceTree exibirá C. elegans nomes sistemáticos para cada núcleo (retângulo verde) após a conclusão das etapas 10.1-10.2. Esta janela (a) fornece informações (retângulo preto) sobre cada célula no traço de linhagem (aba, realçado em azul) que ajudam a orientar os usuários ao rastrear e editar os traços de linhagem. Recomenda-se que os usuários verifiquem e comparem as células linhagens e suas posições à linhagem de células embrionárias C. elegans previamente relatadas por Sulston et al.1 além disso, se os usuários estiverem interessados em localizar células específicas no série de dados seções (veja abaixo, B) digite o nome sistemático C. elegans na barra de pesquisa (retângulo laranja). (B) a série de dados seções do usuário igualmente abre automaticamente em cima da conclusão das etapas 10.1-10.2. Mostrado aqui uma imagem isotropicamente fundida de um embrião do estágio da quatro-pilha com os núcleos etiquetados no vermelho. Durante o rastreamento de um núcleo, os usuários devem alterar a intensidade da imagem (quadrado vermelho) e navegar através do tempo e z usando as teclas de seta em seu teclado (tempo-esquerda/direita, z-up/down). (C) desenhos animados 3D do temporal em (B) com determinadas funcionalidades (retângulo roxo) que permite o 3D-visualização rotatable. Para obter uma visão geral do acetree e sua funcionalidade 3D, consulte as referências5,6,8. Por favor clique aqui para ver uma versão maior desta figura.

Figura 6: dinâmica de desenvolvimento cronometrada de embriões de C. elegans no dispim. Painel superior, imagens diSPIM mostrando a primeira metade do desenvolvimento embrionário para um dos embriões imaged (Strain BV514 ujIs11324). Os embriões foram fotografados continuamente, a cada minuto por 7,5 horas (a 20 ° c). As duas primeiras imagens do painel superior representam embriões de 4 e 8 células com núcleos (vermelhos) e posições de corpos polares (esferas vermelhas densas, marcadas com asteriscos azuis). Cada imagem representa uma projeção de intensidade máxima de visualização única do embrião imaged. Barras de escala = 10 μm. A linha do tempo (barra horizontal) representa minutos após a fertilização (m.p.f.) do desenvolvimento de embriões de C. elegans . Nós validamos que os parâmetros de nosso protocolo para a aquisição de diSPIM não induzem nenhuma fototoxicidade detectável aos embriões imaged como avaliados pela viabilidade, o sincronismo de divisões de pilha, o sincronismo da eclosão e o sincronismo de Marcos desenvolventes (veja referências 1. º , 25 anos de , 26). notamos que o timing dos marcos de desenvolvimento foi reprodutível em embriões com os nossos parâmetros de imagem (SEM ± 8,174 minutos para sessões de imagens de 6,4 horas longas; n = 10). Por favor clique aqui para ver uma versão maior desta figura.

Figura 7. Identificação celular e caracterização de células únicas da dinâmica de conseqüência neurite no desenvolvimento de embriões de C. elegans . Imagem latente da duplo-cor de uma tensão feita cruzando BV514 ujIs11324 (para o lineaging) e DCR7692 (olaex4655), uma tensão transgênicas do nematoide que expresse GFP fora do neuropeptídeo FLP-19 promotor em um subconjunto de pilhas não identificadas. (A-H) Seguindo as etapas do protocolo delineado aqui, determinamos que as células não identificadas correspondem aos neurônios motores RMDDL e RMDDR (setas amarelas), à célula do canal excretor (setas azuis), e a duas células musculares (setas brancas). (I) quantificação da dinâmica de crescimento dos neurônios RMDDL e RMDDR usando o plugin Fiji "rastreamento de neurite simples" e aplicando-o a reconstruções 3D de volumes isotropicamente fundidos. Observe como RMDDL e RMDDR apresentam dinâmicas estereotipantes de crescimento, cada uma estendendo-se sincronicamente para um comprimento total de 11,0 ± 0,6 μm (média ± SEM; n = 12 neurites) e encontro no ápice dorsal do anel nervoso (ver também filme S1). Por favor clique aqui para ver uma versão maior desta figura.

Figura 8: exame das imagens do diSPIM isotrópico de morfologias neuronais em embriões de C. elegans . Visualização isotrópica de neurônios AVHL e AVHR (setas amarelas). Usando o diSPIM, as morfologias neuronais podem ser capturadas produzindo imagens de quatro dimensões (4D) com resolução espacial isotrópica de aproximadamente 330 nm. O diSPIM permite que os usuários praticamente giram volumes de imagem com resolução idêntica em todas as direções. As imagens em a-D são projeções de intensidade máxima do mesmo volume de imagem de dispim isotropicamente fundido a partir de rotações distintas em torno do eixo longo do embrião. Barras de escala = 5 μm. por favor clique aqui para ver uma versão maior desta figura.
Complementar filme S1: C. elegans embrião desenvolvendo de 280 a 434 minutos pós-fertilização. Filme isotrópico de estirpe DCR7692 (olaex4655) expressando ujIs113 ubiquitously com DACR2819 neuritos rmdd de rotulagem escassa (Figura 7a-D, setas amarelas). DACR2819 também rotula duas células musculares (Figura 7a-d, setas brancas) e células de canal excretora (Figura 7a-d, seta azul) durante o desenvolvimento embrionário (Figura 7a-d). Barras de escala = 10 μm. por favor clique aqui para baixar este arquivo.
Discussion
C. elegans destaca-se como o único organismo com as posições finais e conectividade de cada neurônio adulto conhecido27. Entretanto, a dinâmica de desenvolvimento que conduz à organização dos circuitos e das redes de funcionamento que compõe o conectoma do C. elegans permanece desconhecida. Com base nas oportunidades emergentes dos avanços na microscopia de luz, podemos agora capturar e analisar as posições celulares, a morfogênese e a neurogênese em todo o desenvolvimento embrionário de C. elegans .
O procedimento que nós descrevemos e que nós usamos rotineiramente no laboratório rende imagens 4D-isotropic de neurônios e de núcleos etiquetados para o Cell-lineaging em embriões de C. elegans . Mais importante ainda, otimizamos as condições de imagem de longo prazo com as capacidades de lineaging diSPIM e acopladas semiautomatizadas com imagens de alta resolução para melhorar a velocidade e a precisão da análise da embriogênese C. elegans . Este protocolo integrado permitirá que os usuários visualizem e identifiquem pilhas e quantificar características tridimensionais tais como a migração do neurite e o fasciculação do AXON através do início de twitching adiantado. Este procedimento pode prontamente ser adaptado em toda a facilidade com um sistema do diSPIM de ASI, e nós recomendamos este sistema especificamente para este protocolo. Outras formulações SPIM oferecidas comercialmente podem diferir da configuração de ASI na câmara de amostra e nas propriedades ópticas. No entanto, os dados exportados de outras plataformas também podem ser colocados através do nosso pipeline de dados. Portanto, a avaliação de seu valor na lineaging, um teste exigente de qualidade de imagem e estabilidade do instrumento, é viável. Mesmo que nós usamos ativamente o diSPIM para imagem regularmente outros espécimes (tais como embriões de Drosophila e de zebrafish), a análise de lineaging descrita e detalhada dos embriões é limitada ainda atualmente às espécies do nematóide. Para amostras maiores ou grossas, optamos por usar abordagens de varredura de estágio, que digitaliza as amostras através de uma folha de luz estacionária. Kumar et al. demonstraram anteriormente este melhor seccionamento de diSPIM para produzir imagens de alta qualidade de amostras grossas sem modificações adicionais no diSPIM10.
As etapas críticas dentro do protocolo incluem a montagem de embriões C. elegans na cobertura revestida de poli-L-lisina, aquisição de dados e processamento de dados. Colheita e montagem de embriões C. elegans na cobertura de vidro pode ser desafiador para usuários inexperientes, mas aqui nós fornecemos um protocolo detalhado de passos-chave para facilitar a aprendizagem. Se a imagem latente a longo prazo é desejada, nós obtemos os melhores resultados que colhem embriões Four-Cell ou mais adiantados de 8-10 adultos novos28. Note-se que os idosos são menos desejáveis para a colheita de embriões fase inicial, porque eles tendem a conter embriões mais velhos no útero e ovos não fertilizados. No que diz respeito à montagem de embriões, problemas como o bloqueio no aspirador montado (pipeta bucal) ou um muito grande de uma abertura na pipeta microcapilar pode impedir a montagem adequada e orientação de embriões. Para se preparar para imagens ideais, realizamos testes de pré-aquisição em embriões pré-twitching precoces e atrasados para verificar o desempenho das folhas de luz, câmeras, objetivos e autofoco. Obtemos melhores resultados quando todas essas operações são testadas e produzem imagens de alta qualidade durante nossos testes de pré-aquisição. Isso é particularmente relevante para a geração de imagens com resolução espacial isotrópica, para as quais as imagens brutas adquiridas de ambos os pontos de vista (objetivos) devem ser de alta qualidade. Após a aquisição, os volumes adquiridos para cada vista são processados para produzir imagens isotrópicas. É importante usar um cartão de processamento gráfico apropriado (GPU) como descrito neste protocolo (veja abaixo). Isto melhora a velocidade de processamento em que as imagens isotropicamente fundidas são geradas, encurtando o tempo para análises de dados. Também é imperativo que os usuários estão executando a versão mais recente do CytoSHOW e estão usando os parâmetros fornecidos com o nosso pacote de download para StarryNite auto-lineaging. Se os usuários estão interessados em usar auto-lineaging para outras amostras (por exemplo, zebrafish, Drosophila etc.) então a otimização adicional aos parâmetros usados no starrynite será exigida (veja referências3,4).
Embora nosso protocolo integrado forneça imagens e resultados de lineaging no embrião pre-twitching, os usuários devem estar cientes que o lineaging automatizado no embrião post-twitching não é atualmente praticável: as posições nucleares mudam na ordem dos segundos no embrião pós-twitching, muito rapidamente para permitir o rastreamento de linhagem. No entanto, o dispim demonstrou, de fato, uma capacidade promissora para captar eventos de neurodesenvolvimento e rastrear algumas posições celulares nos estágios pós-espasmódios da embriogênese23,29. Se os usuários estão interessados em examinar o embrião pós-contração, o diSPIM fornece a velocidade para obter instantâneos volumétricos e rastrear eventos de neurodesenvolvimento finos, como o crescimento do neurite, em embriões em rápida mudança.
Este protocolo será fundamental para a conclusão célula a célula do Atlas de WormGUIDES30, pois fornecerá uma abordagem integrada com imagens isotrópicas de alta resolução para identificar e capturar morfologias 3D de neurônios rotulados durante os primeiros 430 minutos de embriogênese. Como está, o protótipo do Atlas WormGUIDES fornece posições nucleares de células no embrião em desenvolvimento e visa capturar a dinâmica de desenvolvimento de um subconjunto de neurônios embrionários. Este protocolo será uma chave para a integração de neurônios em desenvolvimento adicionais no Atlas de WormGUIDES30.
Nosso protocolo integrado também simplificará a exploração de novos perfis de expressão gênica no embrião C. elegans . Em C. eleganstransgénicos, muitos promotores Cell-specific espacialmente e controlam temporally a expressão do transgene. Embora os padrões de expressão da maioria dos genes tenham sido extensivamente caracterizados no animal adulto31,32,33,34, quase todos ainda têm de ser caracterizados no desenvolvimento (especialmente fase tardia) embrionário. O promoterome de C. elegans foi um recurso útil à comunidade do sem-fim para conduzir a expressão pilha-específica do transgene, assim como determina se a função do gene é pilha-autônoma ou não-autônoma. A captura de padrões de expressão dinâmica e de alta resolução isotrópicas de genes e a identificação precisa de células expressadas por meio de lineaging serão valiosas para muitos na comunidade científica.
A embryogênese compreende dois processos principais entrelaçados, diferenciação celular e morfogênese tecidual. Um grande negócio é sabido sobre os mecanismos e as moléculas que definem tipos distintos da pilha durante o desenvolvimento de C. elegans. No entanto, pouco se sabe sobre os mecanismos importantes para a migração celular, adesão celular e forma celular no embrião C. elegans . Com a linhagem de células invariantes C. elegans conhecida, nosso protocolo nos permite discernir prontamente a 3D-microanatomia catalogada do embrião durante a morfogênese em novos níveis de detalhe: por exemplo, fasciculação do axão, sinaptogênese e atividade neuronal. Ardiel et al. demonstraram previamente o poder do diSPIM para capturar transientes de cálcio ao nível de um único neurônio em embriões de C. elegans 23. Muitos outros aspectos da fisiologia do desenvolvimento estão maduros para o inquérito por estes métodos.
Finalmente, este protocolo é em grande parte automatizado e reduz sistematicamente o tempo que leva para gerar imagens deconvolução e realizar a lineagem celular via StarryNite e Acetree. As estratégias de software utilizadas neste protocolo podem ser aplicadas a muitas questões de biologia distantes dos campos muito específicos em que os demonstramos aqui.
Detalhes sobre compatibilidade de software e acesso de download
Informações sobre micro-Manager e plugins para imagens diSPIM estão disponíveis em http://dispim.org/software/micro-manager e https://micro-manager.org/wiki/ASIdiSPIM_Plugin.
O pipeline de processamento de dados atualmente requer um sistema operacional Windows. Nós empacotamos um único arquivo de arquivamento para simplificar a instalação de todos os programas de processamento de dados necessários e arquivos de suporte. Está disponível para download em http://dispimlineage.wormguides.org.
CytoSHOW (http://run.cytoshow.org/) baseia-se na plataforma de análise de imagem de código aberto e amplamente utilizado, ImageJ (v1). O Java deve ser instalado e atualizado no computador para usar o CytoSHOW, e as atualizações do CytoSHOW são implantadas automaticamente via Java Web Start. Muitas funções baseadas em ImageJ do CytoSHOW são descritas e ilustradas em https://imagej.nih.gov/ij/docs/examples/index.html. O CytoSHOW foi personalizado para exibir dados brutos multidimensionais do ASI diSPIM, bem como outros softwares de imagem que criam a saída TIFF. Em princípio, outros sistemas de imagiologia SPIM multivista podem ser apoiados por pequenas modificações do CytoSHOW para permitir que este protocolo seja realizado em diferentes sistemas de microscópio.
SpimFusion foi escrito em/C++ usando o Visual Studio 2013 com o kit de ferramentas do cubo v 7.5. Executar SpimFusion requer hardware de computador específico: uma placa de processamento de gráficos NVIDIA (GPU) com capacidade de computação de 1,0 ou superior e um mínimo de 2 GB de memória de placa gráfica. No momento da publicação do nosso protocolo, SpimFusion é inédita (min Guo e Hari Shroff), mas disponível no arquivo de pacote de software mencionado acima.
Uma versão de linha de comando especialmente construída do StarryNite requer que o runtime do compilador do MATLAB disponível livremente esteja instalado, mas não requer uma licença para o software MATLAB comercial. O MATLAB Compiler Runtime está incluído no arquivo de pacote de software mencionado acima. O código para StarryNite como usado neste protocolo é essencialmente inalterado daquele usado para imagens confocal6. No entanto, várias questões operacionais na criação de imagens de entrada para o processamento de StarryNite e o manuseio de resultados de StarryNite foram abordadas aqui por métodos no Citoshow que permitem um pipeline de processamento contínuo de dados para diSPIM isotrópico fundido Volumes. Essas alterações são automatizadas pelo código CytoSHOW que lida com essas etapas de pré e pós-processamento. O CytoSHOW também edita um parâmetro pré-otimizado de modelo específico de diSPIM StarryNite definido para ajustar automaticamente o algoritmo de segmentação à intensidade de fluorescência dos núcleos nos dados imaged. Os parâmetros exclusivos usados pelo StarryNite em cada conjunto de dados diSPIM são salvos em um arquivo junto com a imagem de saída e os dados de lineaging.
Uma versão personalizada do AceTree que funciona com imagens de 16 bits e mantém A compatibilidade com a renderização Java3D é mais adequada para este protocolo. Ele também está incluído no arquivo de pacote de software mencionado acima.
Disclosures
Os autores não têm nada a revelar.
Acknowledgments
Agradecemos a John Murray por estirpe integrada, ujIs113, para gerar estirpe de lineaging BV514; Brandon Harvey (NIBIB) para obter ajuda com o teste do protocolo; Jon Daniels e Gary Rondeau (Instrumentação científica aplicada) para assistência com micro-gerente e diSPIM instrumento; e Andrew York e Hank Eden por seu feedback crítico sobre o sistema diSPIM. Agradecemos também o programa centro de pesquisa para instituições minoritárias e o Instituto de Neurobiología Jose del Castillo (Universidad de Puerto Rico) por fornecer uma plataforma de reunião e brainstorming. Grande parte deste trabalho foi conduzido no laboratório biológico marinho em Woods Hole através do programa Whitman. Este trabalho foi apoiado pelos programas de pesquisa intramural do NIH National Institute of Biomedical Imaging and BioEngineering e pela NIH Grant no. U01-HD075602 e não. R24-OD016474. Mark W. Moyle foi apoiado por F32-NS098616 e Leighton H. Duncan foi apoiado por um suplemento de diversidade para R24-OD016474.
Materials
| Name | Company | Catalog Number | Comments |
| Steps 1-4 | |||
| Concavity slides | ThermoFisher Scientific | 1519006 | 5-18mm diameter, 0.6-0.8mm deep, 1.2-1.5mm |
| Dissecting microscope with 10×–50× zoom range | Motic | SMZ-171 | |
| E. coli (OP50) | Caenorhabditis Genetics Center (CGC) | OP50 | |
| Glass coverslips, no. 1.5, 24 × 50 mm | VWR International | 48393-241 | |
| M9 Buffer | Stiernagle, T. Maintenance of C. elegans. WormBook. 1-11, doi:10.1895/wormbook.1.101.1, (2006). | ||
| Methyl cellulose | Sigma-Aldrich | H7509-25G | |
| Microcapillary pipette aspirator tube | Sigma-Aldrich | A5177 | |
| Microcapillary pipettes, 0.4-mm i.d | Drummond Scientific | 1-000-800 | |
| Needle, no. 18G x 1 ½ (1.2mm x 40mm) | BD Precision Glide | 305196 | |
| NGM plates | prepared as described by Brenner (1974) | ||
| O-ring for imaging chamber | O-Rings West | M1.5X40 | |
| Pasteur pipette | Corning/Sigma-Aldrich | CLS7095D5X | |
| Platinum wire, 0.5-mm diameter | Sigma-Aldrich | 267201 | |
| Poly-L-lysine | Sigma-Aldrich | P1524 | |
| Stainless steel rectangular chamber (76.0 mm x 50.5 mm) | Applied Scientifics Instrumentations (ASI) | I2450 | |
| Worm Eyelash Pick | Hart, A. C. Behavior. WormBook. (2006). | ||
| Worm Pick | Stiernagle, T. Maintenance of C. elegans. WormBook. 1-11, doi:10.1895/wormbook.1.101.1, (2006). | ||
| Name | Company | Catalog Number | Comments |
| Steps 5-6 | |||
| 488 nm long-pass filter | Semrock | LP02-488 RU-2 | |
| 561-nm notch filter | Semrock | NF03-561E-25 | |
| BLP02-561R-25, quantity 2 | Semrock | 561 nm EdgeBasic best-value long-pass edge filter | |
| Control software for bottom camera | Jenoptik | ProgRes CapturePro | |
| diSIPM assembly video | Applied Scientifics Instrumentations (ASI) | https://youtu.be/TAgbr6IrTqw ; http://www.asiimaging.com | |
| diSPIM alignment video | Applied Scientifics Instrumentations (ASI) | https://youtu.be/qnOrg30NNuE | |
| diSPIM imaging PC | Intel | Intel Xeon CPU E5-2630 2.6GHz, 12 cores in total, 64 GB memory, Windows 7 | |
| FF01-525/45-25, quantity 2 | Semrock | 525/45 nm BrightLine single-band bandpass filter | |
| FF555-DI03-25X36, quantity 2 | Semrock | 555 nm edge BrightLine single-edge dichroic beamsplitter | |
| Imaging PC Graphics Card | NVIDIDA | NVIDIA GeForce GTX 1080 Ti graphics cards | |
| Kumar et al diSPIM Setup | Applied Scientifics Instrumentations (ASI) | Instrument setup for this protocol is identical to Kumar et al 10,11 diSPIM, which makes use of 40x 0.8NA water immersion lenses for imaging. (See steps 5.1 and note) | |
| Micro Manager | Micro-Manager | https://micro-manager.org/ | |
| Modifications to Kumar et al diSPIM Setup (see below) | |||
| Optical table with isolators, 4 feet × 6 feet × 12 inches | TMC | 784-651-02DR and 14-416-34 | |
| Name | Company | Catalog Number | Comments |
| Steps 7-10 | |||
| Analysis PC | Intel | Intel Core i7-8700K CPU 3.70GHz, 6 cores in total, 64 GB memory, Windows 10 | |
| Analysis PC Graphics Card | NVIDIDA | NVIDIA GeForce GTX 1080 Ti graphics cards | |
| Installation instructions | Software bundle | http://dispimlineage.wormguides.org/diSPIMlineaging_InstallationInstructions.htm | |
| Software bundle | Software bundle | http://dispimlineage.wormguides.org |
References
- Sulston, J. E., Schierenberg, E., White, J. G., Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology. 100 (1), 64-119 (1983).
- Bao, Z., et al. Automated cell lineage tracing in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 103 (8), 2707-2712 (2006).
- Santella, A., Du, Z., Bao, Z. A semi-local neighborhood-based framework for probabilistic cell lineage tracing. BMC Bioinformatics. 15, 217 (2014).
- Santella, A., Du, Z., Nowotschin, S., Hadjantonakis, A. K., Bao, Z. A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC Bioinformatics. 11, 580 (2010).
- Boyle, T. J., Bao, Z., Murray, J. I., Araya, C. L., Waterston, R. H. AceTree: a tool for visual analysis of Caenorhabditis elegans embryogenesis. BMC Bioinformatics. 7, 275 (2006).
- Katzman, B., Tang, D., Santella, A., Bao, Z. AceTree: a major update and case study in the long term maintenance of open-source scientific software. BMC Bioinformatics. 19 (1), 121 (2018).
- Murray, J. I., et al. Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nature Methods. 5 (8), 703-709 (2008).
- Murray, J. I., Bao, Z., Boyle, T. J., Waterston, R. H. The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nature Protocols. 1 (3), 1468-1476 (2006).
- Wu, Y., et al. Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nature Biotechnology. 31 (11), 1032-1038 (2013).
- Kumar, A., et al. Using Stage- and Slit-Scanning to Improve Contrast and Optical Sectioning in Dual-View Inverted Light Sheet Microscopy (diSPIM). The Biological Bulletin. 231 (1), 26-39 (2016).
- Kumar, A., et al. Dual-view plane illumination microscopy for rapid and spatially isotropic imaging. Nature Protocols. 9 (11), 2555-2573 (2014).
- Wu, Y., Christensen, R., Colon-Ramos, D., Shroff, H. Advanced optical imaging techniques for neurodevelopment. Current Opinion in Neurobiology. 23 (6), 1090-1097 (2013).
- Wu, Y., et al. Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 108 (43), 17708-17713 (2011).
- Huisken, J., Stainier, D. Y. Selective plane illumination microscopy techniques in developmental biology. Development. 136 (12), 1963-1975 (2009).
- Stelzer, E. H. Light-sheet fluorescence microscopy for quantitative biology. Nature Methods. 12 (1), 23-26 (2015).
- Winter, P. W., Shroff, H. Faster fluorescence microscopy: advances in high speed biological imaging. Current Opinion in Chemical Biology. 20, 46-53 (2014).
- Lucy, L. B. An iterative technique for the rectification of observed distributions. Astronomical Journal. 76 (6), 745-754 (1974).
- Richardson, W. H. Bayesian-Based Iterative Method of Image Restoration. JOSA. 62 (1), 55-59 (1972).
- Stiernagle, T. Maintenance of C. elegans. WormBook. , 1-11 (2006).
- Hart, A. C. Behavior. WormBook. , (2006).
- Edelstein, A., Amodaj, N., Hoover, K., Vale, R., Stuurman, N. Computer control of microscopes using microManager. Current Protocols in Molecular Biology. Chapter 14, Unit14 20 (2010).
- Gualda, E. J., et al. SPIM-fluid: open source light-sheet based platform for high-throughput imaging. Biomedical Optics Express. 6 (11), 4447-4456 (2015).
- Ardiel, E. L., et al. Visualizing Calcium Flux in Freely Moving Nematode Embryos. Biophysical Journal. 112 (9), 1975-1983 (2017).
- Walton, T., et al. The Bicoid class homeodomain factors ceh-36/OTX and unc-30/PITX cooperate in C. elegans embryonic progenitor cells to regulate robust development. PLoS Genetics. 11 (3), e1005003 (2015).
- Altun, Z. F. WormAtlas. , (2002).
- Wood, W. B. Embryology: In the nematode C. elegans. Cold Spring Harbor Laboratory Press. Chapter 8, 215-241 (1988).
- White, J. G., Southgate, E., Thomson, J. N., Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical Transactions of the Royal Society B: Biological Sciences. 314 (1165), (1986).
- Bao, Z., Murray, J. I. Mounting Caenorhabditis elegans embryos for live imaging of embryogenesis. Cold Spring Harb Protoc. (9), (2011).
- Christensen, R. P., et al. Untwisting the Caenorhabditis elegans embryo. eLife. 4, (2015).
- Santella, A., et al. WormGUIDES: an interactive single cell developmental atlas and tool for collaborative multidimensional data exploration. BMC Bioinformatics. 16, 189 (2015).
- Dupuy, D., et al. A first version of the Caenorhabditis elegans Promoterome. Genome Research. 14 (10B), 2169-2175 (2004).
- Reece-Hoyes, J. S., et al. Insight into transcription factor gene duplication from Caenorhabditis elegans Promoterome-driven expression patterns. BMC Genomics. 8 (27), (2007).
- WormBase. , Available from: https://www.wormbase.org (2019).
- Lee, R. Y. N., et al. WormBase 2017: molting into a new stage. Nucleic Acids Research. 46 (D1), D869-D874 (2018).
