

Summary
Aqui, descrevemos uma estratégia simples e acessível para visualizar, quantificar e mapear células imunes em seções de tecido tumoral parafina fixada em formalina. Essa metodologia combina técnicas de imagem e análise digital existentes com o objetivo de ampliar a capacidade de multiplexação e a análise multiparâmetro de ensaios de imagem.
Abstract
A paisagem imunológica do microambiente tumoral (TME) é um fator determinante na progressão do câncer e na resposta à terapia. Especificamente, a densidade e a localização das células imunes no TME têm importantes valores diagnósticos e prognósticos. O perfil multiômico do TME aumentou exponencialmente nossa compreensão das numerosas redes celulares e moleculares que regulam a iniciação e a progressão tumoral. No entanto, essas técnicas não fornecem informações sobre a organização espacial das células ou interações células. Técnicas de multiplexação acessíveis, acessíveis e fáceis de executar que permitem a resolução espacial de células imunes em seções teciduais são necessárias para complementar tecnologias de alto throughput baseadas em células únicas. Aqui, descrevemos uma estratégia que integra imagens seriais, rotulagem seqüencial e alinhamento de imagens para gerar slides multiparâmetrovirtuais de seções de tecidos inteiros. Os slides virtuais são posteriormente analisados de forma automatizada usando protocolos definidos pelo usuário que permitem a identificação, quantificação e mapeamento de populações celulares de interesse. A análise de imagem é feita, neste caso, utilizando os módulos de análise Tissuealign, Author e HISTOmap. Apresentamos um exemplo em que aplicamos essa estratégia com sucesso a uma amostra clínica, maximizando as informações que podem ser obtidas a partir de amostras limitadas de tecidos e fornecendo uma visão imparcial do TME em toda a seção de tecidos.
Introduction
O desenvolvimento do câncer é resultado de um processo multietapa envolvendo interações recíprocas entre células malignas e o TME. Além das células tumorais, o TME é composto por células não-malignas, células estrômicas, populações de células imunes e matriz extracelular (ECM)1. A organização espacial dos diferentes componentes celulares e estruturais do tecido tumoral e a troca dinâmica entre o câncer e as células não cancerosas vizinhas, em última análise, modulam a progressão do tumor e a resposta à terapia2,3,4. Foi demonstrado que a resposta imune no câncer é regulada espacialmente5,6. Diferentes populações de células imunes infiltrando-se na lesão neoplásica e no tecido adjacente exibem padrões de distribuição espacial distintos e estados variados de ativação e diferenciação associados a diferentes funções (por exemplo, pró- versus antitumor). Essas diferentes populações imunológicas e seus parâmetros coevoluem horas extras com o tumor e os compartimentos estrômicos.
O surgimento de tecnologias que permitem o perfil multimômico de células únicas aumentou exponencialmente nossa compreensão das inúmeras redes celulares e moleculares que regulam a carcinogênese e a progressão tumoral. No entanto, a maioria das ferramentas analíticas de alto throughput baseadas em células únicas requerem interrupção tecidual e isolamento de células únicas, resultando em perda de informações sobre a organização espacial das células e interaçõescélulas-células 7. Como a localização e o arranjo de células imunes específicas no TME têm valor diagnóstico e prognóstico, as tecnologias que permitem a resolução espacial são um complemento essencial das técnicas de perfil imunológico baseadaem em células únicas.
Tradicionalmente, técnicas de imagem como imunohistoquímica (IHC) e imunofluorescência multiplex (mIF) têm sido restritas a um pequeno número de biomarcadores que podem ser visualizados simultaneamente. Essa limitação tem dificultado o estudo da dinâmica espacial das células imunes infiltradas em tumores, que são tipicamente definidas por vários marcadores fetotípicos. Os recentes avanços em imagens e ferramentas analíticas ampliaram as possibilidades de multiplexação. Novas tecnologias de rotulagem baseadas em anticorpos, como a citometria da histocitometria e a citometria de massa de imagem, têm sido usadas para separar espacialmente até 12 e 32 biomarcadores, respectivamente8,9. A imagem de espectrometria de massa, uma técnica que não requer rotulagem, tem o potencial de visualizar milhares de biomarcadores simultaneamente em uma única seção tecidual10,11. Embora essas técnicas já tenham mostrado grande potencial para dissecar a paisagem imunológica do tecido no câncer, elas usam equipamentos e softwares altamente sofisticados e caros e não são facilmente acessíveis à maioria dos pesquisadores.
Alternativamente, a capacidade de multiplexação do IHC tradicional e do IFM foi expandida através do uso de imagens seriais, rodadas seqüenciais de rotulagem e imagens espectrais7,,12,,13,14,15,16. Essas técnicas geram múltiplas imagens a partir das mesmas ou de seções de tecido serial que podem ser consolidadas em slides de multiparâmetros virtuais usando software de análise de imagem. Como resultado, o número de marcadores que podem ser visualizados e analisados simultaneamente aumenta.
Aqui, propomos uma estratégia para o design racional de ensaios multiplex de tecido usando reagentes disponíveis comercialmente, equipamentos de microscopia acessíveis e software fácil de usar(Figura 1). Essa metodologia integra imagens seriais, rotulagem multiplex sequencial, imagem de tecido inteiro e alinhamento de tecidos para gerar slides multiparâmetrovirtuais que podem ser usados para quantificação automatizada e mapeamento de células imunes em seções teciduais. Usando essa estratégia, criamos um slide virtual composto por 11 biomarcadores mais duas manchas histológicas frequentemente utilizadas: hematoxilina e eosina (H&E) e picrosirius vermelho (PSR). Várias populações de células imunes foram identificadas, localizadas e quantificadas em diferentes compartimentos teciduais e sua distribuição espacial resolvida usando mapas térmicos teciduais. Essa estratégia maximiza as informações que podem ser obtidas a partir de amostras clínicas limitadas e é aplicável a amostras de tecido saqueado por parafina (FFPE) embutidas por parafina (FFPE), incluindo tecido inteiro, biópsias de agulhas e micromatrizes de tecido. Propomos essa metodologia como um guia útil para a concepção de ensaios personalizados para identificação, quantificação e mapeamento de populações de células imunes no TME.
Protocol
Três seções fseriais de hepatite B ressecadas do vírus da hepatite B (HBV) associadas ao carcinoma hepatocelular humano foram obtidas do Centre hospitalier de l'Université de Montréal (CHUM) Hepatopancreatobiliary Cancer Clinical Database and Biological Specimen Repositório (HBP Biobank). Os pacientes que participam deste banco de tecidos forneceram consentimento informado. Este estudo foi aprovado pelo comitê de ética institucional (Protocolo nº 09.237) e realizado de acordo com a Declaração de Helsinque.
1. Protocolo de coloração de hematoxilina e eosina (H&E)
NOTA: A coloração de H&E foi realizada pela instalação do núcleo de patologia molecular do Centre de Recherches du Centre hospitalier de l'Université de Montréal (CRCHUM) utilizando o manobrista robótico shandon multiprograma usando o seguinte programa.
- Para deparafinação, mergulhe slides 3x para 2,5 min cada em substituto de xileno.
ATENÇÃO: Os substitutos de xileno são inflamáveis, irritantes para a pele e prejudiciais se inalados. - Para reidratação, mergulhe em 100% de etanol 3x por 2,5 min cada. Lave por 1 min em água destilada dupla (ddH2O) para reidratar.
- Incubar por 1 min em hematoxilina. Lave 3x por 1 min cada em ddH2O.
- Incubar para 5 s com eosin. Lave 30 s com 95% de etanol. Lave 2x por 1 min com 100% de etanol.
ATENÇÃO: O etanol é inflamável e um olho irritante. Eosin é um irritante para os olhos. - Para desidratação, mergulhe 3x para 1,5 min cada no substituto de xileno. Montar slides manualmente.
NOTA: O tempo estimado para a execução desta parte do protocolo é de 30 min.
2. Protocolo de coloração de imunofluorescência multiplex para seções de FFPE
NOTA: Este protocolo foi adaptado de Robertson et al.17.
- Deparaffinização e reidratação
NOTA: Antes da rotulagem mediada por anticorpos das seções FFPE pelo IHC ou mIF, a parafina deve ser removida. A falha em remover eficientemente a parafina resulta em coloração abaixo do ideal.- Coloque a seção de tecido FFPE de 4 μm desliza em porta-lâminas de vidro. o capô de fumaça, mergulhe os slides em um frasco de Coplin contendo 37°C de xileno pré-aquecido por 10 min.
ATENÇÃO: O xileno é inflamável, irritante da pele e prejudicial se inalado. - Agitar manualmente os slides por 10 s a cada 2 min. Repita 1x em xileno fresco por mais 5 min.
- Na capa química, mergulhe as lâminas sequencialmente por 5 min em cada uma das seguintes soluções: 1) xileno : etanol (1v/v); 2) 100% etanol; 3) 70% de etanol; 4) 50% de etanol; 5) 30% de etanol; 6) solução salina tampoada por fosfato (PBS).
NOTA: Mantenha os slides na PBS até que esteja pronto para realizar a recuperação do antígeno. Mantenha as seções depiladas hidratadas o tempo todo. A secagem causará a ligação de anticorpos não específicas e, portanto, a coloração de fundo elevada.
- Coloque a seção de tecido FFPE de 4 μm desliza em porta-lâminas de vidro. o capô de fumaça, mergulhe os slides em um frasco de Coplin contendo 37°C de xileno pré-aquecido por 10 min.
- Recuperação de antígenos induzidas pelo calor
NOTA: Os antígenos podem ser mascarados mediante fixação de formalina, impedindo a ligação de anticorpos e, consequentemente, a visualização. O uso de tampões e procedimentos de desmascaramento de antígenos restabelece parcialmente a conformação nativa de epítopos e, assim, restaura o reconhecimento de anticorpos. O tipo de tampão de recuperação de antígenos e duração deve ser otimizado para as condições específicas de ensaio (por exemplo, alvo, anticorpo, tecido, etc.).- Imersão slides depilados em um frasco de Coplin contendo a solução de recuperação de antígenos (receita em Tabela de Materiais).
- Coloque a jarra Coplin fechada em uma panela de pressão elétrica com água da torneira. O nível de água não deve exceder metade da altura do frasco para que a água não se misture com a solução de recuperação de antígenos.
- Feche a tampa e a válvula de pressão da panela. Selecione alta pressão por 10 min e comece. Quando estiver pronto, desligue a panela, solte a pressão, abra a tampa e mantenha o frasco dentro da panela por 30 min, permitindo que os slides esfriem.
- Bloqueio de vinculações não específicas
- Transfira o rack com os slides para um frasco coplin cheio de PBS. Enxágüe o buffer de recuperação de antígenos com PBS 2x por 5 min cada.
- Circunda as seções teciduais com uma caneta PAP para criar uma barreira hidrofóbica. Mergulhe os slides em um frasco de Coplin contendo 0,1 M de glicina na PBS. Incubar por 15 min à temperatura ambiente (RT).
NOTA: A glicina satura os grupos de aldeídos gerados durante a recuperação do antígeno. Esses grupos poderiam ligar anticorpos primários e secundários inespecificamente. - Enxágüe a solução de glicina lavando 2x com PBS por 5 min. Coloque os slides em uma câmara de umidade e adicione solução de bloqueio suficiente para cobrir todas as seções de tecido. Evite transbordar a barreira hidrofóbica. Incubar por 30 min no RT.
NOTA: A receita para a solução de bloqueio pode ser encontrada na Tabela de Materiais. A solução de bloqueio deve conter uma proteína (por exemplo, BSA) para bloquear locais de ligação não específicos. Também pode incorporar detergentes como Triton X-100 ou Tween 20 que reduzem interações hidrofóbicas entre anticorpos e alvos teciduais, tornando assim o reconhecimento de antígenos mais seletivo. A adição de 10% de soro total da espécie de onde o tecido vem bloquearia os receptores Fc e, assim, reduziria a ligação de anticorpos inespecíficos. Finalmente, a adição de 10% do soro da espécie em que os anticorpos secundários foram levantados minimizaria a fixação direta e inespecífica de anticorpos secundários à seção de tecidos.
- Rotulagem de imunofluorescência
- Enxágüe com PBS-Tween (0,1% v/v) 2x por 5 min cada e coloque os slides de volta na câmara de umidade.
- Adicione o coquetel de anticorpos primários resuspenso na solução de bloqueio. Incubar durante a noite a 4 °C. Os anticorpos primários e secundários utilizados para este estudo estão listados na Tabela de Materiais.
NOTA: O coquetel de anticorpos primários deve conter anticorpos criados em espécies diferentes, ou da mesma espécie, mas de diferentes isótipos. Para uma lista dos pares de anticorpos secundários primários utilizados neste estudo consulte a Tabela 2. Os detalhes de todos os anticorpos utilizados estão na Tabela de Materiais e na Tabela 2. - Enxágüe com PBS-Tween (0,1% v/v) 3x por 5 min e coloque os slides de volta na câmara de umidade. No escuro, adicione o coquetel de anticorpos secundários e incubar por 1h no RT.
NOTA: Quando os anticorpos primários são de espécies diferentes, os anticorpos secundários devem ser selecionados para que cada um deles se ligue apenas a um dos anticorpos primários e não um ao outro. Isso é comumente conseguido usando anticorpos secundários todos criados na mesma espécie, desde que esta espécie difere das espécies onde os anticorpos primários foram gerados. Nos casos em que os anticorpos primários foram criados na mesma espécie, mas têm diferentes isótipos, anticorpos secundários específicos do isótipo devem ser usados. - Enxágüe com PBS-Tween (0,1% v/v) 3x para 5 min cada. Enxágüe com ddH2O. Remova o excesso de líquido e monte na mídia de montagem com DAPI. O volume utilizado depende do tamanho da seção. Normalmente 40 μL é suficiente para cobrir a superfície de um slide de microscopia regular.
- Coloque a lâmina de cobertura sobre a seção e esprema suavemente a mídia de montagem em excesso evitando a formação de bolhas. Deixe os slides secarem por 20 min no RT no escuro e armazene a 4 °C até ficar pronto para aquisição.
- Adquira imagens para todos os canais usando todo o scanner de slides (ver Tabela de Materiais).
NOTA: Os anticorpos foram validados usando o tecido hepatocelular humano como controle positivo. Para cada anticorpo primário, três seções seriais foram manchadas com anticorpo primário, controle de isótipo ou apenas solução de bloqueio, respectivamente, sem variação no resto do protocolo de coloração. As imagens adquiridas foram comparadas para estabelecer a especificidade da coloração. A coloração foi considerada específica quando o sinal na seção incubada com anticorpo primário tinha o padrão esperado e era facilmente distinguível do fundo. Anticorpos primários que dão um sinal de fundo elevado ou componentes de tecido de rotulagem no isótipo e nenhuma seção de anticorpos primários foram considerados inespecíficos. O tempo estimado para completar esta parte do protocolo é de 2 dias. Os controles necessários incluem: (1) Controle de isótipo para estabelecer a contribuição da vinculação não específica do anticorpo primário ao sinal de fundo. Uma seção está manchada da mesma forma que os outros tecidos amostrais, exceto que é incubada com um anticorpo com o mesmo isótipo e origem do anticorpo primário, mas específica para um alvo ausente na seção de tecidos. Se o anticorpo de controle de isótipo apropriado não estiver disponível, ele pode ser substituído por IgG total da mesma espécie em que o anticorpo primário foi criado; (2) Nenhum controle primário de anticorpos (ou seja, controle negativo) para estabelecer a especificidade da coloração e estimar a contribuição da vinculação inespecífica de anticorpos secundários ao sinal de fundo. Neste caso, a seção de controle está manchada da mesma forma que as outras seções, exceto que nenhum anticorpo primário é adicionado; (3) Controle positivo para estabelecer que a coloração funciona. Neste caso, a coloração é realizada em uma seção de tecido que é conhecida por expressar o marcador reconhecido pelo anticorpo primário.
3. Picro-sirius vermelho (PSR)/protocolo de coloração verde rápida
NOTA: O objetivo desta coloração é visualizar as colagens fibrilais I e III nas seções de tecidos FFPE. Este protocolo foi adaptado a partir de Segnani et al.18. Todas as etapas são realizadas em uma coifa química.
- Realizar a deparafinação e reidratação de seções teciduais semelhantes ao protocolo de coloração de imunofluorescência multiplex para as seções de FFPE (seção 2.1).
NOTA: Se a seção a ser manchada tiver sido usada anteriormente para rotulagem de imunofluorescência e a parafina já tiver sido removida, as etapas de desparafinação-reidratação são úteis para remover a mídia de montagem. O DAPI não é removido usando este procedimento, mas não interfere com a coloração psr. - Mergulhe as lâminas em um frasco contendo a solução verde vermelha/rápida picro-sirius (receita em Tabela de Materiais)e incuba r$ 30 min em RT (mais de 30 min resulta em coloração inespecífica dos núcleos de hepatocitos).
- Lave rapidamente em ddH2O (5 mergulhos). Em seguida, lave rapidamente o etanol 100% (5 mergulhos). Lave para 30 s em xileno-100% etanol (1:1 v/v). Lave por 30 s em xileno. Monte com a mídia de montagem (ver Tabela de Materiais)antes que o xileno tenha evaporado totalmente (isso ajuda na montagem).
NOTA: O tempo estimado para execução desta parte do protocolo é de 1h.
4. Elução de anticorpos de seções teciduais
NOTA: Para reutilizar seções teciduais em ensaios de rotulagem seqüencial, é necessária a remoção completa de anticorpos primários e secundários. Os anticorpos ligados foram despidos como descrito anteriormente13.
Pré-aqueça um banho de água a 56 °C. Coloque as seções dentro de um frasco contendo tampão de descascamento (receita em Tabela de Materiais),feche a tampa e sele-a com fita de filme de parafina para evitar vazamentodurante a agitação.
- Coloque o pote dentro do banho de água e incubapor por 30 min com agitação.
- Lave 4x por 15 min cada em ddH2O em RT. Enxágue com PBS-Tween (0,1% v/v).
- Mantenha as seções hidratadas em PBS-Tween ou água até que esteja pronta para ressonar a seção com a segunda rodada de anticorpos primários.
NOTA: O tempo estimado para execução desta parte do protocolo é de 2h. - Verifique a eficiência do procedimento de elução de anticorpos.
NOTA: Antes de utilizar o protocolo para elução de anticorpos em um ensaio de rotulagem seqüencial, deve-se verificar a eficiência da remoção de anticorpos primários e secundários.- Realizar a coloração e a aquisição de imagens de uma seção com um dado par de anticorpos secundários primários, conforme indicado no protocolo de coloração de imunofluorescência multiplex para seções de FFPE (seções 2.1-2.4.6).
- Após a aquisição da imagem, realize a elução de complexos de anticorpos secundários ligados ao tecido, conforme indicado nas seções 4.1-4.3.
- Incubar a seção com o mesmo anticorpo secundário e as mesmas condições utilizadas na etapa 2.4.3.
- Execute as etapas de lavagem, montagem e aquisição de imagens, conforme indicado em 2.4.4-2.4.6.
- Compare as imagens lado a lado adquiridas antes e depois da descascação, a fim de estabelecer se o sinal específico desapareceu ou não.
NOTA: A comparação das imagens antes e depois da remoção de anticorpos validará a eficiência do procedimento de elução. No entanto, é normal ver um aumento no sinal de fundo em todos os canais, bem como a difusão de DAPI. Isso limita o número de rodadas de descascamento que podem ser executadas na mesma seção de tecido. Três rodadas de strip-tease parecem ser o máximo.
5. Aquisição de imagens
- Gerar imagens usando um scanner de slides inteiro.
- Use uma lente objetiva 20x 0.75NA e uma resolução de 0,3225 μm/pixel.
6. Análise de imagens
NOTA: O método aqui descrito refere-se ao exemplo atual. Consulte a Tabela 1 e o texto para se adaptar a outras amostras específicas.
- Realizar alinhamento tecidual utilizando o módulo Tissualign do software de análise de imagem (VIS neste protocolo, ver Tabela de Materiais).
- Abra o software de análise de imagem e clique na guia Tissuealign.
- Importe as imagens para serem alinhadas na bandeja de slides indo para Arquivo | Banco de dados e selecione a primeira imagem a ser alinhada. Volte para a guia Tecnocone e carregue a imagem clicando no botão Carregar na bandeja de slides. A imagem aparecerá na bandeja de slides e no espaço de trabalho.
NOTA: Apenas a pilha de juros deve ser carregada na bandeja de slides. - Repita a etapa 6.1.2 para todas as imagens a serem alinhadas, carregando-as uma a uma. Uma vez que todas as imagens de interesse sejam carregadas na bandeja de slides, continue a vincular as imagens pressionando Next nas etapas do fluxo de trabalho na fita.
- Em seguida, arraste e solte a segunda imagem em cima da primeira imagem. A primeira e segunda imagens estão agora ligadas. Repita este passo para que as outras imagens sejam alinhadas, uma a uma, de forma ordenada. O nome da primeira imagem mudará, indicando que ela foi ligada às outras imagens. Simultaneamente, as imagens vinculadas serão exibidas no espaço de trabalho à direita da bandeja de slides.
- Neste ponto, alinhe as imagens usando alinhamento automático, alinhamento semiautomático ou alinhamento manual. É sempre preferível tentar o alinhamento automático primeiro. Para o alinhamento automático, pressione o botão Seguinte nas etapas do fluxo de trabalho (etapa 3) na fita.
- Revise o alinhamento automático navegando em diferentes locais do tecido e verificando visualmente se as estruturas correspondentes em diferentes imagens estão dispostas da mesma forma nas duas dimensões da imagem.
- Se o resultado do alinhamento automático não for satisfatório, melhore-o usando pinos (use um mínimo de três pinos por imagem) indicando características de tecido homólogo nas imagens ligadas. Uma vez que os pinos são colocados em locais homólogos nas imagens vinculadas, o usuário tem duas opções: alinhamento semiautomático ou alinhamento manual. Para o alinhamento semiautomático clique no botão Alinhamento automático com base nos pontos atuais na fita. Para o alinhamento manual, clique no botão Aplicar pinos na fita.
- Quando satisfeito com o alinhamento clique no botão Seguinte nas etapas do fluxo de trabalho e salve a imagem composta no banco de dados.
NOTA: O alinhamento de seis slides abrangendo 11 marcadores mais as imagens H&E e e e PSR levaram 15 minutos na análise apresentada.
- Realize a detecção de tecidos usando o protocolo de análise de protocolo definido pelo usuário Pacote 1 (APP 1, Tabela 1).
- Abra o módulo de análise de imagem do software clicando na guia Análise de imagens na fita.
- Importe a imagem composta (alinhada) indo para Arquivo | Banco de dados e selecionando a imagem de interesse e clicando de volta na guia Análise de Imagens.
- Abra a caixa de diálogo de seleção do APP clicando no ícone Do APP Aberto e selecione qual pacote de protocolo de análise (APP) usar. Neste caso, selecione APP 1 para detecção de tecido.
- Uma vez que o APP 1 seja aberto, confirme se o APP1 está funcionando corretamente indo para um local de tecido selecionado e clicando no botão Visualização. Se os resultados forem satisfatórios, vá para o próximo passo.
- Clique para executar o APP 1 e processar a imagem usando o APP selecionado.
- Exportar os dados (por exemplo, imagens, medições, etc.) quando a análise é feita clicando em Arquivo/Exportação.
NOTA: O APP 1 cria uma região de interesse (ROI) delineando o tecido (Tecido ROI) e calcula a área do tecido. - Salvar a imagem modificada com o ROI recém-criado indo para Arquivo | Salve, salve.
NOTA: Detectar o tecido e criar um ROI com APP 1 no exemplo fornecido levou 5 min na estação de análise de imagem descrita. A área do tecido processado foi de 3,2 cm2.
- Realizar segmentação de tecidos em Stroma e Parenchyma usando app 2 (Tabela 1).
NOTA: O APP 2 funciona no tecido de ROI predefinido. App 2 segmenta o tecido nos ROIs Stroma e Parenchyma.- Abra o módulo de análise de imagem clicando na guia Análise de imagens na fita.
- Importe a imagem contendo o tecido DE ROI indo para Arquivo | Banco de dados e seleção da imagem salva na etapa 6.2.7. Volte para a guia Análise de imagens e carregue a imagem clicando no botão Carregar na bandeja de slides. A imagem aparecerá na bandeja de slides e no espaço de trabalho.
- Abra o APP 2 usando a caixa de diálogo de seleção do APP como em 6.2.3.
- Visualizar APP 2 processando em um campo de exibição selecionado. Se os resultados forem satisfatórios, execute o APP 2 na imagem completa clicando no botão Executar. Como saída do APP 2, o tecido de ROI é segmentado nos ROIs Stroma e Parenchyma e suas respectivas áreas determinadas. Os resultados das exportações foram de 6.2.6. Salve a imagem modificada como em 6.2.7.
NOTA: Segmentar o tecido em Stroma e Parenchyma utilizando o APP 2 levou 4h na estação de análise apresentada. A área do tecido processado foi de 3,2 cm2.
- Identifique e quantifique as células FoxP3hiCD4+ usando o protocolo definido pelo usuário APP 3 (Tabela 1).
NOTA: O APP 3 funciona nos ROIs stroma e parenchyma predefinidos.- Abra o módulo de Análise de Imagens e importe a imagem contendo os ROIs Stroma e Parenchyma como em 6.3.1 e 6.3.2. Abra o APP 3 usando a caixa de diálogo de seleção do APP como em 6.2.3.
- Visualizar o processamento do APP 3 em um campo de visão selecionado enriquecido em células FoxP3hiCD4+. Se os resultados forem satisfatórios, execute o APP 3 na imagem completa. Como saída do APP 3, todos os objetos FoxP3hiCD4+ individuais serão rotulados e suas coordenadas teciduais armazenadas. As densidades dos objetos FoxP3hiCD4+ nos ROIs Stroma e Parenchyma serão determinadas. Exportar os resultados em 6.2.6.
- Realize o mapeamento térmico do tecido de objetos rotulados FoxP3hiCD4+.
- Abra o protocolo foxp3hiCD4+ MAP definido pelo usuário usando a caixa de diálogo de seleção do APP como em 6.2.3.
NOTA: FoxP3hiCD4+ MAP usa as coordenadas de objetos rotulados FoxP3hiCD4+ para gerar mapas de calor de densidade. Identificar e contar objetos rotulados FoxP3hiCD4+ usando o APP 3 levou 25 min na estação de análise de imagem descrita. A área do tecido processado foi de 3,2 cm2. - Execute FoxP3hiCD4+ MAP pressionando o botão Executar. Exportar o mapa de calor do tecido clicando em Arquivo | Exportação | Área de trabalho.
NOTA: O mapeamento de objetos rotulados FoxP3hiCD4+ usando FoxP3hiCD4+ MAP levou 5 min na estação de análise de imagem descrita.
- Abra o protocolo foxp3hiCD4+ MAP definido pelo usuário usando a caixa de diálogo de seleção do APP como em 6.2.3.
- Identificar e quantificar objetos CD8+, CD68+, MPO+, αSMA e CD34 + utilizando os protocolos definidos pelo usuário APP 4, APP5, APP6, APP7 e APP 8, respectivamente(Tabela 1),conforme feito na seção 6.4 a 6.4.3.2 carregando o APP de interesse em cada caso.
NOTA: As APPs 4 a 8 trabalham nos ROIs stroma e parenchyma predefinidos.
Representative Results
Visão geral da estratégia de visualização, quantificação e mapeamento de populações celulares de interesse no TME
Para quantificar as populações celulares de interesse (COIs) em diferentes compartimentos teciduais (TCs) e caracterizar sua organização espacial, projetamos um fluxo de trabalho que integra técnicas acessíveis e fáceis de usar e maximiza as informações posicionais que podem ser obtidas a partir de preciosas amostras clínicas de FFPE(Figura 1). Em primeiro lugar, as seções de FFPE de tecido inteiro serial foram manchadas para visualização de COIs (por exemplo, células imunes) e TCs (por exemplo, estroma versus parenchyma) (Figura 1, passo 1). O número de seções consecutivas a serem manchadas deve ser mantido ao mínimo que permita a visualização das células de interesse ou características teciduais necessárias para abordar a questão da pesquisa. Quanto menor o número de seções seriais, maior a semelhança da arquitetura tecidual e a concordância entre seções contíguas. Além disso, a capacidade de multiplexação pode ser expandida através da reutilização de seções manchadas fluorescentes através de técnicas de descascamento e ressarcimento19.
Uma vez que as etapas de coloração foram feitas, um scanner de slides inteiro foi usado para digitalizar as imagens. As imagens adquiridas a partir de seções seriais foram alinhadas e consolidadas em um slide multiplex virtual de forma automatizada(Figura 1, seção 2). Em seguida, um ROI para o tecido foi delineado com um protocolo definido pelo usuário que identificou pixels associados ao tecido (TAPs) (Figura 1, passo 3). Posteriormente, o tecido de ROI foi segmentado em TCs definidos como ROIs adicionais. (Figura 1, passo 4). Em seguida, os protocolos definidos pelo usuário detectaram e quantificaram cois em diferentes TCs(Figura 1, etapa 5). Finalmente, foram gerados mapas térmicos de tecidos de COIs com base em suas densidades e suas coordenadas teciduais (Figura 1, passo 6).
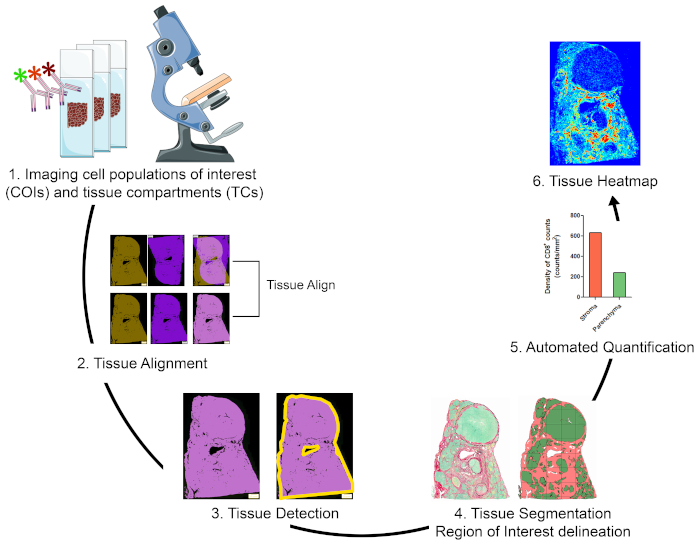
Figura 1: Representação esquemática da estratégia de visualização, quantificação e mapeamento de células imunes no TME. (1) As seções de tecido sumidérdia foram manchadas para rotular COIs e TCs. As seções de tecido inteiros manchadas foram digitalizadas usando um scanner slide inteiro. (2) As imagens adquiridas a partir de seções seriais foram vinculadas, alinhadas e co-registradas de forma automatizada, utilizando um módulo de análise Tissuealign. Uma imagem composta foi gerada a partir do alinhamento de alta precisão de imagens individuais. (3) Foi utilizado um protocolo definido pelo usuário para detecção automatizada de pixels associados ao tecido (TAPs) na imagem composta. (4) O tecido foi segmentado em TCs (por exemplo, estroma e parenchyma) definidos como ROIs. (5) Foram utilizados protocolos definidos pelo usuário para a detecção e quantificação automatizada de COIs em diferentes TCs. (6) Foram gerados mapas térmicos de COIs de tecido. Clique aqui para ver uma versão maior desta figura.
COIs e TCs de imagem
Três seções de tecido inteiro de FFPE serial de tumor ressecado de um sujeito com carcinoma hepatocelular associado ao HBV foram manchadas em uma ou mais rodadas de coloração como na Figura 2A. A seção I foi manchada com H&E para mostrar a arquitetura tecidual, a morfologia celular e determinar parâmetros clinicamente relevantes, como tipo de malignidade, grau de tumor e avaliação geral da infiltração imunológica(Figura 2C). Na seção Contígua II, foram utilizadas duas rodadas de IFM foram utilizadas para rotular células parênquias hepáticas e não parênquias(Figura 2A). Na primeira rodada, os vasos normais e tumorais foram visualizados utilizando-se a coloração de CD34 de células endoteliais. Além disso, as células epitelial (hepatócitos e colangiocitos) foram identificadas por meio de citoqueratina 8/18, e as células hepáticas hepáticas ativadas fibrogênicas foram identificadas como células alfa lisas do músculo (αSMA+)(Figura 2C). Após a aquisição da imagem, as seções teciduais foram despojadas e rescadas com anticorpos contra macrófagos (CD68) e miofibroblastos (desmin). Para melhor caracterizar o infiltrado imunológico tumoral, a seção serial adjacente III foi manchada usando duas rodadas de IFM para os marcadores celulares CD3, CD4, CD8, bifurcação P3 (FoxP3) e mieloperoxidase (MPO). Em todos os casos, o DAPI foi usado como contra-mancha nuclear. Finalmente, a seção III foi manchada com mancha PSR e contrariada com verde rápido para visualizar o colágeno fibrilar e segmentar o tecido em estroma e parenchyma(Figura 2C).
Todo um scanner slide equipado com uma lente objetiva de 20X foi usado para digitalizar seções manchadas e criar slides virtuais. Seis imagens foram adquiridas a partir das três seções seriais (Figura 2B) e os slides virtuais posteriormente analisados utilizando o software VIS de acordo com a representação esquemmática na Figura 1.
Análise de Imagens
A análise da imagem compreendeu cinco etapas: 1) alinhamento tecidual; 2) detecção de tecido; 3) segmentação de tecidos; 4) quantificação automatizada de COIs; e 5) mapeamento térmico do tecido. Todos os protocolos para análise de imagem foram desenvolvidos utilizando o módulo Autor do software de análise de imagem e são referidos no texto como APP.
Alinhamento tecidual
Seis slides virtuais de três seções seriais, abrangendo 11 marcadores mais manchas de H&E e PSR, foram carregados no módulo Tissualign do software de análise de imagens. Em seguida, as imagens foram vinculadas, alinhadas e co-registradas de forma automatizada, gerando uma imagem composta virtual de 11 plex mais H&E e PSR, contendo todas as camadas das imagens individuais(Figuras 2A-C). O alinhamento foi preciso no caso de imagens originárias de seções seriais adjacentes, mostrando estruturas teciduais correspondentes posicionadas e dispostas de forma homóloga no alinhamento(Figura 2C e Figura S1A). Além disso, o alinhamento foi preciso no nível individual da célula para imagens originárias da mesma seção(Figura S1B). O tempo para alinhamento automático depende do número, tamanho, complexidade e semelhança das imagens a serem alinhadas. O alinhamento dos seis slides virtuais acima mencionados levou 15 minutos em nossa estação VIS.

Figura 2: Coloração de seções de tecido serial e alinhamento de imagem. (A) Resumo das manchas feitas em três seções seriais para visualização de COIs e TCs. Os números entre parênteses indicam a designação da imagem. Para as seções II e III, os tecidos foram despidos e ressonados com um segundo coquetel de anticorpos. (B) Visão geral de seis imagens individuais de tecido inteiro antes e depois do alinhamento do tecido (esquerda e direita, respectivamente). Barra de escala = 3.500 μm.(C) Visão ampliada de imagens alinhadas. Barra de escala = 80 μm. Clique aqui para ver uma versão maior desta figura.
Detecção de tecidos
Uma vez que as imagens foram vinculadas e alinhadas, procuramos identificar os TAPs(Figura 3A). Para projetar um APP para a detecção automatizada de TAPs (APP 1, Tabela 1),aproveitamos duas propriedades que diferenciam os TAPs de pixels não associados ao tecido. Primeiro, o sinal DAPI (banda azul) é restrito aos núcleos, que estão localizados exclusivamente no tecido, o que significa que todos os pixels DAPI+ são um subconjunto de TAPs. Em segundo lugar, os TAPs têm maior sinal de autofluorescência nas faixas verde e amarela em comparação com pixels não associados ao tecido. Consequentemente, desenvolvemos o APP 1 para detecção de tecidos(Tabela 1),que detecta os TAPs com base no sinal de linha de base nesses canais usando técnicas simples de limiar. Limiares para as faixas azul, verde e amarela foram definidos de modo que os TAPs tinham valores de intensidade de fundo acima dos limiares, enquanto os pixels não associados ao tecido tinham valores abaixo. O APP 1 para detecção de tecido foi aplicado à imagem IIA, que contém camadas nos canais azul, verde e amarelo(Figura 3A). Como saídas do APP 1, uma máscara verde brilhante foi colocada no topo dos TAPs, e um ROI chamado "Tissue" foi delineado (saída, Figura 3A). Além disso, a área do tecido foi determinada como variável de saída quantitativa. Como o APP 1 não incorpora os pixels não associados ao tecido no tecido DO ROI, eles foram excluídos da análise subseqüente com base neste ROI(Figura 3A). A precisão do APP 1 na identificação de TAPs é mostrada na Figura 3A.
Segmentação de tecidos e delineamento de ROIs para TCs
Em seguida, começamos a definir diferentes compartimentos dentro do tecido de ROI segmentando o tecido em estroma versus parenchyma. Utilizou-se a imagem manchada de PSR (IIIC, Figura 2C), onde o estroma pode ser definido como a área associada à deposição de colagens fibrilais (banda vermelha), a parenchyma como a área onde faltam as colagens fibrilas, e prevalece o rápido tine de contracoloração verde (faixa verde)(Figura 3B). Criamos o APP 2(Tabela 1)para delimitar digitalmente os TCs Stroma e Parenchyma. Este APP funciona no tecido DE ROI predefinido (saída, Figura 3A) e utiliza áreas representativas de estroma e parenchyma para treinar a ferramenta Classifier integrada no módulo de Análise de Imagens. O Classificador treinado atribui os pixels a um estroma ou a um rótulo parenchyma (salmão e verde, respectivamente, Figura 3B). Após a classificação dos pixels, o APP 2 executou operações morfológicas com o objetivo de definir os ROIs Stroma e Parenchyma(Figura 3B e Tabela 1). O desempenho do APP 2 na classificação de pixels e na geração dos respectivos ROIs é mostrado na Figura 3B. Além disso, o APP 2 quantifica a área do estroma e do parenchyma. Finalmente, mesmo que a segmentação seja feita usando a seção manchada de PSR, as regiões de stroma e parenchyma delineadas podem ser transferidas para qualquer imagem alinhada à imagem PSR.

Figura 3: Detecção/segmentação automatizada de tecidos e geração de respectivos ROIs. (A) A Imagem IIA foi utilizada para identificar os TAPs (imagem esquerda, barra de escala = 6.000 μm). Uma máscara verde brilhante foi atribuída aos TAPs usando app 1 (Tabela 1) gerando um ROI chamado Tissue (saída 1). Certo, inset mostra visualização ampliada demonstrando a precisão do APP 1 na detecção de TAPs. Barra de escala = 350 μm.(B) O tecido de ROI (saída 1) é segmentado em estroma e parenchyma usando o APP 2. A imagem à esquerda mostra uma visão do tecido roi segmentado em estromo roi (salmão) e parenchyma ROI (verde). Barra de escala = 4.500 μm. À direita, vistas ampliadas do inset para o ROI Tissue, a coloração psr original (imagem IIIC), e o estromo rois e parenchyma. Barra de escala = 250 μm. Clique aqui para ver uma versão maior desta figura.
Quantificação automatizada de COIs
Em seguida, passamos a identificar, localizar e quantificar COIs nos ROIs Stroma e Parenchyma. ApPs 3 a 8(Tabela 1) foram criados para localizar e contar os seguintes COIs: CD4+FoxP3+, CD8+, CD68+, MPO+, αSMA+, e células CD34+, respectivamente. O APP 3 foi projetado para localizar e contar células CD4+FoxP3+ (imagem IIIA, Figura 2C) como marcadores substitutos de células T regulatórias (Tregs). Este protocolo detecta a colocalização do sinal a partir do fator de transcrição nuclear FoxP3 (banda vermelha) e do dna de rotulagem de dna DAPI (banda azul). Dado que as células T recentemente ativadas regulam o FoxP3, para enriquecer para Tregs, estabelecemos limites para a pré-seleção apenas de células FoxP3+ brilhantes (FoxP3hi). Em seguida, de todas as células deoi DAPI+FoxP3 pré-selecionadas, apenas aquelas que estavam cercadas por sinais CD4 em forma de anel brilhante (banda verde) foram rotuladas e contadas como células FoxP3hiCD4+ (etiqueta rosa, Figura 4A). A densidade das células FoxP3hiCD4+ nos ROIs Stroma e Parenchyma foram determinadas como variáveis de saída quantitativa satisfatoida de APP 3 (Figura 4A).
Da mesma forma, as APPs 4 a 6 foram projetadas para a detecção de células CD8+, CD68+e MPO+. Esses APPs compartilham o mesmo desenho de linha de base para detectar e quantificar COIs. Especificamente, os COIs são identificados com base na intensidade do sinal do biomarcador específico da população celular e, em seguida, várias etapas morfológicas pós-processamento são executadas para delinear células individuais(Tabela 1). As células individuais ou COIs são rotuladas, contadas e suas coordenadas teciduais registradas. As APPs 4 a 6 também determinam a densidade dos COIs nos ROIs Stroma e Parenchyma (Figura 4B-D).
A qualidade da nossa coloração DAPI não foi boa o suficiente para integrar a segmentação de núcleos em APPs 3 a 6, por isso não podemos garantir que todos os objetos rotulados individualmente sejam células individuais. Por essa razão, expressamos a densidade das células em contagens de objetos rotulados/mm2 (Figura 4). No entanto, os agregados celulares foram separados com sucesso em células individuais nas etapas de pós-processamento incorporadas em APPs 3 a 6, e uma extensa inspeção visual mostrou que a maioria dos objetos rotulados correspondia a células únicas.
Para detectar a área αSMA+ e CD34+, desenvolvemos APPs 7 e 8, respectivamente (Tabela 1). Ambas as APPs detectam o sinal específico com base em limiares e determinam a porcentagem de área positiva nos ROIs Stroma e Parenchyma (Figura 4E-F).
Uma das possibilidades mais interessantes de gerar slides multiplex virtuais é a análise da expressão de colocalização. Geramos o APP 10 para detectar a colocalização entre αSMA e desmin, dois marcadores co-expressos por miofibroblastos no fígado. O APP 10 usa limiares para encontrar pixels positivos para αSMA, desmin e αSMA plus desmin(Tabela 1). Como variáveis de saída quantitativas, o APP 10 determina a área αSMA+, a área de desmin+ e a área de expressão colocalizada desses dois marcadores(Figura S3).

Figura 4: Identificação e quantificação de COIs no estroma e parenchyma. (A-F) Detecção e quantificação automatizadas de CoIs CD4+FoxP3+, CD8+, CD68+, MPO+, αSMA+, e CD34+ COIs nos ROIs Stroma e Parenchyma utilizando os protocolos 3, 4, 5, 6, 7 e 8, respectivamente(Tabela 1). Mostradas à esquerda as imagens originais, no meio as imagens processadas, e à direita as quantificações. Para figuras 4A-D, barra de escala = 40 μm. Para as figuras 4E e F, barra de escala = 350 μm. Clique aqui para ver uma versão maior desta figura.
Como alternativa para quantificar os COIs nos TCs Stroma e Parenchyma, determinamos a densidade de células imunes nos diferentes nódulos malignos nomeados 1 a 4(Figura 5A, He I). O ROI para cada nódulo foi delineado manualmente conforme indicado na Figura 5A. As assinaturas imunológicas teciduais distintas caracterizaram cada nódulo, revelando ainda mais a heterogeneidade intrínseca do TME.
Mapas térmicos de tecidos
Como mencionado acima, as APPs 3 a 8 armazenam as coordenadas teciduais de cada objeto rotulado individualmente. Este recurso permite a geração automatizada de mapas teciduais onde regiões de alta densidade de uma determinada população celular são exibidas como pontos quentes (vermelho), e regiões com densidade relativamente baixa como pontos frios (azul escuro). Os valores de densidade intermediária são atribuídos cores de acordo com a escala de cor mostrada na Figura 5. Mapas térmicos de tecidos foram gerados por APPs que dividiram as imagens em círculos de 50 μm de diâmetro e atribuíram uma cor de acordo com a densidade relativa de um dado COI dentro do círculo. Como mostrado na Figura 5B-G,os padrões de posicionamento e a distribuição de intensidade dos diferentes COIs no TME foram bastante variados. Além disso, ao nível dos nódulos individuais, o arranjo de diferentes populações na área tecidual foi único (Figura S2A-C). Para dar um exemplo do poder dessa técnica e visualizar a organização espacial de pontos quentes de diferentes populações no mesmo nódulo, os pontos quentes de tipos de células individuais foram extraídos manualmente e mapeados juntos no contorno do nódulo 2 (Figura S2, Figura De Figura E).

Figura 5: Mapas de calor de tecido de COIs no TME. (A)Coloração vermelha picrosirius mostrando localização dos nódulos 1, 2, 3 e 4. (B-G) Mapas de calor de tecido para COIs CD4+FoxP3+, CD8+, CD68+, MPO+, CD34+e αSMA+, respectivamente. Azul escuro indica densidade relativa baixa, e vermelho indica relativa alta densidade. Os valores de densidade intermediária são atribuídos cores de acordo com a escala de cor mostrada. (H e I) Quantificação de COIs nos nódulos 1, 2 e 3 + 4 organizados por tipo celular e por nódulo, respectivamente. Clique aqui para ver uma versão maior desta figura.

Figura Suplementar S1: Validação do alinhamento tecidual. (A) A coloração CD34 (em vermelho) feita na seção II (entrada 1) é usada para gerar uma máscara CD34 em verde (saída 1). A máscara verde (saída 1) é sobreposta na imagem H&E da seção serial alinhada I (entrada 2). A imagem de fusão mostra correspondência perfeita de estruturas vasculares. Barra de escala = 50 μm. (B) Imagem IIIA mostrando a fusão de DAPI, CD4 e FoxP3 (entrada 1) foi usada para gerar uma etiqueta para células CD4+FoxP3+ (saída 1 em magenta). O rótulo de saída 1 foi transferido para a imagem alinhada IIIB (entrada 2) e mostra correspondência perfeita entre os pares FoxP3/DAPI e CD4/CD3 na imagem de mesclagem. Barra de escala = 15 μm. Clique aqui para ver uma versão maior desta figura.

Figura suplementar S2: Visão ampliada dos mapas de calor do tecido. (A–C) Mapas de calor tecidual para células CD4+FoxP3+, CD8+, CD68+e MPO+ em nódulos 1-4. As barras de escala nos nódulos 1, 2 e 3 + 4 representam 1.500 μm, 700 μm e 500 μm, respectivamente. (D) Contorno do nódulo 2 com linha sólida preta. (E) Os pontos quentes para as células CD4+FoxP3+, CD8+, CD68+e MPO+ no nódulo 2 foram extraídos e mapeados juntos no contorno do nódulo 2 definido em D. Clique aqui para ver uma versão maior desta figura.

Figura Suplementar S3: Análise de Colocalização. (A) À esquerda e no meio estão imagens da etiqueta αSMA em verde e etiqueta de smin em vermelho, respectivamente. À direita está uma área dupla positiva αSMA/desmin em amarelo. (B) Quantificação da área αSMA+, área de desmin + e αSMA/desmin área dupla positiva. Barra de escala = 150 μm. Clique aqui para ver uma versão maior desta figura.
| App | Propósito | Classificação | Classificação | Etapas pós-processamento | Variáveis de saída |
| Método | Características | ||||
| (valor de pixel) | |||||
| 1 | Detecção de tecido | Limite | Canal DAPI (150) | o Rotular objetos com valores acima do limiar colocalizados para os 3 canais | o TECIDO ROI |
| Canal FITC/A488 (120) | o Fechar objeto positivo 5 pixels | o Área de Tecido | |||
| Canal TRITC/A568 (40) | o Criar tecido de ROI | ||||
| 2 | Segmentação de tecidos | Floresta de Decisão | Mediana RGB-R | o Preencher buracos | o ROI Stroma |
| Mediana RGB-G | o Criar ROI Stroma | o Área de Stroma | |||
| Mediana RGB-B | o Criar ROI Parenchyma | o ROI Parenchyma | |||
| Mediana do IHS-S | o Parenchyma Área | ||||
| H&E Eosin mediana | |||||
| 3 | Para localizar e quantificar células CD4+ FoxP3+ | Limite | Canal DAPI (>600) | o Etiquetar objetos com colocalização de DAPI e Cy5/A647, cercados pelo sinal FITC/A488 | o Contagem e densidade de células CD4+FoxP3+ em ROIs Stroma e Parenchyma |
| Canal FITC/A488 poli suavização (>850) | o Limpar objetos menores que 7 μm2 | o Coordenadas de células CD4+FoxP3+ individuais | |||
| Canal Cy5/A647(>800) | |||||
| 4 | Para localizar e quantificar células CD8+ | Limite | Canal DAPI (<1200) | o Limpar objetos positivos menores que 15 μm2 | o Contagem e densidade de células CD8+ em ROIs Stroma e Parenchyma |
| Canal Cy5/A647 mediana (>80) | o Fechar objetos positivos 2 pixels | o Coordenadas de células individuais | |||
| o Objetos separados | |||||
| 5 | Para localizar e quantificar células CD68+ | Limite | Canal FITC/A488 (>200) | o Limpar objetos positivos menores que 20 μm2 | o Contagem e densidade de células CD68+ em ROIs Stroma e Parenchyma |
| o Dilate objetos positivos 3 pixels | o Coordenadas de células CD68+ individuais | ||||
| o Objetos separados | |||||
| 6 | Para localizar e quantificar células MPO+ | Limite | Canal DAPI (>400) | o Limpar objetos menores que 5 μm2 | o Contagem e densidade de células MPO+ em ROIs Stroma e Parenchyma. |
| Canal TRITC/A568 (900-4000) | o Dilado 3 pixels objetos positivos | o Coordenadas de células MPO+ individuais. | |||
| o Objetos separados | |||||
| 7 | Para localizar e quantificar a área αSMA+ | Limite | Canal TRITC/CF568 (>1050) | o Limpar objetos positivos menores que 25 μm2 | o Contagem e densidade da área αSMA+ em ROIs Stroma e Parenchyma |
| o Dilado 3 pixels objetos positivos | o Coordenadas de pixels αSMA+ | ||||
| 8 | Para localizar e quantificar a área CD34+ | Limite | Canal DAPI (<5000) | o Limpar objetos positivos menores que 25 μm2 | o Contagem e densidade da área CD34+ em ROIs Stroma e Parenchyma |
| Canal Cy5/A647 mediana (>120) | o Dilado 3 pixels objetos positivos | o Coordenadas de pixels CD34+ | |||
| 9 | Crie mapas de calor de tecido para uma determinada população celular | Mapa de calor do objeto | Mapa de calor do objeto | o Mapa de Calor | |
| Raio de desenho 50 μm | --- | ||||
| 10 | Quantificar a colocalização entre αSMA e Desmin | Limite | Canal TRITC (CF568) (>1050) | o Rotular objetos com valores de limite acima para TRITC (CF568) | o Quantificar expressão co-localizada de αSMA e Desmin |
| Canal Cy5 (A647) (>1000) | o Rotular objetos com valores de limite acima para Cy5 (A647) | ||||
| o Rotular objetos com colocalização de valores limiares acima para TRITC (CF568) e Cy5 (A647) | |||||
| o Limpar objetos positivos menores que 25 μm2 |
Tabela 1: Parâmetros gerais utilizados para o desenho de APPs empregados para análise de imagem. Os parâmetros especificados nesta tabela são ajustados às características únicas das imagens utilizadas nesta análise (por exemplo, fundo, artefatos, etc.) e podem não ser aplicáveis a outras imagens. Como as etapas de pós-processamento mencionadas foram definidas para as imagens específicas analisadas neste estudo, elas não são intencionalmente detalhadas. O usuário deve personalizar as APPs para as imagens a serem analisadas.
| Seção/Coloração | Anticorpo Primário | Anticorpo Secundário |
| Seção II/1st Coloração | Mouse IgG2a anti-humano αSMA Mouse IgG1 anti-humano CD34 Coelho anti-humano Cytokeratin 8/18 |
Cabra anti-rato IgG2a CF568 Rato anti-rato IgG1 A647 Burro anti-coelho A488 |
| Seção II/2de Coloração | Coelho anti-humano Desmin CD68 anti-humano do rato |
Burro anti-coelho A647 Burro anti-mouse DyLight 755 |
| Seção III/1st Coloração | CD anti-humano do rato Coelho anti-humano FoxP3 MPO anti-humano de cabra |
Burro anti-mouse A488 Burro anti-coelho A647 Burro anti-cabra A568 |
| Seção III/2de Coloração | Coelho anti-humano CD3 CD anti-humano do rato |
Burro anti-mouse DyLight 755 Burro anti-coelho A647 |
Tabela 2: Pares de anticorpos secundários primários para if
Discussion
Técnicas simples, acessíveis e fáceis de executar multiplexantes que permitam a resolução espacial de células imunes em seções teciduais são necessárias para mapear a paisagem imunológica em câncer e outras doenças imunológicas. Aqui, descrevemos uma estratégia que integra técnicas de rotulagem e análise digital amplamente disponíveis para ampliar a capacidade de multiplexação e avaliação multidimensional dos ensaios de imagem12,,13,,17,,19. A coloração de três seções seriais para diferentes marcadores, e o reaproveitamento de seções através de técnicas de descascamento e ressarência, nos permitiu visualizar 11 parâmetros, além de manchas de H&E e PSR. Seis imagens dessas seções foram alinhadas de forma automatizada usando o módulo de alinhamento de tecidos. O alinhamento foi preciso no nível individual da célula para imagens originárias da mesma seção e altamente concordante para imagens originárias de seções vizinhas. O multiplexação virtual nos permitiu determinar como os marcadores visualizados em uma seção se relacionam espacialmente com marcadores visualizados em outra seção contígua. Enquanto algumas das manchas rotularam COIs, outras rotularam TCs, permitindo-nos quantificar COIs nos diferentes TCs. O uso de ferramentas de software para a quantificação automatizada de COIs simplificou e acelerou muito o processamento de imagens. Além disso, a análise digital foi aplicada em seções de tecidos inteiros em vez de campos de visão selecionados, resultando em uma representação imparcial do TME. Além disso, como as coordenadas teciduais dos COIs foram registradas, foi possível gerar mapas de calor tecidual.
Existem várias áreas neste protocolo onde a solução de problemas pode ser necessária. Em primeiro lugar, a má recuperação de antígenos pode afetar a qualidade do IFM, portanto, o tipo de buffer de recuperação de antígenos e a duração devem ser otimizados para as condições específicas de ensaio/biomarcador utilizados. Em segundo lugar, o tipo de solução de bloqueio utilizada deve ser adaptado aos tecidos/antígeno/espécie de anticorpos primários e secundários. Em nossas mãos, a adição de 10% do soro total da espécie onde o tecido vem de receptores Fc bloqueados, e, portanto, reduziu a ligação de anticorpos inespecíficos. A adição de 10% do soro da espécie em que os anticorpos secundários foram levantados minimizaria a fixação direta e inespecífica de anticorpos secundários à seção tecidual. Em terceiro lugar, a validação da especificidade dos anticorpos primários e secundários utilizando os controles positivos e negativos adequados é essencial. Em quarto lugar, o aumento da autofluorescência em alguns canais e a difusão de DAPI sobre a desmontagem de anticorpos primários também são comuns. Para abordar a autofluorescência aprimorada, usamos pares de anticorpos primários/secundários onde o sinal específico tinha valores de intensidade pelo menos 5x os do fundo. Finalmente, alguns anticorpos de alta afinidade não podem ser eluidos com procedimentos regulares de desmontagem. Neste caso, recomendamos o uso desses anticorpos na última rodada de rotulagem. O usuário pode ter que tentar diferentes seqüências de coloração para encontrar a configuração ideal para os anticorpos de interesse. A eficiência da descascamento deve ser confirmada antes de prosseguir para uma segunda ou terceira rodada de rotulagem.
A principal limitação e desafio dessa estratégia é encontrar as combinações certas de anticorpos fluorescentes primários e secundários para os marcadores de interesse. Encontrar anticorpos primários criados em diferentes espécies ou com diferentes iótipos que poderiam ser usados simultaneamente é limitado pelo que está comercialmente disponível. A maioria dos scanners de slides inteiros são equipados com lâmpadas e filtros que permitem a imagem de no máximo cinco canais, e anticorpos secundários nas espécies certas e fluoróforos certos nem sempre estão disponíveis. Superamos parcialmente essas limitações usando manchas seriais e rotulagem seqüencial. Várias combinações de anticorpos podem precisar ser testadas para chegar à melhor combinação para os marcadores de interesse. Outra limitação é a qualidade da coloração da DAPI, pois a descascamento e a resondagem podem nem sempre permitir a realização da segmentação de núcleos.
O módulo de alinhamento de tecidos requer treinamento mínimo e nenhuma habilidade de programação dos usuários. O software teoricamente permite o alinhamento de um número ilimitado de imagens. No entanto, o alinhamento preciso depende da relação das seções, onde seções mais próximas que são mais histologicamente concordantes estão mais alinhadas com mais precisão. Utilizou-se o módulo Autor do VIS para a geração dos APPs. O conhecimento básico da análise de imagens é necessário para a criação de APPs, mas este é igualmente o caso ao usar qualquer outro software de análise de imagem. As vantagens únicas do VIS em comparação com outros softwares de análise de imagens incluem alinhamento automatizado de imagens de seções preparadas usando diferentes métodos (por exemplo, IF, histoquímica, IHC). Isso permite estudos de colocalização de múltiplos marcadores de interesse usando multiplexação virtual. Além disso, o design flexível e fácil de usar de APPs permite personalização específica do usuário. Quantificação e mapeamento automatizados, e a possibilidade de processar seções de tecidos inteiros, economiza tempo e reduz o viés em comparação com a contagem manual por inspeção visual.
Essa estratégia é uma ferramenta de pesquisa muito útil para a imunologia tecidual no contexto do câncer e da autoimunidade, mas permanece invalidada para uso clínico. Com padronização e validação adicionais, pode ser usado no futuro para múltiplas aplicações (por exemplo, mapear a paisagem imunológica do câncer para prever e monitorar a resposta a agentes imunoterapêuticos). Também pode ser adaptado a diferentes condições inflamatórias (por exemplo, doença inflamatória intestinal) para combinar avaliação patológica com biomarcadores prognósticos.
As principais etapas críticas deste protocolo são a eficiência/especificidade da rotulagem e a robustez dos APPs projetados para o uso pretendido ou biomarcador. Por isso, a validação regular por inspeção visual, especialmente na concepção de um novo APP, é essencial. O uso eficiente de múltiplas rodadas de descascamento e resplanagem ou diferentes tipos de manchas na mesma seção são componentes críticos e podem ser específicos do tecido ou da seção. Verificar a eficiência desses processos antes de prosseguir com a análise de grandes lotes é fundamental.
Em resumo, fornecemos uma estratégia que maximiza as informações quantitativas e espaciais que podem ser obtidas a partir de valiosas amostras de tecido clínico. Os recursos, equipamentos e conhecimentos necessários para implementar essa metodologia são amplamente acessíveis. Propomos essa metodologia como um guia útil para o planejamento de ensaios com o objetivo de identificar, quantificar e mapear populações de células imunes no TME.
Disclosures
Os autores não declaram conflitos de interesse.
Acknowledgments
Agradecemos ao participante do estudo. Agradecemos a Louise Rousseau, coordenadora do biobanco hbp pela recuperação das amostras de tecidos e todas as informações clínicas associadas. Reconhecemos as instalações do núcleo de patologia molecular e imagem celular no CRCHUM e Michael Persch de Visiopharm para excelente assistência técnica. Financiamento: Este estudo foi apoiado por subsídios da Canadian Liver Foundation, Fonds de recherche du Québec-Santé (FRQS) AIDS e Infectious Disease Network (Réseau SIDA-MI), e da Rede Canadense de Hepatite C (CanHepC). O CanHepC é financiado por uma iniciativa conjunta dos Institutos Canadenses de Pesquisa em Saúde (CIHR) (NHC-142832) e da Agência de Saúde Pública do Canadá. M.F.M. recebeu bolsas da Université de Montréal, Bourse Gabriel Marquis, e do FRQS. T.F. recebeu bolsas de doutorado do CIHR e canHepC. S.T. ocupa a Cadeira Roger-Des-Groseillers em cirurgia oncológica hepatobiliária e pancreática, Université de Montréal.
Contribuições do autor: M.F.M. projetou, realizou experimentos e analisou dados. T.F. projetou experimentos. A.C.B. forneceu orientação técnica. A G.S. realizou toda a avaliação patológica do sujeito do estudo e forneceu informações sobre todos os aspectos patológicos. A L.M. realizou a coloração de H&E, otimizou e realizou a aquisição de imagens. M.N.A. realizou a mancha PSR e forneceu informações técnicas valiosas. N.B. contribuiu para a análise da imagem. S.T. é o principal investigador do biobanco HBP e é responsável por supervisionar a operação geral do biobanco. Ele também forneceu informações inestimáveis sobre todos os aspectos do projeto e suas implicações clínicas. M.F.M., T.F., e N.H.S. conceituaram e projetaram o estudo. N.H.S. supervisionou o trabalho e obteve financiamento. M.F.M., T.F., A.C-B e N.H.S. escreveram o manuscrito. Todos os autores revisaram e aprovaram o manuscrito.
Materials
| Name | Company | Catalog Number | Comments |
| Antigen Retrieval Solution: Sodium Citrate Buffer (10 mM Sodium Citrate, 0.05% v/v Tween 20, pH 6.0) | |||
| Blocking Solution: 1 % BSA, 10 % filtered human serum, 10 % filtered donkey serum, 0.1 % Tween 20, and 0.3% Triton in PBS | |||
| Bovine serum albumin (BSA) | Multicell | 800-095-EG | |
| Coplin jars (EASYDIP SLIDE STAINING SYSTEM) | Newcomersupply | 5300KIT | |
| Cover slides | Fisherbrand | 12-545E 22*50 | |
| Direct Red 80 | Sigma Aldrich | 365548 | |
| Donkey Serum | Sigma Aldrich | D9663 | |
| Ethanol 100% | |||
| Electric pressure cooker | Salton | ||
| Eosin | Leica Biosystems | 3801600 | CAUTION, eye irritation |
| Fast Green FCF | Sigma Aldrich | F7252 | CAUTION, harmful by inhalation, ingestion and skin absortion |
| FFPE section (4μm) slides | |||
| Glycine 0,1 M in PBS | |||
| Hematoxylin Stain Solution, Gil 1. Formulation, Regular Strength | Ricca Chemical Company | 3535-32 | |
| Holder (EasyDip Staining Jar Holder) | Newcomersupply | 5300RK | |
| Human Serum | Gemini | 22210 | |
| Humidity chamber | Millipore Sigma | Z670138-1EA | |
| Pap pen | abcam | ab2601 | |
| PBS | |||
| PBS-Tween 20 (0.1% v/v) | |||
| Permount Mounting Media | Fisher Chemical | SP15-500 | |
| Picric Acid 1.3 % | Sigma Aldrich | P6744 | CAUTION, skin and eye irritation |
| Picro-Sirius Red/Fast Green solution: Fast Green 0.1 % w/v + Sirius Red 0.2 % w/v in 1,3 % picric acid solution | |||
| Primary Antibody Anti-αSMA | Mouse IgG2a 1A4 | Sigma A2547 | Dilution 1/100 |
| Primary Antibody Anti-CD34 | Mouse IgG1 HPCA1/763 | Novus Biologicals NBP2-44568 | Dilution 1/250 |
| Primary Antibody Anti-Cytokeratin 8/18 | Rabbit EP17/EP30 | Agilent IR09461-2 | Ready to use |
| Primary Antibody Anti-CD68 | Mouse KP1 | Abcam ab955 | Dilution 1/200 |
| Primary Antibody Anti-Desmin | Rabbit Polyclonal | Invitrogen PA5-16705 | Dilution 1/200 |
| Primary Antibody Anti-CD4 | Mouse N1UG0 | Affymetrix 14-2444 | Dilution 1/250 |
| Primary Antibody Anti-FoxP3 | Rabbit 1054C | R & D MAB8214 | Dilution 1/100 |
| Primary Antibody Anti-MPO | Goat Polyclonal | R & D Systems AF3667 | Dilution 1/250 |
| Primary Antibody Anti-CD3 | Rabbit SP7 | Abcam ab16669 | Dilution 1/200 |
| Primary Antibody Anti-CD8 | Mouse C8/144B | Invitrogen 14-0085-80 | Dilution 1/200 |
| Secondary Antibody Donkey anti-mouse A488 | Polyclonal | Invitrogen A-21202 | Dilution 1/500 |
| Secondary Antibody Donkey anti-Rabbit A488 | Polyclonal | Invitrogen A-21206 | Dilution 1/500 |
| Secondary Antibody Donkey anti-goat A568 | Polyclonal | Invitrogen A-11057 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit A647 | Polyclonal | Invitrogen A-31573 | Dilution 1/500 |
| Secondary Antibody Rat anti-mouse IgG1 A647 | RMG1-1 | Biolegend 406618 | Dilution 1/500 |
| Secondary Antibody Goat anti-mouse IgG2a CF568 | Polyclonal | Sigma Aldrich SAB4600315 | Dilution 1/500 |
| Secondary Antibody Donkey anti-mouse DyLight 755 | Polyclonal | Invitrogen SA5-10171 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit DyLight 755 | Polyclonal | Invitrogen SA5-10043 | Dilution 1/500 |
| SDS | BioShop | SDS001,500 | CAUTION, oral skin and eye toxicity |
| Shandon multi-program robotic slide stainer | LabX | 11384903 | |
| Shandon Xylene Substitute, | Thermo Fisher Scientific | CA89413-336 | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Shaking water bath | |||
| SlowFade Gold antifade reagent with DAPI | Invitrogen | S36938 | |
| Sodium Citrate Dihydrate | Millipore Sigma | 1545801 | CAUTION, eye irritation |
| Stripping Buffer: mix 20 ml 10% w/v SDS with 12.5 ml 0.5 M Tris-HCl (pH 6.8), and 67.5 ml ultra-pure water. Under a fume hood, add 800 uL of 2-mercapto ethanol (114,4 mM final concentration) | |||
| Triton X-100 | Sigma Aldrich | T8787-50ML | |
| Tris-HCl | BioShop | 77-86-1 | |
| Tween 20 | Fisher Scientific | BP337-500 | |
| VIS Software | Visiopharm | ||
| Whole slide scanner Olympus BX61VS | Olympus | Microscope: Olympus Slide Scanner BX61VS, 5 slides scanner, motorized stage, autofocus. Camera: Lightsource: Xcite-120. Filters: BrightLine® Sedat filter set (# LED-DA/FI/TR/Cy5-4X4M-B-000, Semrock) | |
| Xylene | Sigma Aldrich | 214736-4L | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Xylene : Ethanol solution (1:1 v/v) | |||
| 2-mercaptoethanol | Sigma | M6250 | CAUTION, harmful by ingestion, inhalation, fatal if sking absortion. Eye irritation. Use fume hood |
References
- Greten, F. R., Grivennikov, S. I. Inflammation and Cancer:Triggers, Mechanisms, and Consequences. Immunity. 51 (1), 27-41 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Binnewies, M., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine. 24 (5), 541-550 (2018).
- Taube, J. M., et al. Implications of the tumor immune microenvironment for staging and therapeutics. Modern Pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 31 (2), 214-234 (2018).
- Bindea, G., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 39 (4), 782-795 (2013).
- Galon, J., et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. The Journal of Pathology. 232 (2), 199-209 (2014).
- Finotello, F., Eduati, F. Multi-Omics Profiling of the Tumor Microenvironment: Paving the Way to Precision Immuno-Oncology. Frontiers in Oncology. 8, 430 (2018).
- Gerner, M. Y., Kastenmuller, W., Ifrim, I., Kabat, J., Germain, R. N. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 37 (2), 364-376 (2012).
- Giesen, C., et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nature Methods. 11 (4), 417-422 (2014).
- Porta Siegel, T., et al. Mass Spectrometry Imaging and Integration with Other Imaging Modalities for Greater Molecular Understanding of Biological Tissues. Molecular Imaging and Biology : MIB: the official publication of the Academy of Molecular Imaging. 20 (6), 888-901 (2018).
- Buchberger, A. R., DeLaney, K., Johnson, J., Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Analytical Chemistry. 90 (1), 240-265 (2018).
- Pirici, D., et al. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. Journal of Histochemistry and Cytochemistry. 57 (6), 567-575 (2009).
- Gendusa, R., Scalia, C. R., Buscone, S., Cattoretti, G. Elution of High-affinity (>10-9 KD) Antibodies from Tissue Sections: Clues to the Molecular Mechanism and Use in Sequential Immunostaining. Journal of Histochemistry and Cytochemistry. 62 (7), 519-531 (2014).
- van der Loos, C. M. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. Journal of Histochemistry and Cytochemistry. 56 (4), 313-328 (2008).
- Stack, E. C., Wang, C., Roman, K. A., Hoyt, C. C. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 70 (1), 46-58 (2014).
- Toth, Z. E., Mezey, E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. Journal of Histochemistry and Cytochemistry. 55 (6), 545-554 (2007).
- Robertson, D., Savage, K., Reis-Filho, J. S., Isacke, C. M. Multiple immunofluorescence labeling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell Biology. 9, 13 (2008).
- Segnani, C., et al. Histochemical Detection of Collagen Fibers by Sirius Red/Fast Green Is More Sensitive than van Gieson or Sirius Red Alone in Normal and Inflamed Rat Colon. PloS One. 10 (12), 0144630 (2015).
- Bolognesi, M. M., et al. Multiplex Staining by Sequential Immunostaining and Antibody Removal on Routine Tissue Sections. Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
