

Summary
Este artículo tiene como objetivo proporcionar la metodología para la transgénesis lentiviral en embriones de rata utilizando múltiples inyecciones de una suspensión de virus en el espacio perivitelina cigota. Las ratas hembra que están apareadas con una cepa masculina fértil con un color de piel dominante diferente se utiliza para generar madres adoptivas pseudoembarazadas.
Abstract
Los modelos animales transgénicos son fundamentalmente importantes para la investigación biomédica moderna. La incorporación de genes extraños en embriones de ratón o rata tempranos es una herramienta invaluable para el análisis de la función génica en organismos vivos. El método de transgénesis estándar se basa en la microinyección de fragmentos de ADN extraños en un pronúcleo de un ovocitos fertilizados. Esta técnica es ampliamente utilizada en ratones, pero sigue siendo relativamente ineficiente y técnicamente exigente en otras especies animales. El transgén también se puede introducir en embriones de una fase celular a través de una infección lentiviral, proporcionando una alternativa eficaz a las inyecciones pronucleares estándar, especialmente en especies o cepas con una estructura embrionaria más desafiante. En este enfoque, se inyecta una suspensión que contiene vectores lentivirales en el espacio perivelinal de un embrión de rata fertilizada, que es técnicamente menos exigente y tiene una mayor tasa de éxito. Se demostró que los vectores lentivirales incorporan eficientemente el transgén en el genoma para determinar la generación de líneas transgénicas estables. A pesar de algunas limitaciones (por ejemplo, requisitos de nivel 2 de bioseguridad, límites de tamaño de fragmentos de ADN), la transgénesis lentiviral es un método de transgénesis rápido y eficiente. Además, el uso de ratas hembra que se aparean con una cepa masculina fértil con un color de piel dominante diferente se presenta como una alternativa para generar madres adoptivas pseudoembarazadas.
Introduction
Durante muchos años, los roedores de laboratorio, como ratas y ratones, se han utilizado para modelar condiciones fisiológicas y patológicas humanas. La investigación en animales ha dado lugar a descubrimientos que eran inalcanzables por cualquier otro medio. Inicialmente, estudios genéticos se centraron en el análisis de trastornos y fenotipos que ocurren espontáneamente y que se consideran para imitar de cerca la condición humana1. El desarrollo de métodos de ingeniería genética permitió la introducción o eliminación de genes específicos para obtener un fenotipo deseado. Por lo tanto, la generación de animales transgénicos es reconocida como una técnica fundamental en la investigación moderna que permite estudios de la función génica en organismos vivos.
La tecnología animal transgénica se ha hecho posible a través de una combinación de logros en embriología experimental y biología molecular. En la década de 1960, el embriólogo polaco A. K. Tarkowski publicó el primer trabajo sobre manipulación de embriones de ratón durante las primeras etapas del desarrollo2. Además, los biólogos moleculares desarrollaron técnicas para generar vectores de ADN (es decir, portadores) para la introducción, entre otras cosas, de ADN extraño en el genoma del animal. Estos vectores permiten la propagación de genes seleccionados y su modificación adecuada, dependiendo del tipo de investigación que se lleve a cabo. El término "animal transgénico" fue introducido por Gordon y Ruddle3.
La primera especie ampliamente aceptada que se utilizó en neurobiología, fisiología, farmacología, toxicología, y muchos otros campos de las ciencias biológicas y médicas fue la rata noruega, Rattus norvegicus4. Sin embargo, debido a la dificultad en la manipulación de embriones de rata, el ratón de la casa Mus musculus se ha convertido en la especie animal dominante en la investigación genética5. Otra razón para la primacía del ratón en dicha investigación fue la disponibilidad de tecnología de células madre embrionarias para generar animales noqueantes para esta especie. La técnica más utilizada de transgénesis (2–10% de la descendencia transgénica en relación con todos los animales nacidos) es la microinyección de fragmentos de ADN en un pronúcleo de un ovocitos fertilizados. En 1990, este enfoque, que se introdujo por primera vez en ratones, fue adaptado para ratas6,,7. La transgénesis de ratas por inyección pronuclear se caracteriza por una menor eficiencia8 en comparación con los ratones, que está estrictamente relacionada con la presencia de plasma elástico y membranas pronucleares9. Aunque la supervivencia de los embriones después de la manipulación es un 40-50% menor que en ratones, esta técnica se considera un estándar en la generación de ratas modificadas genéticamente10. Se han investigado enfoques alternativos que pueden garantizar una incorporación eficiente de transgenes y mayores tasas de supervivencia de los cigotos inyectados.
El determinante clave de la expresión transgénica estable y la transmisión a la progenie es su integración en el genoma de la célula huésped. Los lentivirus (LV) tienen la característica distintiva de ser capaz de infectar células divisorias y no divisorias. Su uso como herramienta para la incorporación de genes hetólogos en embriones demostró ser altamente eficiente11,y los individuos transgénicos se caracterizan por una expresión estable del fragmento de ADN incorporado. Se ha confirmado la eficacia de los vectores lentivirales para la modificación genética de ratones12,,13,ratas12,,14y otras especies11. En este método, la suspensión LV se inyecta bajo la zona pellucida del embrión en la etapa de dos pronuclei. Esta técnica garantiza esencialmente el 100% de supervivencia de los embriones porque el oolemma no se ve afectado. La producción de suspensiones LV de alta calidad y relativamente altamente concentradas son factores cruciales. Sin embargo, las concentraciones más bajas de suspensiones DE LV pueden ser superadas por inyecciones repetidas11,lo que aumenta la cantidad de partículas virales en la superficie del óvulo sin afectar a la integración de la membrana. Los embriones sometidos a inyecciones repetidas en el espacio perivelinal se desarrollan aún más, y los resortes transgénicos pueden transmitir el transgén a través de la línea germinal. La eficiencia de la generación de ratas transgénicas por transgénesis lentiviral puede ser tan alta como 80%12.
Aquí, describimos la producción de lentivirus recombinante derivado del VIH-1 que fue pseudotipofado con el virus de la estomatitis vesicular (VSV) G proteína de envoltura. El uso del pseudotipo VSV del sistema de envasado de segunda generación determina la amplia infectividad de partículas virales y permite la producción de vectores altamente estables que pueden ser concentrados por ultracentrifugación y crioconservado. Después de la verificación del diezmo, los vectores están listos para ser utilizados como un vehículo para la entrega de transgénicos en los cigotes albinos de rata Wistar. Después de una serie de inyecciones, los embriones pueden ser cultivados durante la noche y transferidos en la etapa de dos células a madres adoptivas. En este punto, se puede considerar uno de los dos enfoques alternativos. El procedimiento estándar utiliza hembras pseudoembarazadas como receptoras de embriones. Sin embargo, cuando la tasa de embarazo es baja después del apareamiento con machos vasectomizados, los embriones se pueden implantar en hembras embarazadas de Wistar/Sprague-Dawley (SD) que se aparean con ratas macho fértiles con un color de piel oscura (por ejemplo, ratas Brown Norway [BN]). El color del pelaje permite distinguir la descendencia del embarazo natural de la descendencia que se origina a partir de los embriones manipulados transferidos.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
La producción y aplicación de vectores virales se ajustaba a las directrices de nivel 2 de seguridad de la bioseguridad y fue aprobada por el Ministerio de Medio Ambiente de Polonia. Todos los procedimientos animales experimentales que se describen a continuación fueron aprobados por el Comité Ético Local. Los animales fueron alojados en jaulas ventiladas individualmente a una temperatura estable (21–23 oC) y humedad (50-60%) con acceso ad libitum al agua y alimentos bajo un ciclo de luz/oscuridad de 12 h/12 h.
1. Producción de vectores lentivirales
- Transfección de células HEK 293T
NOTA: El protocolo que se presenta aquí está diseñado para la transfección de veinte platos de cultivo de 10 cm que produce aproximadamente 200 ml de sobrenadante vectorial crudo.- Cultivo de células HEK 293T en medio DMEM que se complementa con suero bovino fetal (10%, v/v) en una incubadora de CO2 humidificada a 37oC. Para la transfección, prepare veinte placas de 10 cm de diámetro y semillas 1.5–2 x 106 HEK 293T células por plato.
- Cuando la confluencia alcanza el 70 %, transfectar las células utilizando reactivo de polietilenimina (PEI), pH 7,0, a una proporción de 3 g de PEI por 1 g de ADN.
- Preparar la mezcla de transfección para cinco platos (preparar el número de repeticiones de acuerdo con el número total de platos). A 1 ml del medio de águila modificado de Dulbecco (DMEM; sin suero), añadir la mezcla de tres plásmidos para que alcancen una cantidad final de 25 g de plásmido VSVg, 50 g de Delta R8.2 y 50 g de plásmido de codificación.
- Pipetear hacia arriba y hacia abajo, y añadir 125 ml de PEI a una concentración de 3 g/L. Incubar a temperatura ambiente durante 15 minutos, invirtiendo el tubo tres veces durante la incubación. Añadir 200 ml de la mezcla de transfección por plato. A continuación, incubar las placas en una incubadora de CO2 humidificada a 37oC.
- Concentración de vectores lentivirales
- Cuarenta y ocho horas después de la transfección, cosechan el medio que contiene partículas LV. Utilice tubos cónicos de 50 ml.
NOTA: Cuando se utiliza un plásmido con una etiqueta fluorescente, las celdas se pueden visualizar en este punto para verificar la eficiencia de la transfección. Se puede agregar una nueva porción del medio DMEM, y las células pueden ser incubadas por 24 h adicionales. El rendimiento de LV es comparable cuando se recoge en los puntos de tiempo de 48 y 72 h después de la transfección. - Centrifugar el medio a 3.000 x g durante 5 min y temperatura ambiente para eliminar las células separadas.
- Filtrar el sobrenadante (0,45 m) y verterlo en tubos nuevos.
NOTA: Este paso se puede omitir. - Añadir DNase I (libre de RNase, 1 g/ml) y MgCl2 (1 mM), e incubar en un baño de agua a 37 oC durante 15 minutos.
- Transfiera los tubos de polietileno medio a desechables, y ultracentrifugar en un rotor oscilante a 115.000 x g y 4 oC durante 1,5 h.
- Después de la centrifugación, drenar suavemente las paredes de los tubos de los residuos medios.
- Remoje el pellet con solución salina estéril con fosfato (PBS; 70-80 l por tubo).
- Incubar durante 2 h a 4-8oC.
- Resuspenda los vectores virales en PBS mediante pipeteo suave.
ADVERTENCIA: Evite la formación de espuma. - Pasar a un tubo centrífugo de 1,5 ml y centrífuga a 7.000 x g y 4 oC durante 30 s. Transfiera el sobrenadante a un tubo nuevo. Repita este paso hasta que no haya ningún pellet de desechos celulares visible.
- Aliquot y congelar a -80oC. Evite volver a congelar la alícuota LV.
- Cuarenta y ocho horas después de la transfección, cosechan el medio que contiene partículas LV. Utilice tubos cónicos de 50 ml.
- Determinación del morte del virus mediante la reacción cuantitativa en cadena de la polimerasa
NOTA: La valoración de los vectores virales se realiza utilizando PCR cuantitativo (qPCR). Este método se basa en la amplificación de un fragmento de ADN de 84 bp de largo dentro de la larga región de repetición terminal del genoma viral15.- Prepare la curva estándar haciendo diluciones en serie del plásmido de codificación LV: 1:500, 1:1,000, 1:5,000, 1:10,000, 1:100,000 y 1:1,000,000. Determine el número de copias del plásmido que se utiliza para la curva estándar. Utilice la siguiente fórmula: número de copias/ L (concentración [g/L] x 6,02 x 1023 [número/mol]) / (660 [g/mol] x tamaño de plásmido [bp]), donde 6,02 x 1023 número/mol es el número de Avogadro, y 660 g/mol es el peso de la presión.
NOTA: Se pueden utilizar calculadoras de números de copia en línea. - Preparar diluciones de la suspensión lentiviral: 1:100, 1:500 y 1:1,000.
- Preparar la mezcla de reacción (volúmenes por pozo): 10 ml de qPCR Mastermix, 1 l de imprimación directa de 10 m, 1 l de imprimación inversa de 10 oM y 7 ml de H2O. Pipetear la mezcla en los pocillos de las placas de 96 pocillos.
NOTA: Imprimación delantera: 5'-AGCTTGCCTTGAGTGCTTCA. Imprimación inversa: 5'-TGACTAAAAAGGTGAGGAGGGA. - Añadir 1 l de cada dilución estándar y suspensión lentiviral en triplicado.
- Ejecuta el qPCR de acuerdo con los siguientes parámetros: 50oC durante 2 min, 96oC durante 5 min y 35 ciclos de 96oC para 20 s, 60oC para 40 s y 70oC durante 1 min, seguido de la etapa de curva de fusión: 95oC para 1 min y 60oC a 30 s.
- Analice los resultados comparando el número de moléculas que se reciben para cada dilución con la curva estándar. Determinar la concentración de moléculas vectoriales como el promedio de tres réplicas para cada dilución.
NOTA: La cuantificación presentada proporciona la concentración física de las partículas virales. No debe tratarse como un titer funcional.
- Prepare la curva estándar haciendo diluciones en serie del plásmido de codificación LV: 1:500, 1:1,000, 1:5,000, 1:10,000, 1:100,000 y 1:1,000,000. Determine el número de copias del plásmido que se utiliza para la curva estándar. Utilice la siguiente fórmula: número de copias/ L (concentración [g/L] x 6,02 x 1023 [número/mol]) / (660 [g/mol] x tamaño de plásmido [bp]), donde 6,02 x 1023 número/mol es el número de Avogadro, y 660 g/mol es el peso de la presión.
2. Generación de ratas transgénicas
- Superovulación y recolección de embriones fertilizados
- Administrar gonadotropinas.
NOTA: Para aumentar el número de embriones recogidos (aproximadamente 30 por mujer), utilice hembras Wistar inmaduras de 5 semanas para la estimulación hormonal.- En el día 1 (12 PM–1 PM), inyectar por vía intraperitoneal gonadotropina sérica de la yegua embarazada (PMSG; 25 UI por mujer). Preparar 1 ml de alícuotas de solución de trabajo a una concentración de 125 UI/ml mediante la disolución de la hormona en polvo en 0,9% NaCl. Conservar a -20oC durante un máximo de 1 mes o -80oC durante un máximo de 6 meses.
- El día 3 (12 PM–1 PM), inyectar por vía intraperitoneal gonadotrofina coriónica humana (hCG; 30 UI por mujer). Preparar 1 ml de alícuotas de solución de trabajo (150 UI/ml) mediante la disolución de la hormona en polvo en 0,9% NaCl. Conservar a -20oC durante un máximo de 1 mes o -80oC durante un máximo de 6 meses.
- Después de la administración de hCG, aparee a las hembras 1:1 con machos sexualmente fértiles (3-10 meses de edad).
- A la mañana siguiente (día 4 a las 8-10 AM), revise a las hembras para ver si hay un tapón vaginal. Compruebe la abertura vaginal para la presencia de un tapón de apareamiento blanquecino, que para una mejor visualización debe ser revisado temprano en la mañana después de la noche de apareamiento. Para la recolección de embriones, utilice sólo hembras con un enchufe visible.
- Recoger embriones a las 10 AM. Sacrificar a los animales para extirpar los oviductos, y recoger los oviductos en un plato con medio M2 precalentado.
- Transfiera los oviductos a un plato de 35 mm que contenga un medio M2 precalentado con hialuronidasa de testículos bovinos a una concentración de 0,5 mg/ml.
- Abra las paredes del oviducto usando fórceps finos bajo un estereomicroscopio y presione la ampolla (es decir, la parte hinchada del oviducto que contiene embriones fertilizados que están rodeados por células cúmulos) hasta que los embriones se liberen.
NOTA: La hialuronidasa digiere enzimáticamente las células cúmulosas, liberando embriones.
ADVERTENCIA: La exposición prolongada a la hialuronidasa es perjudicial para los embriones; por lo tanto, este paso no debe durar más de 5 min. - Para facilitar la liberación de embriones de células cúmulos, pipetee suavemente hacia arriba y hacia abajo utilizando una pipeta de transferencia de vidrio que esté conectada a un tubo aspirador operado por la boca.
- Para producir la pipeta de transferencia, tire de una pipeta Pasteur de vidrio sobre una llama para producir una punta recta de 5-10 cm. Romper la pipeta dejando una punta de 4 cm.
- Lavar los embriones varias veces en medio M2 para eliminar la hialuronidasa y los desechos celulares. Transfiera los embriones a un plato de 60 mm que contenga gotas de medio M16 preequilibrado, cubierto por parafina líquida o aceite mineral, en una incubadora humidificada de 37 oC con una atmósfera deCO2 del 5%.
- Administrar gonadotropinas.
- Microinyección de vectores lentivirales en un embrión de una fase celular bajo la zona pellucida
NOTA: Utilice embriones de una etapa celular con dos pronúcleos visibles para la microinyección(Figura 1).- Descongelar la alícuota LV a temperatura ambiente y centrifugar a 10.000 x g y RT durante 2 minutos para peletizar los restos celulares restantes.
- Configuración de microinyección
- Prepare pipetas de sujeción de vidrio (capilar de vidrio de borosilicato) utilizando una microforja. Tire del capilar de vidrio sobre una llama para producir una punta de 5-10 cm. Romper la pipeta dejando una punta de 4 cm. El diámetro exterior debe ser de 80–120 m.
NOTA: Asegúrese de que la punta de la pipeta esté perfectamente recta y lisa. - Montar la pipeta tirada en una microforja con la punta delante del filamento de calentamiento. Calentar el filamento muy cerca de la punta de la pipeta y permitir que se encoja a un diámetro de 15 m (aproximadamente un 20 % del tamaño del embrión). Coloque la pipeta perpendicularmente al filamento de calentamiento, 2-3 mm desde la punta de la pipeta, y comience a calentar. El vidrio se ablandará. Calienta hasta que alcance un ángulo de 15o.
- Prepare capilares de vidrio de borosilicato de microinyección con un filamento utilizando un tirador de pipeta. Inserte el capilar en la cámara de tracción. Ejecute una prueba de rampa (por primera vez para vidrio nuevo y cada vez después de cambiar el filamento). Establezca el Heat en el valor de rampa -10, Pull en 100, Velocity en 150 y Time en 100.
NOTA: Modifique los parámetros para obtener un capilar de inyección óptimo. - Bajo una campana de flujo laminar de bioseguridad, cargue aproximadamente 2 ml de la solución viral en la pipeta de microinyección con una punta de microcargador.
- Preparar un plato de microinyección (tapa de 60 mm de plato Petri) con una gota de 100 ml de medio M2 (en el medio), cubierto por parafina líquida o aceite mineral.
- Monte la pipeta de sujeción y el capilar de microinyección cargado sin solución viral en un micromanipulador y una antena de microinyección bajo un microscopio invertido.
- Prepare pipetas de sujeción de vidrio (capilar de vidrio de borosilicato) utilizando una microforja. Tire del capilar de vidrio sobre una llama para producir una punta de 5-10 cm. Romper la pipeta dejando una punta de 4 cm. El diámetro exterior debe ser de 80–120 m.
- Realice la microinyección.
- Transfiera entre 15 y 20 embriones de una etapa celular a la gota M2 en la placa de microinyección. Sostenga el embrión con una pipeta de sujeción.
- Usando el aumento 400x, inyecte la solución LV bajo la zona pellucida al espacio perivelinal usando el capilar de vidrio que está conectado a un inyector automático. Sostenga el capilar bajo la zona pellucida por un momento.
NOTA: Con una presión positiva suave, la solución viral fluirá continuamente fuera del capilar de inyección, pero el volumen de la suspensión que se entrega no se puede controlar. - Usando una pipeta fina, devuelva los embriones al plato de cultivo en la incubadora a 37 oC en una atmósfera deCO2 del 5%. El número de inyecciones de un cigota puede variar y puede adaptarse en función de la concentración de vectores virales.
NOTA: Los embriones inyectados pueden ser transferidos a madres adoptivas en la etapa de una célula o incubados O/N en el medio M16 antes de ser transferidos en la etapa de dos células. Debe evitarse el cultivo prolongado in vitro de embriones de rata.
- Transferencia de embriones inyectados a madres adoptivas
- Preparar a las madres adoptivas apareando hembras SD sexualmente maduras con machos fértiles de BN o con machos vasectomizados de SD (el procedimiento de vasectomía se describe en la sección 3 a continuación) el día 3 (para transferir embriones en la etapa de una célula) o el día 4 (para transferir embriones en la etapa de dos células).
NOTA: Para la transferencia de oviductos, utilice 0,5 días después del coito (dpc) hembras. - A la mañana siguiente, revisa a las hembras SD en busca de un tapón vaginal, y usa solo aquellas con un tapón visible.
- Realizar transferencia de embriones.
NOTA: Realice el procedimiento quirúrgico con instrumentos estériles bajo un estereomicroscopio. Antes del día de la cirugía, tijeras de autoclave, fórceps finos, soporte de aguja y soporte para bisturí.- Anestesiar una hembra con administración i.p. de ketamina (50 mg/kg) y medetomidina (0,5 mg/kg) solución. Prueba de reflejos para confirmar la anestesia antes de iniciar el procedimiento quirúrgico.
- Inyectar al animal por vía subcutánea con ácido tolefenámico (2 mg/kg), tartrato de butorphanol (1 mg/kg) y enrofloksacina (5-10 mg/kg) para prevenir la inflamación, el dolor y la infección, respectivamente.
- Aplique lubricación por pomada oftálmica en ambos ojos para evitar el secado de la córnea. Afeitar el pelaje de la espalda, y esterilizar la piel con exfoliación quirúrgica seguido de 70% de alcohol usando almohadillas estériles no adherentes. Deje que la piel se seque.
- Inyectar el animal por vía subcutánea con 100 ml de bupivacaína al 0,25% (anestésico local) en el lugar de la incisión. Transfiera al animal en una posición propensa a una superficie limpia en una almohadilla de calentamiento bajo el objetivo de un microscopio quirúrgico. Cubra la rata con una cortina estéril con un pequeño agujero cortado sobre la parte inferior de la espalda.
- Realizar una incisión cutánea de aproximadamente 2 cm, paralela a la columna vertebral lumbar.
- Usando tijeras afiladas, haz un corte en la pared abdominal. Coge una almohadilla de grasa ovárica usando fórceps, y saca el ovario y el oviducto y colócalos en una gasa que esté mojada con 0.9% NaCl.
- Aspirar el medio M2, tres burbujas de aire, y los embriones en el capilar de transferencia. Número total recomendado de embriones a transferir (unilaterales o bilaterales): mujer embarazada (15-16 embriones), pseudoembarazada (30 embriones).
- Haga una pequeña incisión en el oviducto (entre el infundibulum y la ampolla) usando microtijeras, e inserte la pipeta de transferencia en el oviducto.
- Expulse suavemente embriones y burbujas de aire desde la pipeta hasta el oviducto. Con fórceps contundentes, vuelva a colocar el tracto reproductivo en la cavidad abdominal.
- Suturar la pared abdominal con suturas absorbibles de ácido poliglicólico y cerrar la incisión cutánea con clips de herida. Dependiendo del número de embriones disponibles, repita este procedimiento para el otro oviducto.
- Inyectar al animal por vía intraperitoneal con atipamezol (0,5 mg/kg) para revertir el efecto de la anestesia.
- Transfiera al animal a una jaula limpia y guárdelo en una placa de calentamiento para recuperarse completamente de la anestesia. La entrega en ratas se produce después de 21 días.
NOTA: Cuando se utilizan ratas BN macho para el apareamiento, sólo los cachorros blancos son potencialmente transgénicos; cachorros marrones son de embarazo natural. - Recoger fragmentos de tejido (preferiblemente del oído) a cachorros genotipo de 3 semanas de edad.
- Preparar a las madres adoptivas apareando hembras SD sexualmente maduras con machos fértiles de BN o con machos vasectomizados de SD (el procedimiento de vasectomía se describe en la sección 3 a continuación) el día 3 (para transferir embriones en la etapa de una célula) o el día 4 (para transferir embriones en la etapa de dos células).
3. Vasectomía
NOTA: Antes del día de la cirugía, tijeras de autoclave, fórceps finos y soporte de aguja.
- Anestesiar una rata SD macho de 5 semanas de edad con administración i.p. de ketamina (50 mg/kg) y medetomidina (0,5 mg/kg). Prueba de reflejos para confirmar la anestesia antes de iniciar el procedimiento quirúrgico.
- Administrar ácido tolfenamico (2 mg/kg), tartrato de butorphanol (1 mg/kg) y enrofloksacina (5-10 mg/kg) por vía subcutánea para prevenir la inflamación, el dolor y la infección, respectivamente.
- Aplique lubricación por pomada oftálmica en ambos ojos para evitar el secado de la córnea. Coloque la columna vertebral de la rata sobre una superficie limpia sobre una almohadilla de calentamiento y esterilice la piel en los testículos con exfoliación quirúrgica seguida de un 70% de alcohol utilizando almohadillas estériles no adherentes. Deje que la piel se seque. Cubra la rata con una cortina estéril con un pequeño agujero cortado sobre los testículos. Presione suavemente el abdomen para exponer los testículos en el saco escrotal.
- Usando tijeras quirúrgicas, haga una incisión de 0,5 cm en el centro del saco escrotal. Localice la pared de la línea media (línea blanquecina) entre los testículos.
- Haga una incisión de 5 mm en la membrana testis cerca del lado izquierdo de la pared de la línea media.
- Empuje cuidadosamente los testículos hacia la izquierda y localice los vasos deferentes (entre los testículos y la línea media) como un conducto blanco con un solo vaso sanguíneo.
- Tire suavemente del vasa deferente del saco escrotal usando los fórceps de un relojero. Sostenga el vas deferente con un par de fórceps, y córtelo con tijeras finas (o cauterice con puntas al rojo-caliente de un segundo par de fórceps). Retire un fragmento de 1 cm del conducto.
NOTA: Si se realiza la cauterización, sostenga la punta del segundo par de fórceps en la llama. - Repita el procedimiento anterior para los otros testículos. Suturar la piel con suturas absorbibles de ácido poliglicólico e inyectar al animal por vía intraperitoneal con atipamezol (0,5 mg/kg).
- Coloque la rata en una jaula limpia en una placa de calentamiento hasta que el animal se recupere de la anestesia.
NOTA: Los machos se pueden utilizar en los apareamientos de prueba después de un período de recuperación de 2 semanas. Después de que se confirme la esterilidad, se pueden utilizar para la inducción del pseudoembarazo.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Usando el protocolo descrito en el presente documento, se produjeron vectores lentivirales que llevaban la construcción Syn-TDP-43-eGFP (titer físico de LV a 3,4 x 108/L) y luego se podrían utilizar para inyecciones subzonales de embriones en etapa de una célula. Sólo se sometieron al procedimiento embriones con dos pronúcleos visibles. El número de inyecciones de suspensiones virales se determinó experimentalmente. La alta eficiencia de implantación y la falta simultánea de descendencia transgénica se consideraron indicadores de un número insuficiente de partículas virales para una transducción exitosa. En este caso, se aumentó el número de inyecciones. La administración única de LV dio lugar al nacimiento de 20 ratas de generación F0, ninguna de las cuales fue transgénica. Un aumento en el número de inyecciones en un orden de magnitud no dio lugar al nacimiento de ratas, pero el 100% de los embriones desarrollados a la etapa de dos células. En experimentos posteriores, el número de inyecciones se incrementó en uno en comparación con el valor para el que se obtuvieron las crías. Para la variante de dos inyecciones, nacieron ocho ratas, tres de las cuales fueron confirmadas para transportar el transgén (resumido en la Tabla 1). Uno de los fundadores no transfirió el transgén a la descendencia. El número de embriones que se inyectaron y transfirieron en cada variante experimental fue de 48 en las variantes LV x1 y LV x2 y 45 en LV x10. Se utilizaron tres hembras adoptivas para cada configuración experimental. El enfoque elegido permitió la generación de líneas de ratas transgénicas estables que expresaban la proteína de fusión TDP-43-eGFP bajo el control del promotor neuronal Synapsin-1 en todo el sistema nervioso central(Figura 2A,B)14. La transgénesis basada en Lentivirus dio lugar a una única inserción de copia del transgén, como lo demuestra qPCR(Figura 2C).
En la configuración experimental que se describe anteriormente, la tasa de supervivencia de los embriones inyectados fue del 95%. Se obtuvieron resultados similares cuando se utilizó el mismo método para otros vectores lentivirales, tal como se resume en el Cuadro 2. El porcentaje de embriones que sobrevivieron a las inyecciones pronucleares fue significativamente menor (29-45%). En el Cuadro 2 se resumen los resultados representativos de la eficiencia de implantación de los cigotes manipulados, teniendo en cuenta la transferencia de pseudoembarazadas frente a mujeres embarazadas. El uso de embriones no manipulados junto con embriones inyectados se notificó previamente16. Nuestros resultados generales sugieren que las ratas hembra embarazadas se pueden utilizar como madres adoptivas con una eficiencia comparable. Obtuvimos un porcentaje similar de implantación de embriones extranjeros en ratas embarazadas y pseudoembarazadas (promedio general para varias configuraciones experimentales: 15% vs. 16%). Sin embargo, la tasa de implantación fue mayor cuando los embriones se sometieron a una manipulación más sutil, lo que significa una inyección subzonal (10% frente a 21%). En particular, los datos numéricos que se analizaron para rondas individuales de microinyección indicaron que la eficacia de la implantación dependía del número de inyecciones de un embrión(Tabla 1,última columna) e indirectamente dependía de la carga viral.
| Vector | número de inyecciones/embrión | número de embriones inyectados | número de cachorros | número de madres adoptivas | número de fundadores transgénicos | Eficiencia de implantación para cada variante |
| Syn-TDP-43WTLV | 1 | 48 | 20 | 3 | 0 | 42% |
| 10 | 45 | 0 | 3 | 0 | 0% | |
| 2 | 48 | 8 | 3 | 3 | 17% |
Tabla 1: Resumen del número de inyecciones subzonales de cigotes con vectores lentivirales Syn-TDP-43WT.lentiviral vectors.
| Método | Vector | Titer/ Concentración | Número de embriones inyectados | Embriones sobreviven | Tasa de supervivencia | Número de madres adoptivas | Número de cachorros | Eficiencia de implantación | Embarazo (P) /Pseudoembarazo (PP) |
| Pni | TTYH1-Thy1-EGFP | 1 ng/L | 1083 | 424 | 39% | 16 | 54 | 13% | Pp |
| Pni | H3mCherry | 0.5-2 ng/L | 2229 | 647 | 29% | 29 | 67 | 10% | Pp |
| Pni | Syn-TDP-43-A315T | 2 ng/L | 1256 | 562 | 45% | 31 | 42 | 7% | Pp |
| Lv | Syn-TDP-43-A315T | 8.7 x 108 | 115 | 106 | 92% | 7 | 18 | 17% | P |
| Lv | Syn-TDP-43 WT | 3.4 x 108 | 152 | 141 | 93% | 9 | 28 | 20% | P |
| Lv | LVH3mcherry | 1.3 x 107 | 504 | 450 | 89% | 13 | 115 | 26% | Pp |
Tabla 2: Tasa de supervivencia del embrión y eficiencia de implantación, dependiendo del método de inyección utilizado y la inducción del embarazo frente al pseudoembarazo. PNI, inyección pronuclear; LV, inyección subzonal de vector escenórviral.

Figura 1: Fotografía microscópica del embrión de rata de una etapa celular que se preparó para la inyección de vectores lentivirales subzonales. El embrión fue inmovilizado con una pipeta de retención. Dos pronúcleos que contenían material genético materno y paterno y el cuerpo polar son visibles. Barra de escala a 20 m. Haga clic aquí para ver una versión más grande de esta figura.
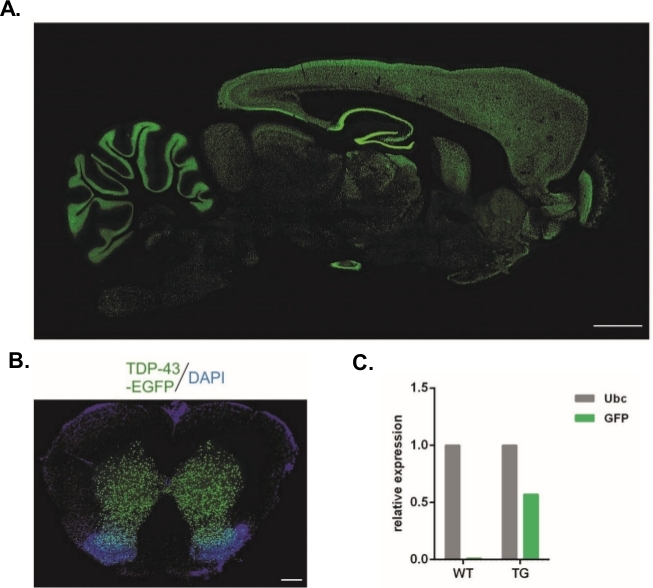
Figura 2: Generación de líneas de ratas transgénicas estables que expresaron la proteína de fusión TDP-43-eGFP bajo control del promotor neuronal Synapsin-1 en todo el sistema nervioso central. (A) Patrón de expresión hTDP-43-eGFP impulsado por Synapsin-1 (Syn) en una sección sagital del cerebro de rata transgénica. Barra de escala de 3 mm.(B) Sección coronal de la médula espinal de una rata transgénica donde la fluorescencia eGFP, contrarrevida con DAPI, se limitaba a la materia gris de la médula espinal. Barra de escala a 250 m. (C) Expresión relativa de la transcripción transgénea GFP en comparación con la transcripción de referencia de ubiquitina C. n 2 tipo salvaje. n 2 transgénicos. La cifra fue modificada a partir de14. Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Los avances en tecnologías transgénicas han hecho de los modelos de roedores una herramienta invaluable en la investigación biomédica. Proporcionan la oportunidad de estudiar las relaciones genotipo-fenotipo in vivo. Aquí, presentamos una alternativa ampliamente disponible para la transgénesis convencional por inyecciones pronucleares. El uso de la transducción génica lentiviral evita la necesidad de microinyecciones exigentes porque se pueden inyectar vectores virales bajo la zona pellucida. Este enfoque no afecta a la integridad del embrión, lo que esencialmente garantiza una tasa de supervivencia del 100% para los cigotos inyectados. El transgén que se incorpora mediante vectores lentivirales se integra establemente en el genoma del huésped, permitiendo la expresión a largo plazo y la transmisión de líneas germinales. Además, presentamos dos técnicas alternativas para la transferencia de embriones modificados a madres adoptivas. Una técnica utiliza la transferencia de embriones a hembras pseudoembarazadas que se preparan previamente mediante el apareamiento con machos fértiles vasectomizados. La otra técnica se basa en el uso de hembras embarazadas naturalmente que están apareadas con machos fértiles pero con un color de piel diferente (es decir, ratas BN). Este curso más fisiológico del embarazo permite el correcto desarrollo de embriones que se someten a modificaciones genéticas desafiantes16.
Los primeros intentos exitosos de generar ratas transgénicas se notificaron en 19907. Sin embargo, debido a las dificultades en la transgénesis de ratas17, un número relativamente pequeño de líneas de ratas transgénicas se han generado en las últimas décadas9. Se observan varias diferencias principales entre la transgénesis del ratón y la rata mediante microinyecciones. Para las ratas, principalmente las líneas de raza (por ejemplo, Wistar y SD) se utilizan para la transgénesis. Para ratones, los investigadores utilizan principalmente el cruce F1 de cepas endogámicas debido a su mayor fertilidad, mejor respuesta a la superovulación hormonal y un desarrollo relativamente fácil de embriones in vitro desde la etapa de una célula a blastocistos18. La inducción de la superovulación en ratas es mucho menos eficiente que en ratones que utilizan estimulación hormonal PMSG/hCG estándar. Por esta razón, se ha intentado desarrollar protocolos alternativos para administrar estas hormonas en ratas que utilizan perfusión continua de FSH en lugar de una sola administración de PMSG19. Sin embargo, se ha demostrado que la superovulación causada por PMSG/hCG o FSH/hCG tiene una eficiencia comparable20. En nuestra opinión, el factor más crítico que afecta a la eficacia de la superovulación es la edad de las hembras seleccionadas. Sin embargo, los parámetros exactos deben ser probados para cada cepa de rata, laboratorio, etc.
El procedimiento para inyectar solución de ADN en el pronúcleo de un embrión de una sola célula es similar para ambas especies de roedores. Sin embargo, los pronúcleos de los cigotes de rata no tienen formas tan regulares como en ratones y tienden a ser más difíciles de definir en el citoplasma de la célula. Además, la membrana celular del cigota de rata y la membrana pronuclear son más elásticas y viscosas, complicando así la inserción de un micropipeta de vidrio que está cargado con solución de ADN. Estos factores conducen a tasas más bajas de supervivencia del huevo de rata después de la microinyección (31-65% frente a 80% en ratones) y explican la menor eficiencia de transgénesis en ratas9. Además, la manipulación mecánica intensiva del embrión también puede afectar a la eficiencia de implantación, que en muchos laboratorios, incluido el nuestro, alcanza un máximo del 10%. Este rendimiento relativamente bajo se observa incluso después de la implantación de un número adecuado de embriones21.
Un método que supera las dificultades antes mencionadas es la infección de embriones unicelulares con retrovirus. Los retrovirus contienen material genético en forma de ARN, que al entrar en la célula infectada se transcribe en el ADN por transcriptasa inversa del virus. El ADN se transporta a través de los poros nucleares al núcleo celular, donde se integra en el genoma de la célula en forma de provirus. Se han utilizado vectores lentivirales para generar ratones transgénicos y ratas12,,14,,22. Los embriones unicelulares que carecen de una zona pellucida se pueden incubar en una solución con un vector lentiviral, o el vector se puede inyectar bajo la zona pellucida en el espacio periviellina. La principal ventaja de este método es su extremadamente alta eficiencia, alcanzando más del 80% de la descendencia transgénica. Después de la infección con el vector lentiviral, muchas copias en diferentes sitios pueden integrarse en el genoma cigota, en contraste con el método de transgénesis por microinyección pronuclear, en el que generalmente se observa un sitio de integración12. En la descendencia del fundador transgénico que se hace utilizando vectores lentivirales, las copias individuales del transgén se segregan, que pueden ser manifestadas por diferentes perfiles de expresión del transgén en cada una de las progenies. Sin embargo, esto puede aumentar la probabilidad de recibir un sujeto con el perfil de expresión deseado que se deriva del transgén. Las restricciones se aplican principalmente al tamaño del transgén, que está limitado a aproximadamente 8 kb23.
Otra dificultad en la transgénesis de ratas es la generación de hembras que sirven como madres sustitutas para embriones modificados genéticamente. En el procedimiento estándar, las hembras se cruzan con machos vasectomizados estériles para inducir el pseudoembarazo. En ratas, la técnica de evaluación de pseudoembarazo es mucho más difícil que en ratones, por lo que la estimulación con el agonista de la hormona liberadora de gonadotropina se utiliza a veces unos días antes de aparearse con los hombres. Por estas razones, en el protocolo descrito proporcionamos dos enfoques alternativos para obtener madres adoptivas. La eficiencia general de implantación de cigotes manipulados cuando se utilizan hembras embarazadas o pseudoembarazadas es similar. Sin embargo, la presencia de embriones naturales no manipulados junto con los manipulados puede mejorar la tasa de embarazo16. Aunque la principal diferencia en la tasa de implantación es la técnica de manipulación (es decir, PNI frente a LV, 10% frente a 20%; ver Tabla 2), el uso de mujeres pseudoembarazadas como madres adoptivas puede ser beneficioso para algunos experimentos.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
El autor (W.K.) tiene derecho a la patente "Método de producción de un animal transgénico", de la oficina de patentes de la República de Polonia (n.o P 355353; 21.03.2008).
Acknowledgments
Este estudio fue apoyado por el proyecto ANIMOD dentro del programa Team Tech Core Facility Plus de la Fundación para la Ciencia Polaca, cofinanciado por la Unión Europea en el marco del Fondo Europeo de Desarrollo Regional a WK.
Materials
| Name | Company | Catalog Number | Comments |
| 7500 Real Time PCR System | Applied Biosystems | ||
| Aerrane (isoflurane) | Baxter | FDG9623 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177-5EA | |
| Atipam 5 mg/ml | Eurovet Animal Health BV | N/A | 0.5 mg/kg |
| Baytril 25 mg/ml (enrofloksacin) | Bayer | N/A | 5-10 mg/kg |
| Borosilicate glass capillaries with filament GC100TF-15 | Harvard Apparatus Limited | 30-0039 | injection capillary |
| Bupivacaine 25 mg/ml | Advanz Pharma | N/A | 0.25% in 0.9% NaCl |
| Butomidor 10 mg/ml (butorphanol tartrate) | Orion Pharma | N/A | 1 mg/kg |
| CELLSTAR Tissue Cell Culture Dish 35-mm | Greiner Bio-One | 627160 | |
| CELLSTAR Tissue Cell Culture Dish 60-mm | Greiner Bio-One | 628160 | |
| CellTram Oil | Eppendorf | 5176 000.025 | |
| Cepetor (Medetomidine) 1 mg/ml | cp-pharma | N/A | 0.5 mg/kg |
| Chorulon, Human Chorionic Gonadotrophin | Intervet | N/A | 150 IU/ ml ml 0.9% NaCl |
| DMEM low glucose | Sigma Aldrich | D6048 | |
| DNase, RNase-free | A&A Biotechnology | 1009-100 | |
| EmbryoMax Filtered Light Mineral Oil | Sigma | ES-005-C | |
| Envelope protein coding plasmid for lentiviral vectors (VSVg plasmid) | ADDGENE | 14888 | |
| FemtoJet | Eppendorf | 4i /5252 000.013 | |
| Fetal Bovine Serum | Sigma Aldrich | F9665-500ML | |
| Folligon, Pregnant Mare’s Serum Gonadotropin | Intervet | N/A | 125 IU/ml in .9% NaCl |
| HEK 293T cells | ATCC | ATCC CRL-3216 | |
| Hyaluronidase from Bovine Testis | Sigma | H4272-30MG | 0.5 mg/ml in M2 medium |
| Inverted Microscope | Zeiss | Axiovert 200 | |
| Ketamine 100mg/ml | Biowet Pulawy | N/A | 50 mg/kg |
| Liquid Paraffin | Merck Millipore | 8042-47-5 | |
| M16 medium EmbryoMax | Sigma | MR-016-D | |
| M2 medium | Sigma | M7167 | |
| Magnesium Chloride 1M | Sigma Aldrich | 63069-100ML | |
| Microforge | Narishige | MF-900 | |
| Mineral Oil | Sigma | M8410-500ML | |
| NaCl 0.9% | POLPHARMA OTC | N/A | sterile, 5ml ampules |
| Operation microscope | Inami Ophthalmic Instruments | Deca-21 | |
| Packaging system coding plasmid for lentiviral vectors (delta R8.2 plasmid) | ADDGENE | 12263 | |
| PEI reagent (Polyethylenimine, Mw ~ 25,000,), | Polysciences, Inc | 23966-1 | |
| Penicilin-streptomycin | Sigma Aldrich | P0781-100ML | |
| Phosphate Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma Aldrich | 806552-500ML | |
| Puller | Sutter Instrument Co. | P-97 | |
| Reflex Clip Applier/Reflex Clips | World Precision Instruments | 500345/500346 | |
| Safil, polyglycolic acid, braided, coated, absorbable threads | B.Braun Surgical | 1048029 | |
| Stereomicroscope | Olympus | SZX16 | |
| Surgical Sewing Thread | B.Braun | C1048040 | |
| SYBR Green PCR Master Mix | Applied Biosystem | 4334973 | |
| Tolfedine 4% (tolfenamic acid) | Vetoquinol | N/A | 2 mg/kg |
| TransferMan NK2 | Eppendorf | N/A | |
| Trypsin EDTA solution | Sigma Aldrich | T3924-500ML | |
| Ultracentrifuge | Beckman Coulter | Optima L-100 XP | |
| VacuTip | Eppendorf | 5175108.000 | holders capillary |
| Vita-POS | Ursapharm | N/A | eye ointment |
| Warming Plate | Semic | N/A | |
| Watchmaker Forceps | VWR | 470018-868 |
References
- Lazar, J., Moreno, C., Jacob, H. J., Kwitek, A. E. Impact of genomics on research in the rat. Genome Research. 15 (12), 1717-1728 (2005).
- Tarkowski, A. K. Studies on mouse chimeras developed from eggs fused in vitro. National Cancer Institute Monographs. 11, 51-71 (1963).
- Gordon, J. W., Ruddle, F. H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 214 (4526), 1244-1246 (1981).
- Gill, T. J., Smith, G. J., Wissler, R. W., Kunz, H. W. The Rat as an Experimental Animal. Science. 245 (4915), 269-276 (1989).
- Aitman, T. J., et al. Progress and prospects in rat genetics: a community view. Nature Genetics. 40 (5), 516-522 (2008).
- Hammer, R. E., Maika, S. D., Richardson, J. A., Tang, J. P., Taurog, J. D. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 63 (5), 1099-1112 (1990).
- Mullins, J. J., Peters, J., Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 344 (6266), 541-544 (1990).
- Menoret, S., Remy, S., Usal, C., Tesson, L., Anegon, I. Generation of Transgenic Rats by Microinjection of Short DNA Fragments. Rat Genomics: Methods and Protocols. 597, 81-92 (2010).
- Tesson, L., et al. Transgenic modifications of the rat genome. Transgenic Research. 14 (5), 531-546 (2005).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: Technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Ritchie, W. A., Neil, C., King, T., Whitelaw, C. B. Transgenic embryos and mice produced from low titre lentiviral vectors. Transgenic Research. 16 (5), 661-664 (2007).
- Lois, C., Hong, E. J., Pease, S., Brown, E. J., Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295 (5556), 868-872 (2002).
- Pfeifer, A., Ikawa, M., Dayn, Y., Verma, I. M. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proceedings of the National Academy of Sciences of the United States of America. 99 (4), 2140-2145 (2002).
- Koza, P., et al. Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiology of Disease. 130, 104499 (2019).
- Scherr, M., Battmer, K., Blomer, U., Ganser, A., Grez, M. Quantitative determination of lentiviral vector particle numbers by real-time PCR. Biotechniques. 31 (3), 520 (2001).
- Canseco, R. S., et al. Gene transfer efficiency during gestation and the influence of co-transfer of non-manipulated embryos on production of transgenic mice. Transgenic Research. 3 (1), 20-25 (1994).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., Palmiter, R. D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proceedings of the National Academy of Sciences of the United States of America. 82 (13), 4438-4442 (1985).
- Armstrong, D. T., Opavsky, M. A. Superovulation of immature rats by continuous infusion of follicle-stimulating hormone. Biology of Reproduction. 39 (3), 511-518 (1988).
- Popova, E., Krivokharchenko, A., Ganten, D., Bader, M. Comparison between PMSG- and FSH-induced superovulation for the generation of transgenic rats. Molecular Reproduction and Development. 63 (2), 177-182 (2002).
- Johnson, L. W., Moffatt, R. J., Bartol, F. F., Pinkert, C. A. Optimization of embryo transfer protocols for mice. Theriogenology. 46 (7), 1267-1276 (1996).
- van den Brandt, J., Wang, D., Kwon, S. H., Heinkelein, M., Reichardt, H. M. Lentivirally generated eGFP-transgenic rats allow efficient cell tracking in vivo. Genesis. 39 (2), 94-99 (2004).
- Remy, S., et al. The Use of Lentiviral Vectors to Obtain Transgenic Rats. Rat Genomics: Methods and Protocols. 597, 109-125 (2010).
