

Summary
Aquí, describimos una estrategia simple y accesible para visualizar, cuantificar y mapear células inmunitarias en secciones de tejido tumoral incrustado en parafina fija de formalina. Esta metodología combina las técnicas existentes de análisis de imágenes y digitales con el propósito de ampliar la capacidad de multiplexación y el análisis multiparámetro de ensayos de imágenes.
Abstract
El paisaje inmune del microambiente tumoral (TME) es un factor determinante en la progresión del cáncer y la respuesta a la terapia. Específicamente, la densidad y la ubicación de las células inmunitarias en el TME tienen valores diagnósticos y pronósticoimportantes. El perfil multiómico del TME ha aumentado exponencialmente nuestra comprensión de las numerosas redes celulares y moleculares que regulan la iniciación y progresión tumoral. Sin embargo, estas técnicas no proporcionan información sobre la organización espacial de las celdas o las interacciones de celdas. Se necesitan técnicas de multiplexación asequibles, accesibles y fáciles de ejecutar que permitan la resolución espacial de las células inmunitarias en las secciones de tejido para complementar las tecnologías de alto rendimiento basadas en células únicas. Aquí, describimos una estrategia que integra imágenes en serie, etiquetado secuencial y alineación de imágenes para generar diapositivas virtuales multiparámetros de secciones de tejido entero. Las diapositivas virtuales se analizan posteriormente de forma automatizada utilizando protocolos definidos por el usuario que permiten la identificación, cuantificación y mapeo de poblaciones de células de interés. El análisis de imágenes se realiza, en este caso utilizando los módulos de análisis Tissuealign, Author y HISTOmap. Presentamos un ejemplo en el que aplicamos esta estrategia con éxito a una muestra clínica, maximizando la información que se puede obtener de muestras de tejido limitadas y proporcionando una visión imparcial del TME en toda la sección de tejido.
Introduction
El desarrollo del cáncer es el resultado de un proceso de varios pasos que implica interacciones recíprocas entre las células malignas y el TME. Aparte de las células tumorales, el TME se compone de células no malignas, células estromales, poblaciones de células inmunitarias y matriz extracelular (ECM)1. La organización espacial de los diferentes componentes celulares y estructurales del tejido tumoral y el intercambio dinámico entre el cáncer y las células no cancerosas vecinas modulan en última instancia la progresión tumoral y la respuesta a la terapia2,,3,4. Se ha demostrado que la respuesta inmune en el cáncer está regulada espaciotemporalmente5,,6. Diferentes poblaciones de células inmunitarias que se infiltran en la lesión neoplásica y el tejido adyacente presentan patrones de distribución espacial distintivos y estados variados de activación y diferenciación asociados con diferentes funciones (por ejemplo, pro- versus antitumoral). Estas diferentes poblaciones inmunes y sus parámetros coevolucionan las horas extras con el tumor y los compartimentos estromales.
La aparición de tecnologías que permiten el perfilado multiómico de una sola célula ha aumentado exponencialmente nuestra comprensión de las numerosas redes celulares y moleculares que regulan la carcinogénesis y la progresión tumoral. Sin embargo, la mayoría de las herramientas analíticas de alto rendimiento basadas en células simples requieren interrupción del tejido y aislamiento de una sola célula, lo que resulta en la pérdida de información sobre la organización espacial de las células y las interacciones de células celulares7. Debido a que la ubicación y disposición de células inmunitarias específicas en el TME tienen valor diagnóstico y pronóstico, las tecnologías que permiten la resolución espacial son un complemento esencial de las técnicas de perfilado inmune basadas en células únicas.
Tradicionalmente, las técnicas de imagen como la inmunohistoquímica (IHC) y la inmunofluorescencia múltiple (mIF) se han restringido a un pequeño número de biomarcadores que se pueden visualizar simultáneamente. Esta limitación ha obstaculizado el estudio de la dinámica espaciotemporal de las células inmunitarias infiltrantes tumorales, que normalmente se definen mediante varios marcadores fenotípicos. Los recientes avances en herramientas analíticas y de imágenes han ampliado las posibilidades de multiplexación. Se han utilizado nuevas tecnologías de etiquetado basadas en anticuerpos como la histocitometría y la citometría de masas por imágenes para separar espacialmente hasta 12 y 32 biomarcadores, respectivamente8,9. La punción de espectrometría de masas, una técnica que no requiere etiquetado, tiene el potencial de crear imágenes de miles de biomarcadores simultáneamente en una sola sección10de tejido,11. Aunque estas técnicas ya han demostrado un gran potencial para disuader el paisaje inmune del tejido en el cáncer, utilizan equipos y software altamente sofisticados y costosos y no son fácilmente accesibles para la mayoría de los investigadores.
Alternativamente, la capacidad de multiplexación de IHC y mIF tradicionales se ha ampliado mediante el uso de imágenes en serie, rondas secuenciales de etiquetado e imágenes espectrales7,12,13,14,15,16. Estas técnicas generan varias imágenes a partir de la misma o de secciones de tejido serie que se pueden consolidar en diapositivas virtuales multiparámetro utilizando el software de análisis de imágenes. Como resultado, el número de marcadores que se pueden visualizar y analizar simultáneamente aumenta.
Aquí, proponemos una estrategia para el diseño racional de ensayos multiplex de tejidos utilizando reactivos disponibles comercialmente, equipos de microscopía asequibles y software fácil de usar(Figura 1). Esta metodología integra imágenes en serie, etiquetado multiplex secuencial, imágenes de tejido entero y alineación de tejidos para generar diapositivas virtuales multiparámetros que se pueden utilizar para la cuantificación automatizada y el mapeo de células inmunitarias en secciones de tejido. Usando esta estrategia, creamos una diapositiva virtual compuesta por 11 biomarcadores más dos manchas histológicas de uso frecuente: hematoxilina y eosina (H&E) y picrosirius rojo (PSR). Se identificaron, ubicaron y cuantificaron múltiples poblaciones de células inmunitarias en diferentes compartimentos tisulares y su distribución espacial se resolvió utilizando mapas de calor tisulares. Esta estrategia maximiza la información que se puede obtener de muestras clínicas limitadas y es aplicable a muestras de tejido archivado sin parafina (FFPE) fijadas en formalina, incluyendo tejido entero, biopsias de agujas de núcleo y microarrays de tejido. Proponemos esta metodología como guía útil para el diseño de ensayos personalizados para la identificación, cuantificación y mapeo de poblaciones de células inmunitarias en el TME.
Protocol
Se obtuvieron tres secciones seriales de FFPE a partir del carcinoma hepatocelular humano asociado al virus de la hepatitis B (VHB) del Centre hospitalier de l'Université de Montréal (CHUM) Hepatopancreatobiliary Cancer Database (ENhebrabilario Cancer) y Biological Specimen Repositorio (HBP Biobank). Los pacientes que participaron en este banco de tejidos proporcionaron consentimiento informado. Este estudio fue aprobado por el comité de ética institucional (Protocolo número 09.237) y realizado de conformidad con la Declaración de Helsinki.
1. Protocolo de tinción de hematoxilina y eosina (H&E)
NOTA: La tinción de H&E fue realizada por la instalación central de patología molecular del Centre de Recherches du Centre hospitalier de l'Université de Montréal (CRCHUM) utilizando la tinción de diapositivas robótica multiprograma Shandon utilizando el siguiente programa.
- Para la desparafinación, sumerja las diapositivas 3x durante 2,5 min cada una en sustituto de xileno.
ADVERTENCIA: Los sustitutos del xileno son inflamables, irritantes de la piel y dañinos si se inhalan. - Para la rehidratación, sumerja los portaobjetos en etanol 100% 3x durante 2,5 min cada uno. Lavar durante 1 min en agua destilada doble (ddH2O) para rehidratar.
- Incubar durante 1 min en hematoxilina. Lavar 3x durante 1 min cada uno en ddH2O.
- Incubar durante 5 s con eosina. Lavar 30 s con 95% de etanol. Lavar 2x durante 1 min con 100% etanol.
ADVERTENCIA: El etanol es inflamable y un irritante ocular. La eosina es un irritante ocular. - Para la deshidratación, sumerja 3 veces durante 1,5 min cada uno en el sustituto del xileno. Monte las diapositivas manualmente.
NOTA: El tiempo estimado para ejecutar esta parte del protocolo es de 30 min.
2. Protocolo de tinción de inmunofluorescencia múltiple para secciones FFPE
NOTA: Este protocolo fue adaptado de Robertson et al.17.
- Desparafinación y rehidratación
NOTA: Antes de que el etiquetado mediado por anticuerpos de secciones FFPE por IHC o mIF, la parafina debe ser removida. Si no se elimina eficientemente la parafina, se produce una tinción subóptima.- Coloque los portaobjetos de 4 mm de sección de tejido FFPE en portaobjetos de vidrio. Debajo de la campana de humos, sumerja las diapositivas en un frasco de Coplin que contenga xileno precalentado a 37oC durante 10 min.
ADVERTENCIA: El xileno es inflamable, irritante de la piel y dañino si se inhala. - Agitar manualmente las diapositivas durante 10 s cada 2 min. Repetir 1x en xileno fresco durante otros 5 min.
- En la campana química, sumerja las correderas secuencialmente durante 5 minutos en cada una de las siguientes soluciones: 1) xileno: etanol (1:1 v/v); 2) 100% etanol; 3) 70% etanol; 4) 50% etanol; 5) 30% etanol; 6) salina con fosfato (PBS).
NOTA: Mantenga las diapositivas en PBS hasta que estén listas para realizar la recuperación del antígeno. Mantenga las secciones desenceladas hidratadas en todo momento. El secado causará una unión inespecífica de anticuerpos y, por lo tanto, una alta tinción de fondo.
- Coloque los portaobjetos de 4 mm de sección de tejido FFPE en portaobjetos de vidrio. Debajo de la campana de humos, sumerja las diapositivas en un frasco de Coplin que contenga xileno precalentado a 37oC durante 10 min.
- Recuperación de antígenos inducida por calor
NOTA: Los antígenos se pueden enmascarar en el momento de la fijación de la formalina, evitando la unión de anticuerpos y, en consecuencia, la visualización. El uso de tampones y procedimientos de desenmascaramiento de antígenos restablecen parcialmente la conformación nativa de epítopos y, por lo tanto, restaura el reconocimiento de anticuerpos. El tipo de búfer de recuperación de antígenos y la duración deben optimizarse para las condiciones específicas del ensayo (por ejemplo, objetivo, anticuerpos, tejido, etc.).- Sumerja las diapositivas desparasitadas en un frasco de Coplin que contenga la solución de recuperación de antígenos (receta en Tabla de materiales).
- Coloque el frasco cerrado de Coplin en una olla a presión eléctrica con agua del grifo. El nivel del agua no debe exceder la mitad de la altura del frasco para que el agua no se mezcle con la solución de recuperación de antígenos.
- Cierre la tapa y la válvula de presión de la olla. Seleccione alta presión durante 10 min y comience. Cuando haya terminado, desenchufe la olla, suelte la presión, abra la tapa y mantenga el frasco dentro de la olla durante 30 minutos, permitiendo que los portaobjetos se enfríen.
- Bloqueo de enlaces inespecíficos
- Transfiera el bastidor con las diapositivas a un frasco de Coplin lleno de PBS. Enjuague el búfer de recuperación de antígenos con PBS 2x durante 5 min cada uno.
- Rodear las secciones de tejido con una pluma PAP para crear una barrera hidrófoba. Sumerja las diapositivas en un frasco de Coplin que contenga glicina de 0,1 M en PBS. Incubar durante 15 min a temperatura ambiente (RT).
NOTA: La glicina satura los grupos de aldehídos generados durante la recuperación del antígeno. Estos grupos podrían unir los anticuerpos primarios y secundarios de manera inespecífica. - Enjuague la solución de glicina lavando 2x con PBS durante 5 min. Coloque los portaobjetos en una cámara de humedad y agregue suficiente solución de bloqueo para cubrir todas las secciones de tejido. Evite desbordarse la barrera hidrofóbica. Incubar durante 30 min a RT.
NOTA: La receta de la solución de bloqueo se puede encontrar en la Tabla de materiales. La solución de bloqueo debe contener una proteína (por ejemplo, BSA) para bloquear sitios de unión inespecíficos. También puede incorporar detergentes como Triton X-100 o Tween 20 que reducen las interacciones hidrofóbicas entre anticuerpos y dianas tisulares, haciendo que el reconocimiento de antígenos sea más selectivo. La adición de 10% de suero total de la especie de donde proviene el tejido bloquearía los receptores Fc, y por lo tanto reduciría la unión inespecífica de anticuerpos. Finalmente, la adición del 10% del suero de la especie en la que se plantearon los anticuerpos secundarios minimizaría la unión directa inespecífica de anticuerpos secundarios a la sección tisular.
- Etiquetado de inmunofluorescencia
- Enjuague con PBS-Tween (0,1% v/v) 2 veces durante 5 minutos cada uno y vuelva a colocar los portaobjetos en la cámara de humedad.
- Añadir el cóctel de anticuerpos primarios resuspendidos en solución de bloqueo. Incubar durante la noche a 4oC. Los anticuerpos primarios y secundarios utilizados para este estudio se enumeran en la Tabla de materiales.
NOTA: El cóctel de anticuerpos primarios debe contener anticuerpos criados en diferentes especies, o de la misma especie pero de diferentes isotipos. Para obtener una lista de los pares de anticuerpos primarios y secundarios utilizados en este estudio, consulte la Tabla 2. Los detalles de todos los anticuerpos utilizados se encuentran en la Tabla de Materiales y en la Tabla 2. - Enjuague con PBS-Tween (0,1% v/v) 3 veces durante 5 min y vuelva a colocar los portaobjetos en la cámara de humedad. En la oscuridad, añadir el cóctel de anticuerpos secundarios e incubar durante 1 h a RT.
NOTA: Cuando los anticuerpos primarios son de diferentes especies, los anticuerpos secundarios deben seleccionarse de modo que cada uno de ellos solo se une a uno de los anticuerpos primarios y no entre sí. Esto se logra comúnmente mediante el uso de anticuerpos secundarios todos criados en la misma especie, siempre y cuando esta especie difiere de la especie donde se generaron los anticuerpos primarios. En los casos en que los anticuerpos primarios se criaron en la misma especie pero tienen diferentes isotipos, se deben utilizar anticuerpos secundarios específicos de isotipos. - Enjuague con PBS-Tween (0,1% v/v) 3 veces durante 5 min cada uno. Enjuague con ddH2O. Retire el exceso de líquido y monte en los soportes de montaje con DAPI. El volumen utilizado depende del tamaño de la sección. Por lo general, 40 l es suficiente para cubrir la superficie de una diapositiva de microscopía regular.
- Coloque el portaobjetos de la cubierta sobre la sección y apriete suavemente el exceso de medios de montaje evitando la formación de burbujas. Dejar secar las diapositivas durante 20 minutos a RT en la oscuridad y almacenar a 4 oC hasta que estén listas para su adquisición.
- Adquirir imágenes para todos los canales utilizando todo el escáner de diapositivas (consulte Tabla de materiales).
NOTA: Los anticuerpos fueron validados utilizando el tejido del carcinoma hepatocelular humano como un control positivo. Para cada anticuerpo primario, se teñieron tres secciones en serie con anticuerpos primarios, control de isotipos o solo solución de bloqueo respectivamente sin variación en el resto del protocolo de tinción. Las imágenes adquiridas se compararon para establecer la especificidad de la tinción. La tinción se consideró específica cuando la señal en la sección incubada con anticuerpo primario tenía el patrón esperado y era fácilmente distinguible del fondo. Los anticuerpos primarios que dan una señal de fondo alta o componentes de tejido de etiquetado en el isotipo y no se consideraron secciones de anticuerpos primarios inespecíficos. El tiempo estimado para completar esta parte del protocolo es de 2 días. Los controles requeridos incluyen: (1) Control de isotipo para establecer la contribución de la unión inespecífica del anticuerpo primario a la señal de fondo. Una sección se tiñe de la misma manera que los otros tejidos de la muestra, excepto que se incuba con un anticuerpo con el mismo isotipo y origen del anticuerpo primario pero específico para un objetivo que está ausente en la sección del tejido. Si no se dispone del anticuerpo de control de isotipo apropiado, puede sustituirse por el IgG total de la misma especie en la que se planteó el anticuerpo primario; (2) No hay control de anticuerpos primarios (es decir, control negativo) para establecer la especificidad de la tinción y estimar la contribución de la unión inespecífica de anticuerpos secundarios a la señal de fondo. En este caso, la sección de control se tiñe de la misma manera que las otras secciones, excepto que no se añade ningún anticuerpo primario; (3) Control positivo para establecer que la tinción funciona. En este caso, la tinción se realiza en una sección tisular que se sabe que expresa el marcador reconocido por el anticuerpo primario.
3. Protocolo de tinción verde pico-sirius (PSR)/verde rápido
NOTA: El objetivo de esta tinción es visualizar colágenos fibrilares I y III en las secciones de tejido FFPE. Este protocolo fue adaptado de Segnani et al.18. Todos los pasos se realizan en una campana química.
- Realizar la desparafinación y rehidratación de secciones tisulares similares al protocolo de tinción de inmunofluorescencia múltiple para las secciones FFPE (sección 2.1).
NOTA: Si la sección que se va a manchar se ha utilizado previamente para el etiquetado de inmunofluorescencia y la parafina ya se ha eliminado, los pasos de desfinación-rehidratación son útiles para retirar el soporte de montaje. DAPI no se elimina mediante este procedimiento, pero no interfiere perceptiblemente con la tinción de PSR. - Sumerja las diapositivas en un frasco que contenga la solución de color rojo/verde rápido picro-sirius (receta en Tabla de Materiales)e incubar durante 30 min a RT (más de 30 min da como resultado la tinción inespecífica de los núcleos de los hepatocitos).
- Lave los portaobjetos rápidamente en ddH2O (5 inmersiones). Luego, lavar rápidamente en etanol 100% (5 inmersiones). Lavado durante 30 s en etanol de xileno-100% (1:1 v/v). Lavar durante 30 s en xileno. Montar con medios de montaje (ver Tabla de Materiales)antes de que el xileno se haya evaporado totalmente (esto ayuda con el montaje).
NOTA: El tiempo estimado para ejecutar esta parte del protocolo es de 1 h.
4. Elución de anticuerpos de secciones tisulares
NOTA: Para reutilizar secciones de tejido en ensayos de etiquetado secuencial, se requiere la eliminación completa de anticuerpos primarios y secundarios. Los anticuerpos encuadernados fueron despojados como se describió anteriormente13.
Precalentar un baño de agua a 56 oC. Coloque las secciones dentro de un frasco que contenga tampón de desmontaje (receta en Tabla de materiales),cierre la tapa y séllela con cinta de película de parafina para evitar fugas durante el temblor.
- Poner el frasco dentro del baño de agua e incubar durante 30 minutos con agitación.
- Lavar 4veces durante 15 min cada uno en ddH2O en RT. Enjuague con PBS-Tween (0,1% v/v).
- Mantenga las secciones hidratadas en PBS-Tween o agua hasta que estén listas para volver a sondear la sección con la segunda ronda de anticuerpos primarios.
NOTA: El tiempo estimado para ejecutar esta parte del protocolo es de 2 h. - Verifique la eficiencia del procedimiento de elución de anticuerpos.
NOTA: Antes de utilizar el protocolo de elución de anticuerpos en un ensayo de etiquetado secuencial, se debe verificar la eficacia de la eliminación de anticuerpos primarios y secundarios.- Realizar la tinción y la adquisición de imágenes de una sección con un par de anticuerpos primario-secundario determinado de interés como se indica en el protocolo de tinción de inmunofluorescencia multiplex para secciones FFPE (secciones 2.1–2.4.6).
- Tras la adquisición de la imagen, realice la elución de los complejos de anticuerpos primarios secundarios unidos a tejidos, como se indica en las secciones 4.1–4.3.
- Incubar la sección con el mismo anticuerpo secundario y las mismas condiciones utilizadas en el paso 2.4.3.
- Realice los pasos de lavado, montaje y adquisición de imágenes como se indica en 2.4.4–2.4.6.
- Compare las imágenes de lado a lado adquiridas antes y después del desmontaje para establecer si la señal específica ha desaparecido o no.
NOTA: La comparación de imágenes antes y después de la eliminación de anticuerpos validará la eficiencia del procedimiento de elución. Sin embargo, es normal ver un aumento en la señal de fondo en todos los canales, así como la difusión de DAPI. Esto limita el número de rondas de desmontaje que se pueden ejecutar en la misma sección de tejido. Tres rondas de desmontaje parecen ser el máximo.
5. Adquisición de imágenes
- Genere imágenes utilizando un escáner de diapositivas completo.
- Utilice una lente objetivo de 20x 0.75NA y una resolución de 0.3225 m/píxel.
6. Análisis de imágenes
NOTA: El método descrito aquí hace referencia al ejemplo actual. Consulte la Tabla 1 y el texto para adaptarse a otras muestras específicas.
- Realice la alineación de tejidos utilizando el módulo Tissualign del software de análisis de imágenes (VIS en este protocolo, consulte Tabla de materiales).
- Abra el software de análisis de imágenes y haga clic en la pestaña Alineación de tejidos.
- Importe las imágenes que se van a alinear en la bandeja de diapositivas yendo a Archivo . Base de datos y seleccione la primera imagen que desea alinear. Vuelva a la pestaña Alineación de tejidos y cargue la imagen haciendo clic en el botón Cargar de la bandeja de diapositivas. La imagen aparecerá en la bandeja de diapositivas y en el espacio de trabajo.
NOTA: Solo se debe cargar la pila de interés en la bandeja deslizante. - Repita el paso 6.1.2 para todas las imágenes en el orden de alinearse, cargándolas una por una. Una vez que todas las imágenes de interés se cargan en la bandeja de diapositivas, proceda a vincular las imágenes pulsando Siguiente en los pasos de flujo de trabajo de la cinta de opciones.
- A continuación, arrastre y suelte la segunda imagen encima de la primera imagen. La primera y la segunda imagen ahora están vinculadas. Repita este paso para que las otras imágenes se alineen, una por una, de manera ordenada. El nombre de la primera imagen cambiará, lo que indica que se ha vinculado a las otras imágenes. Simultáneamente, las imágenes vinculadas se mostrarán en el espacio de trabajo situado a la derecha de la bandeja de diapositivas.
- En este punto, alinee las imágenes mediante alineación automática, alineación semiautomática o alineación manual. Siempre es preferible probar primero la alineación automática. Para la alineación automática, pulse el botón Siguiente en los pasos del flujo de trabajo (paso 3) de la cinta de opciones.
- Revise la alineación automática navegando por diferentes ubicaciones del tejido y verificando visualmente que las estructuras correspondientes en diferentes imágenes están dispuestas de la misma manera en las dos dimensiones de la imagen.
- Si el resultado de la alineación automática no es satisfactorio, mejore con pines (utilice un mínimo de tres pines por imagen) que indiquen características de tejido homóloga en las imágenes enlazadas. Una vez que los pines se colocan en ubicaciones homólogas en las imágenes vinculadas, el usuario tiene dos opciones: alineación semiautomática o alineación manual. Para la alineación semiautomática, haga clic en el botón Alinear automáticamente en función de los puntos actuales de la cinta de opciones. Para la alineación manual, haga clic en el botón Aplicar pasadores en la cinta de opciones.
- Cuando esté satisfecho con la alineación, haga clic en el botón Siguiente en los pasos del flujo de trabajo y guarde la imagen compuesta en la base de datos.
NOTA: Alinear seis diapositivas que abarcan 11 marcadores más las imágenes de H&E y PSR tardaron 15 minutos en el análisis presentado.
- Realice la detección de tejidos utilizando el protocolo definido por el usuario Analysis Protocol Package 1 (APP 1, Tabla 1).
- Abra el módulo Análisis de imagen del software haciendo clic en la pestaña Análisis de imagen de la cinta de opciones.
- Importe la imagen compuesta (alineada) yendo a Archivo . Base de datos y seleccionar la imagen de interés y hacer clic de nuevo en la pestaña Análisis de imagen.
- Abra el cuadro de diálogo de selección de aplicaciones haciendo clic en el icono Abrir APP y seleccione qué paquete de protocolo de análisis (APP) utilizar. En este caso seleccione APP 1 para la detección de tejidos.
- Una vez que se abre la aplicación 1, confirme que APP1 funciona correctamente yendo a una ubicación de tejido seleccionada y haciendo clic en el botón de vista previa. Si los resultados son satisfactorios, vaya al siguiente paso.
- Haga clic para ejecutar APP 1 y procesar la imagen utilizando la APP seleccionada.
- Exporte los datos (por ejemplo, imágenes, mediciones, etc.) cuando el análisis se realice haciendo clic en Archivo/Exportar.
NOTA: APP 1 crea una región de interés (ROI) delineando el tejido (roiú) y calcula el área del tejido. - Guarde la imagen modificada con el ROI recién creado yendo a Archivo . Ahorra.
NOTA: La detección del tejido y la creación de un ROI con APP 1 en el ejemplo proporcionado tardaron 5 minutos en la estación de análisis de imágenes descrita. El área del tejido procesado fue de 3,2 cm2.
- Realizar la segmentación de tejidos en Stroma y Parenchyma usando APP 2 (Tabla 1).
NOTA: APP 2 funciona en el tejido DE ROI predefinido. APP 2 segmenta el tejido en los ROIs Stroma y Parenchyma.- Abra el módulo Análisis de imagen haciendo clic en la pestaña Análisis de imagen de la cinta de opciones.
- Importe la imagen que contiene el tejido de ROI yendo a Archivo . Base de datos y selección de la imagen guardada en el paso 6.2.7. Vuelva a la pestaña Análisis de imagen y cargue la imagen haciendo clic en el botón Cargar de la bandeja de diapositivas. La imagen aparecerá en la bandeja de diapositivas y en el espacio de trabajo.
- Abra APP 2 utilizando el cuadro de diálogo de selección de APP como en 6.2.3.
- Vista previa de APP 2 mediante el procesamiento en un campo de visión seleccionado. Si los resultados son satisfactorios, ejecute APP 2 en la imagen completa haciendo clic en el botón Ejecutar. Como salida de APP 2, el tejido ROI se segmenta en los ROI Stroma y Parenchyma y sus respectivas áreas determinadas. Resultados de exportación como en 6.2.6. Guarde la imagen modificada como en 6.2.7.
NOTA: La segmentación del tejido en Stroma y Parenchyma utilizando APP 2 tomó 4 horas en la estación de análisis presentada. El área del tejido procesado fue de 3,2 cm2.
- Identifique y cuantifique las células FoxP3hiCD4+ utilizando el protocolo definido por el usuario APP 3 (Tabla 1).
NOTA: APP 3 funciona en los ROIs Stroma y Parenchyma predefinidos.- Abra el módulo Análisis de imagen e importe la imagen que contiene los ROIs Stroma y Parenchyma como en 6.3.1 y 6.3.2. Abra APP 3 utilizando el cuadro de diálogo de selección de APP como en 6.2.3.
- Previsualiza el procesamiento de APP 3 en un campo de visión seleccionado enriquecido en las celdas FoxP3hiCD4+. Si los resultados son satisfactorios, ejecute APP 3 en la imagen completa. Como salida de APP 3, todos los objetos individuales FoxP3hiCD4+ serán etiquetados y sus coordenadas de tejido almacenadas. Se determinarán las densidades de los objetos FoxP3hiCD4+ en los ROIs Stroma y Parenchyma. Exporte los resultados como en 6.2.6.
- Realizar el calentamiento de tejido de FoxP3hiCD4+ objetos etiquetados.
- Abra el protocolo definido por el usuario FoxP3hiCD4+ MAP utilizando el cuadro de diálogo de selección de APP como en 6.2.3.
NOTA: FoxP3hiCD4+ MAP utiliza las coordenadas de los objetos etiquetados FoxP3hiCD4+ para generar mapas de calor de densidad. Identificar y contar los objetos etiquetados FoxP3hiCD4+ usando APP 3 tomó 25 minutos en la estación de análisis de imágenes descrita. El área del tejido procesado fue de 3,2 cm2. - Ejecute FoxP3hiCD4+ MAP pulsando el botón Ejecutar. Exportar el mapa de calor de tejidohaciendo archivo . Exportar ? Zona de trabajo.
NOTA: La asignación de objetos etiquetados FoxP3hiCD4+ utilizando FoxP3hiCD4+ MAP tomó 5 minutos en la estación de análisis de imágenes descrita.
- Abra el protocolo definido por el usuario FoxP3hiCD4+ MAP utilizando el cuadro de diálogo de selección de APP como en 6.2.3.
- Identifique y cuantifique los objetos CD8+, CD68+, MPO+, SMA y CD34 + utilizando los protocolos definidos por el usuario APP 4, APP5, APP6, APP7 y APP 8, respectivamente(Tabla 1) como se hace en la sección 6.4 a 6.4.3.2 cargando la APP de interés en cada caso.
NOTA: Las APPs 4 a 8 funcionan en los ROIs predefinidos Stroma y Parenchyma.
Representative Results
Visión general de la estrategia para visualizar, cuantificar y mapear las poblaciones celulares de interés en el TME
Para cuantificar las poblaciones celulares de interés (COI) en diferentes compartimentos tisulares (TCs) y para caracterizar su organización espacial, diseñamos un flujo de trabajo que integra técnicas asequibles y fáciles de usar y maximiza la información posicional que se puede obtener de especímenes clínicos preciosos FFPE(Figura 1). En primer lugar, se teñieron secciones de FFPE de tejido entero en serie para la visualización de COI (por ejemplo, células inmunitarias) y TCs (por ejemplo, estroma versus parénquima)(Figura 1,paso 1). El número de secciones consecutivas que deben mancharse debe mantenerse al mínimo que permita la visualización de las células de interés o las características tisulares necesarias para abordar la cuestión de la investigación. Cuanto menor sea el número de secciones seriales, mayor será el parecido de la arquitectura tisular y la concordancia en secciones contiguas. Además, la capacidad de multiplexación se puede ampliar mediante la reutilización de secciones manchadas fluorescentes a través de técnicas de desmontaje y reprobing19.
Una vez realizados los pasos de tinción, se utilizó un escáner de diapositivas completo para digitalizar las imágenes. Las imágenes adquiridas a partir de secciones en serie se alinearon y consolidaron en una diapositiva multiplexación virtual de forma automatizada(Figura 1,sección 2). A continuación, se delineó un ROI para el tejido con un protocolo definido por el usuario que identificaba píxeles asociados al tejido (TAP)(Figura 1, paso 3). Posteriormente, el tejido ROI se segmentó en CI definidas como ROI adicionales. (Figura1, paso 4). A continuación, los protocolos definidos por el usuario detectaron y cuantificaron los COI en diferentes CI(figura 1,paso 5). Finalmente, se generaron mapas de calor tisulares de COI basados en sus densidades y sus coordenadas tisulares(Figura 1,paso 6).
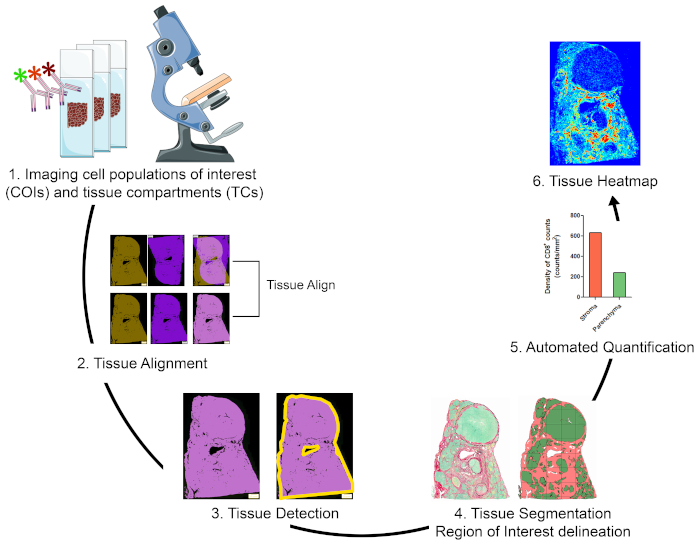
Figura 1: Representación esquemática de la estrategia para visualizar, cuantificar y mapear células inmunitarias en el TME. (1) Se tiñó secciones de tejido entero en serie para etiquetar COI y TCs. Las secciones de tejido entero manchado se digitalizaron utilizando un escáner de diapositivas completo. (2) Las imágenes adquiridas en secciones en serie se vincularon, alinearon y coregistraron de forma automatizada utilizando un módulo de análisis de Tissuealign. Se generó una imagen compuesta a partir de la alineación de alta precisión de imágenes individuales. (3) Se utilizó un protocolo definido por el usuario para la detección automatizada de píxeles asociados a tejidos (SAP) en la imagen compuesta. (4) El tejido se segmentó en TCs (por ejemplo, estroma y parénquima) definidos como ROIs. (5) Se utilizaron protocolos definidos por el usuario para la detección y cuantificación automatizada de COI en diferentes CI. (6) Se generaron mapas de calor de tejidos de COI. Haga clic aquí para ver una versión más grande de esta figura.
COI y TCs de imágenes
Tres secciones seriales de tejido entero FFPE de tumor resecado de un sujeto con carcinoma hepatocelular asociado al VHB se teñieron en una o más rondas de tinción como en la Figura 2A. La sección I se mantuvo con H&E para mostrar la arquitectura del tejido, la morfología celular y para determinar parámetros clínicamente relevantes como el tipo de neoplasia maligna, el grado tumoral y la evaluación general de la infiltración inmunitaria(Figura 2C). En la sección II contigua, se utilizaron dos rondas de mIF para etiquetar células parénquimales hepáticas y no parénquimales(Figura 2A). En la primera ronda, los vasos normales y tumorales se visualizaron utilizando la tinción CD34 de células endoteliales. Además, las células epiteliales (hepatocitos y colangiocitos) se identificaron utilizando citoqueratina 8/18, y las células estelares hepáticas activadas fibrogénicas se identificaron como células de actina muscular alfa suave (AMA+)(Figura 2C). Después de la adquisición de la imagen, las secciones de tejido fueron despojadas y reprobadas con anticuerpos contra macrófagos (CD68) y miofibroblastos (desmin). Para caracterizar mejor el infiltrado inmune tumoral, la sección iii serial adyacente se tiñó usando dos rondas de mIF para los marcadores celulares CD3, CD4, CD8, caja de cabezal de horquilla P3 (FoxP3) y mieloperoxidasa (MPO). En todos los casos, la DAPI se utilizó como contramancha nuclear. Finalmente, la sección III se tiñó con la mancha PSR y se contramancha con verde rápido para visualizar el colágeno fibrilar y segmentar el tejido en estroma y parénquima(Figura 2C).
Se utilizó un escáner de diapositivas completo equipado con una lente objetivo 20X para digitalizar secciones manchadas y crear diapositivas virtuales. Se fusionaron seis imágenes de las tres secciones seriales(Figura 2B)y las diapositivas virtuales se analizaron posteriormente utilizando el software VIS de acuerdo con la representación esquemática de la Figura 1.
Análisis de imágenes
El análisis de imagen comprendía cinco pasos: 1) alineación de tejidos; 2) detección de tejido; 3) segmentación de tejidos; 4) cuantificación automatizada de los COI; y 5) mapeo de calor tisular. Todos los protocolos para el análisis de imágenes se desarrollaron utilizando el módulo Autor del software de análisis de imágenes y se denominan en el texto como APP.
Alineación de tejidos
Se cargaron seis diapositivas virtuales de tres secciones seriales, que abarcan 11 marcadores más las manchas de H&E y PSR, en el módulo Tissualign del software de análisis de imágenes. A continuación, las imágenes se vincularon, alinearon y registraron de forma automatizada, generando una imagen compuesta virtual de 11 plex más H&E y PSR, que contiene todas las capas de las imágenes individuales(Figuras 2A–C). La alineación fue precisa en el caso de imágenes procedentes de secciones seriales adyacentes, mostrando las estructuras de tejido correspondientes posicionadas y dispuestas de manera homóloga tras la alineación(Figura 2C y Figura S1A). Además, la alineación fue precisa a nivel de celda individual para las imágenes procedentes de la misma sección(Figura S1B). El tiempo de alineación automática depende del número, tamaño, complejidad y similitud de las imágenes que se van a alinear. La alineación de las seis diapositivas virtuales mencionadas anteriormente tomó 15 minutos en nuestra estación VIS.

Figura 2: Manchado de secciones de tejido serial y alineación de la imagen. (A) Resumen de las manchas realizadas en tres secciones seriales para la visualización de COI y TCs. Para las secciones II y III, los tejidos fueron despojados y reprobidos con un segundo cóctel de anticuerpos. (B) Descripción general de seis imágenes de tejido entero individuales antes y después de la alineación del tejido (izquierda y derecha, respectivamente). Barra de escala a 3.500 m. (C) Vista ampliada de imágenes alineadas. Barra de escala a 80 m. Por favor, haga clic aquí para ver una versión más grande de esta figura.
Detección de tejidos
Una vez que las imágenes estaban vinculadas y alineadas, buscamos identificar los TAP(Figura 3A). Para diseñar una APP para la detección automatizada de TAPs (APP 1, Tabla 1), aprovechamos dos propiedades que diferencian los TAP de los píxeles no asociados al tejido. En primer lugar, la señal DAPI (banda azul) está restringida a los núcleos, que se encuentran exclusivamente en el tejido, lo que significa que todos los píxeles DAPI+ son un subconjunto de TAP. En segundo lugar, los TAP tienen una mayor señal de autofluorescencia en las bandas verde y amarilla en comparación con los píxeles no asociados con el tejido. En consecuencia, desarrollamos APP 1 para la detección de tejidos(Tabla 1),que detecta los TAPs basados en la señal de línea de base en estos canales utilizando técnicas de umbral simples. Los umbrales para las bandas azul, verde y amarilla se establecieron de modo que los TPA tuvieran valores de intensidad de fondo por encima de los umbrales, mientras que los píxeles no asociados con el tejido tenían valores por debajo. APP 1 para la detección de tejidos se aplicó a la imagen IIA, que contiene capas en los canales azul, verde y amarillo(Figura 3A). Como salidas de APP 1, se estableció una máscara verde brillante enlaciendo encima de los TAP, y un ROI llamado "Tissue" fue delineado (salida, Figura 3A). Además, el área del tejido se determinó como una variable de salida cuantitativa. Debido a que APP 1 no incorpora los píxeles no asociados con el tejido en el tejido ROI, fueron excluidos del análisis posterior basado en este ROI(Figura 3A). La precisión de app 1 en la identificación de los TAP se muestra en la Figura 3A.
Segmentación y delineación de tejidos para las empresas de libre seguridad
A continuación, procedimos a definir diferentes compartimentos dentro del tejido ROI mediante la segmentación del tejido en estroma versus parénquima. Usamos la imagen teñida de PSR (IIIC, Figura 2C),donde el estroma se puede definir como el área asociada con la deposición de colágenos fibrilares (banda roja), el parénquima como el área donde los colágenos fibrilares están ausentes, y prevalecen los tintes de contramancha verde rápido (banda verde)(Figura 3B). Creamos APP 2 (Tabla 1) para delimitar digitalmente los TCs Stroma y Parenchyma. Esta APP funciona en el tejido DE ROI predefinido (salida, Figura 3A)y utiliza áreas representativas de estroma y parénquima para entrenar la herramienta Clasificadora integrada en el módulo Análisis de imagen. El clasificador entrenado asigna los píxeles a un estroma o a una etiqueta de parénquima (salmón y verde, respectivamente, Figura 3B). Tras la clasificación de los píxeles, APP 2 ejecutó operaciones morfológicas destinadas a definir los ROIs Stroma y Parenchyma(Figura 3B y Tabla 1). El rendimiento de APP 2 en la clasificación de píxeles y la generación de los RESPECTIVOs ROI se muestra en la Figura 3B. Además, APP 2 cuantifica el área del estroma y el parénquima. Finalmente, aunque la segmentación se realiza usando la sección manchada PSR, las regiones de estroma y parénquima esbozadas se pueden transferir a cualquier imagen alineada con la imagen PSR.

Figura 3: Detección/segmentación automatizada de tejidos y generación de ROI respectivos. (A) La imagen IIA se utilizó para identificar los TPA (imagen izquierda, barra de escala a 6.000 m). Se asignó una máscara verde brillante a los TAP utilizando APP 1 (Tabla 1) generando un ROI llamado Tejido (salida 1). Correcto, el retén muestra una vista ampliada que demuestra la precisión de la aplicación 1 en la detección de TAPs. Barra de escala a 350 m. (B) El tejido ROI (salida 1) se segmenta en estroma y parénquima utilizando APP 2. La imagen de la izquierda muestra una vista del tejido ROI segmentado en estroma ROI (salmón) y parénquima de ROI (verde). Barra de escala a 4.500 m. A la derecha, las vistas ampliadas de la inserción de ROI Tissue, la tinción PSR original (imagen IIIC) y el trote y parénquima de ROI. Barra de escala a 250 m. Haga clic aquí para ver una versión más grande de esta figura.
Cuantificación automatizada de COI
A continuación, procedimos a identificar, localizar y cuantificar los COI en los ROIs Stroma y Parenchyma. Las APLICACIONES 3 a 8(Tabla 1) se crearon para localizar y contar las siguientes COI: células CD4+FoxP3+, CD8+, CD68+, MPO+, SMA+ y CD34+, respectivamente. APP 3 fue diseñado para localizar y contar células CD4+FoxP3+ (imagen IIIA, Figura 2C)como marcadores suplentes de células T reguladoras (Tregs). Este protocolo detecta la colocación de la señal desde el factor de transcripción nuclear FoxP3 (banda roja) y el colorante de etiquetado de ADN DAPI (banda azul). Dado que las células T activadas recientemente regulan FoxP3, para enriquecer para Tregs establecemos umbrales para preseleccionar solo celdas FoxP3+ brillantes (FoxP3hi). A continuación, de todas las célulasde hi dAPI+FoxP3 preseleccionadas, solo las que estaban rodeadas por señales CD4 en forma de anillo brillante (banda verde) fueron etiquetadas y contadas como células FoxP3hiCD4+ (etiqueta rosa, Figura 4A). La densidad de las células FoxP3hiCD4+ en los ROIs Stroma y Parenchyma se determinó como variables de salida cuantitativas de APP 3(Figura 4A).
Del mismo modo, las APPs 4 a 6 fueron diseñadas para la detección de células CD8+, CD68+ y MPO+. Estas API comparten el mismo diseño de línea base para detectar y cuantificar LAS COI. Específicamente, los COI se identifican en función de la intensidad de la señal del biomarcador específico de la población celular y, a continuación, se ejecutan varios pasos morfológicos de postprocesamiento para delinear células individuales (Tabla 1). Las células individuales o COIs se etiquetan, se cuentan y sus coordenadas tisulares se registran. Las APPs 4 a 6 también determinan la densidad de los COI en los ROI Stroma y Parenchyma(Figura 4B–D).
La calidad de nuestra tinción DAPI no fue lo suficientemente buena para integrar la segmentación de núcleos en las APPs 3 a 6, por lo que no podemos garantizar que todos los objetos etiquetados individualmente sean células individuales. Por esta razón, expresamos la densidad de celdas en recuentos de objetos etiquetados/mm2 (Figura 4). Sin embargo, los agregados de celdas se separaron con éxito en celdas individuales en los pasos de postprocesamiento integrados en las APLICACIONES 3 a 6, y una inspección visual exhaustiva mostró que la mayoría de los objetos etiquetados correspondían a celdas individuales.
Para detectar el área de SMA+ y CD34+, desarrollamos apps 7 y 8, respectivamente(Tabla 1). Ambas APP detectan la señal específica basada en umbrales y determinan el porcentaje de área positiva en los ROI Stroma y Parenchyma(Figura 4E–F).
Una de las posibilidades más interesantes de generar diapositivas multiplexvirtuales virtuales es el análisis de la expresión de colocalización. Generamos APP 10 para detectar la colocalización entre la AMA y la desmina, dos marcadores co-expresados por miofibroblastos en el hígado. APP 10 utiliza umbrales para encontrar píxeles positivos para la aME, la desmina y la AIMA más la desmina(Tabla 1). Como variables de salida cuantitativas, APP 10 determina el área de SMA+, el área desmin+ y el área de expresión colocalizada de estos dos marcadores(Figura S3).

Figura 4: Identificación y cuantificación de los COI en el estroma y el parénquima. (A–F) Detección y cuantificación automatizadas de los COI CD4+FoxP3+, CD8+, CD68+, MPO+, SMA+ y CD34+ en los ROI Stroma y Parenchyma utilizando los protocolos 3, 4, 5, 6, 7 y 8, respectivamente (Tabla 1). A la izquierda se muestran las imágenes originales, en el centro las imágenes procesadas y a la derecha las cuantificaciones. Para las figuras 4A–D, barra de escala a 40 m. Para las figuras 4E y F, barra de escala de 350 m. Haga clic aquí para ver una versión más grande de esta figura.
Como alternativa a la cuantificación de los COI en los TCs Stroma y Parenchyma, determinamos la densidad de las células inmunitarias en los diferentes nódulos malignos denominados 1 a 4(Figura 5A, He I). El ROI para cada nódulo se delineó manualmente como se indica en la Figura 5A. Las firmas inmunitarias de tejido distintivo caracterizaron cada nódulo, revelando aún más la heterogeneidad intrínseca del TME.
Mapas de calor de tejido
Como se mencionó anteriormente, las APPs 3 a 8 almacenan las coordenadas tisulares de cada objeto etiquetado individualmente. Esta característica permite la generación automatizada de mapas de tejido donde las regiones de alta densidad de una población celular determinada se muestran como puntos calientes (rojo) y regiones con densidad relativamente baja como puntos fríos (azul oscuro). A los valores de densidad intermedia se les asignan colores según la escala de color que se muestra en la Figura 5. Los mapas de calor de tejido fueron generados por apps que dividieron las imágenes en círculos de 50 m de diámetro y asignaron un color de acuerdo con la densidad relativa de un COI dado dentro del círculo. Como se muestra en la Figura 5B–G, los patrones de posicionamiento y la distribución de intensidad de los diferentes COI en el TME fueron bastante variados. Además, a nivel de nódulos individuales, la disposición de diferentes poblaciones en el área tisular fue única(Figura S2A–C). Para proporcionar un ejemplo del poder de esta técnica y visualizar la organización espacial de puntos calientes de diferentes poblaciones en el mismo nódulo, los puntos calientes de tipos de celda individuales se extrajeron manualmente y se asignaron juntos en el contorno del nódulo 2(Figura S2, Figura Dy Figura E).

Figura 5: Mapas de calor de tejido de COI en el TME. (A) Mancha roja picrosirius que muestra la ubicación de los nódulos 1, 2, 3 y 4. (B–G) Mapas de calor de tejido para LOS COI CD4+FoxP3+, CD8+, CD68+, MPO+, CD34+ y .SMA+, respectivamente. El azul oscuro indica una densidad relativamente baja, y el rojo indica una densidad relativamente alta. A los valores de densidad intermedios se les asignan colores según la escala de colormostrada. (H e I) Cuantificación de COI en nódulos 1, 2 y 3 + 4 organizados por tipo de celda y por nódulo, respectivamente. Haga clic aquí para ver una versión más grande de esta figura.

Figura suplementaria S1: Validación de la alineación de tejidos. (A) La tinción CD34 (en rojo) realizada en la sección II (entrada 1) se utiliza para generar una máscara CD34 en verde (salida 1). La máscara verde (salida 1) se superpone en la imagen de H&E de la sección serial alineada I (entrada 2). La imagen de fusión muestra una correspondencia perfecta de las estructuras vasculares. Barra de escala a 50 m. (B) La imagen IIIA que muestra la fusión de DAPI, CD4 y FoxP3 (entrada 1) se utilizó para generar una etiqueta para las celdas CD4+FoxP3+ (salida 1 en magenta). La etiqueta de salida 1 se transfirió a la imagen alineada IIIB (entrada 2) y muestra la correspondencia perfecta entre los pares FoxP3/DAPI y CD4/CD3 en la imagen de fusión. Barra de escala a 15 m. Haga clic aquí para ver una versión más grande de esta figura.

Figura suplementaria S2: Vista ampliada de los mapas de calor de tejido. (A–C) Mapas de calor de tejido para células CD4+FoxP3+, CD8+, CD68+ y MPO+ en nódulos 1–4. Las barras de escala en los nódulos 1, 2 y 3 + 4 representan 1.500 m, 700 m y 500 m respectivamente. (D) Esquema del nódulo 2 con línea sólida negra. (E) Los puntos calientes para las celdas CD4+FoxP3+, CD8+, CD68+ y MPO+ en el nódulo 2 se extrajeron y asignaron juntos en el contorno del nódulo 2 definido en D. Haga clic aquí para ver una versión más grande de esta figura.

Figura suplementaria S3: Análisis de colocación. (A) A la izquierda y al centro hay imágenes de la etiqueta de la ameta en verde y la etiqueta de desmin en rojo respectivamente. A la derecha se encuentra un área doble positiva de la AMA/desmina en amarillo. (B) Cuantificación del área de la AMA+, el área de desmin +y el área doble positiva de la AMA/desmina. Barra de escala a 150 m. Haga clic aquí para ver una versión más grande de esta figura.
| Aplicación | Propósito | Clasificación | Clasificación | Pasos posteriores al procesamiento | Variables de salida |
| Método | Funciones | ||||
| (valor de píxel) | |||||
| 1 | Detección de tejidos | Umbral | Canal DAPI (150) | o Etiquetar objetos con valores colocalizados por encima del umbral para los 3 canales | o Roi Tissue |
| Canal FITC/A488 (120) | o Cerrar objeto positivo 5 píxeles | o Zona de tejidos | |||
| Canal TRITC/A568 (40) | o Crear tejido ROI | ||||
| 2 | Segmentación de tejidos | Bosque de decisión | Mediana RGB-R | o Rellenar agujeros | o ROI Stroma |
| Mediana RGB-G | o Crear ROI Stroma | o Stroma Area | |||
| Mediana RGB-B | o Crear ROI Parenchyma | o ROI Parenchyma | |||
| Mediana de IHS-S | o Zona de Parenchyma | ||||
| H&E Eosin mediana | |||||
| 3 | Para localizar y cuantificar células CD4+ FoxP3+ | Umbral | DAPI de canal (>600) | o Etiquetar objetos con colocalización de DAPI y Cy5/A647, rodeados de señal FITC/A488 | o Recuentos y densidad de células CD4+FoxP3+ en ROIs Stroma y Parenchyma |
| Suavizado de polisuavizado DE canal FITC/A488 (>850) | o Borrar objetos de menos de 7 m2 | o Coordenadas de células INDIVIDUALes CD4+FoxP3+ | |||
| Canal Cy5/A647(>800) | |||||
| 4 | Para localizar y cuantificar células CD8+ | Umbral | DAPI de canal (<1200) | o Borrar objetos positivos de menos de 15 m2 | o Recuentos y densidad de células CD8+ en ROIs Stroma y Parenchyma |
| Mediana del canal Cy5/A647 (>80) | o Cerrar objetos positivos 2 píxeles | o Coordenadas de células individuales | |||
| o Objetos separados | |||||
| 5 | Para localizar y cuantificar células CD68+ | Umbral | Canal FITC/A488 (>200) | o Borrar objetos positivos de menos de 20 m2 | o Recuentos y densidad de células CD68+ en ROIs Stroma y Parenchyma |
| o Dilatar objetos positivos 3 píxeles | o Coordenadas de células CD68+ individuales | ||||
| o Objetos separados | |||||
| 6 | Para localizar y cuantificar células MPO+ | Umbral | DAPI de canal (>400) | o Borrar objetos de menos de 5m2 | o Recuentos y densidad de células MPO+ en ROIs Stroma y Parenchyma. |
| Canal TRITC/A568 (900-4000) | o Dilatar objetos positivos de 3 píxeles | o Coordenadas de células MPO+ individuales. | |||
| o Objetos separados | |||||
| 7 | Para localizar y cuantificar el área de sMA+ | Umbral | Canal TRITC/CF568 (>1050) | o Borrar objetos positivos de menos de 25 m2 | o Recuentos y densidad del área de sMA+ en los ROIs Stroma y Parenchyma |
| o Dilatar objetos positivos de 3 píxeles | o Coordenadas de píxeles de la SMA+ | ||||
| 8 | Localizar y cuantificar el área DE CD34+ | Umbral | DAPI de canal (<5000) | o Borrar objetos positivos de menos de 25 m2 | o Recuentos y densidad del área CD34+ en ROIs Stroma y Parenchyma |
| Mediana del canal Cy5/A647 (>120) | o Dilatar objetos positivos de 3 píxeles | o Coordenadas de los píxeles CD34+ | |||
| 9 | Crear mapas de calor de tejido para una población celular determinada | Mapa de calor de objetos | Mapa de calor de objetos | o Mapa de calor | |
| Radio de dibujo 50 m | --- | ||||
| 10 | Cuantificar la colocalización entre el SMA y el Desmin | Umbral | Canal TRITC (CF568) (>1050) | o Etiquetar objetos con valores de umbral superiores para TRITC (CF568) | o Cuantificar la expresión colocalizada de la AMA y desmin |
| Canal Cy5 (A647) (>1000) | o Etiquetar objetos con valores de umbral superiores para Cy5 (A647) | ||||
| o Etiquetar objetos con colocación de valores de umbral superiores para TRITC (CF568) y Cy5 (A647) | |||||
| o Borrar objetos positivos de menos de 25 m2 |
Tabla 1: Parámetros generales utilizados para el diseño de APPs empleadas para el análisis de imágenes. Los parámetros especificados en esta tabla se ajustan a las características únicas de las imágenes utilizadas en este análisis (por ejemplo, fondo, artefactos, etc.) y pueden no ser aplicables a otras imágenes. Debido a que los pasos de postprocesamiento mencionados se definieron para las imágenes específicas analizadas en este estudio, no se detallan intencionalmente. El usuario debe personalizar las APLICACIONES a las imágenes que se analizarán.
| Sección/Mancha | Anticuerpo primario | Anticuerpo secundario |
| Sección II/1st Tining | Ratón IgG2a anti-humanos -SMA Ratón IgG1 antihumano CD34 Conejo anti-humano Cytoqueatin 8/18 |
Cabra anti-ratón IgG2a CF568 Rata anti-ratón IgG1 A647 Burro anticonejo A488 |
| Sección II/2nd Tining | Conejo antihumano Desmin Ratón antihumano CD68 |
Burro anticonejo A647 Donkey anti-ratón DyLight 755 |
| Sección III/1st Tining | Ratón antihumano CD4 Conejo antihumano FoxP3 Cabra anti-humano MPO |
Donkey anti-ratón A488 Burro anticonejo A647 Burro anti-cabra A568 |
| SecciónIII/2nd Tining | Rabbit anti-humano CD3 Ratón antihumano CD8 |
Donkey anti-ratón DyLight 755 Burro anticonejo A647 |
Tabla 2: Pares de anticuerpos primarios-secundarios para mIF.
Discussion
Se necesitan técnicas de multiplexación simples, accesibles y fáciles de ejecutar que permitan la resolución espacial de las células inmunitarias en las secciones de tejido para mapear el paisaje inmunológico en el cáncer y otros trastornos inmunológicos. Aquí, describimos una estrategia que integra técnicas de etiquetado y análisis digital ampliamente disponibles para ampliar la capacidad de multiplexación y evaluación multidimensional de los ensayos de imágenes12,,13,,17,19. La tinción de tres secciones seriales para diferentes marcadores, y la reutilización de secciones a través de técnicas de desmontaje y reproducción, nos permitió visualizar 11 parámetros además de las manchas H&E y PSR. Seis imágenes de estas secciones se alinearon de forma automatizada utilizando el módulo de alineación de tejidos. La alineación fue precisa a nivel de celda individual para las imágenes procedentes de la misma sección y altamente concordante para las imágenes que se originan en secciones vecinas. La multiplexación virtual nos permitió determinar cómo los marcadores visualizados en una sección se relacionan espacialmente con los marcadores visualizados en otra sección contigua. Mientras que algunas de las tinciones etiquetadas como COI, otras etiquetados como TCs, lo que nos permite cuantificar los COI en las diferentes tCs. El uso de herramientas de software para la cuantificación automatizada de COI simplificó y aceleró en gran medida el procesamiento de imágenes. Además, el análisis digital se aplicó a secciones de tejidos enteros en lugar de campos de visión seleccionados, lo que dio lugar a una representación imparcial del TME. Además, debido a que se registraron las coordenadas tisulares de los COI, fue posible generar mapas de calor tisulares.
Hay varias áreas en este protocolo donde puede ser necesario solucionar problemas. En primer lugar, la recuperación deficiente de antígenos puede afectar a la calidad del mIF, por lo que el tipo de búfer de recuperación de antígenos y la duración deben optimizarse para las condiciones específicas de ensayo/biomarcador utilizadas. En segundo lugar, el tipo de solución de bloqueo utilizada debe adaptarse a los tejidos/antígenos/especies de anticuerpos primarios y secundarios. En nuestras manos, la adición de 10% de suero total de la especie donde el tejido proviene de receptores Fc bloqueados, y por lo tanto redujo la unión inespecífica de anticuerpos. La adición del 10% del suero de la especie en la que se plantearon los anticuerpos secundarios minimizaría la unión directa inespecífica de anticuerpos secundarios a la sección tisular. En tercer lugar, la validación de la especificidad de los anticuerpos primarios y secundarios utilizando los controles positivos y negativos adecuados es esencial. En cuarto lugar, el aumento de la autofluorescencia en algunos canales y la difusión de DAPI tras la extracción de anticuerpos primarios también son comunes. Para abordar la autofluorescencia mejorada, utilizamos pares de anticuerpos primarios/secundarios donde la señal específica tenía valores de intensidad al menos 5 veces mayores que el del fondo. Por último, algunos anticuerpos de alta afinidad no se pueden eludar con procedimientos de desmontaje regulares. En este caso, se recomienda el uso de estos anticuerpos en la última ronda de etiquetado. El usuario puede tener que probar diferentes secuencias de tinción para encontrar la configuración óptima para los anticuerpos de interés. La eficiencia del desmontaje debe confirmarse antes de proceder a una segunda o tercera ronda de etiquetado.
La principal limitación y desafío de esta estrategia es encontrar las combinaciones correctas de anticuerpos fluorescentes primarios y secundarios para los marcadores de interés. Encontrar anticuerpos primarios criados en diferentes especies o con diferentes isotipos que podrían ser utilizados simultáneamente está limitado por lo que está disponible comercialmente. La mayoría de los escáneres de diapositivas enteras están equipados con lámparas y filtros que permiten tomar imágenes de un máximo de cinco canales, y los anticuerpos secundarios en las especies adecuadas y el fluoróforo adecuado no siempre están disponibles. Superamos parcialmente estas limitaciones utilizando manchas en serie y etiquetado secuencial. Es posible que sea necesario probar varias combinaciones de anticuerpos para llegar a la mejor combinación para los marcadores de interés. Otra limitación es la calidad de la tinción DAPI, ya que el desmontaje y la reprobing pueden no permitir siempre la realización de la segmentación de núcleos.
El módulo de alineación de tejido requiere un entrenamiento mínimo y ninguna habilidad de programación de los usuarios. El software teóricamente permite la alineación de un número ilimitado de imágenes. Sin embargo, la alineación precisa depende de la relación de las secciones, donde las secciones más cercanas que son más histológicamente concordantes se alinean con mayor precisión. Usamos el módulo Author de VIS para generar las APPs. El conocimiento básico del análisis de imágenes es necesario para crear APPs, pero esto es igualmente el caso cuando se utiliza cualquier otro software de análisis de imágenes. Las ventajas únicas de VIS en comparación con otros programas de análisis de imágenes incluyen la alineación automatizada de imágenes de secciones preparadas utilizando diferentes métodos (por ejemplo, IF, histoquímica, IHC). Esto permite realizar estudios de colocación de múltiples marcadores de interés mediante multiplexación virtual. Además, el diseño flexible y fácil de usar de las aplicaciones permite la personalización específica del usuario. La cuantificación y el mapeo automatizados, y la posibilidad de procesar secciones de tejido entero, ahorran tiempo y reducen el sesgo en comparación con el recuento manual mediante inspección visual.
Esta estrategia es una herramienta de investigación muy útil para la inmunología de tejidos en el contexto del cáncer y la autoinmunidad, pero sigue sin ser validada para uso clínico. Con estandarización y validación adicionales, se puede utilizar en el futuro para múltiples aplicaciones (por ejemplo, para mapear el paisaje inmune en el cáncer para predecir y monitorear la respuesta a los agentes inmunoterapéuticos). También se puede adaptar a diferentes condiciones inflamatorias (por ejemplo, enfermedad inflamatoria intestinal) para combinar la evaluación patológica con biomarcadores de pronóstico.
Los principales pasos críticos de este protocolo son la eficiencia/especificidad del etiquetado y la robustez de las APP diseñadas para el uso previsto o biomarcador. Por lo tanto, la validación regular por inspección visual, especialmente al diseñar una nueva APP, es esencial. El uso eficiente de múltiples rondas de desmontaje y reprobing o diferentes tipos de manchas en la misma sección son componentes críticos y pueden ser tejidoo o sección específica. Verificar la eficiencia de estos procesos antes de continuar con el análisis de lotes grandes es fundamental.
En resumen, proporcionamos una estrategia que maximiza la información cuantitativa y espacial que se puede obtener de valiosas muestras de tejido clínico. Los recursos, equipos y conocimientos necesarios para implementar esta metodología son ampliamente accesibles. Proponemos esta metodología como una guía útil para la planificación de ensayos destinados a identificar, cuantificar y mapear las poblaciones de células inmunitarias en el TME.
Disclosures
Los autores no declaran conflictos de intereses.
Acknowledgments
Agradecemos al participante del estudio. Agradecemos a Louise Rousseau, coordinadora del biobanco HBP por la recuperación de las muestras de tejido y toda la información clínica asociada. Reconocemos las instalaciones de geopatología molecular y núcleo de imágenes celulares en el CRCHUM y Michael Persch de Visiopharm por su excelente asistencia técnica. Financiación: Este estudio fue apoyado por subvenciones de la Canadian Liver Foundation, Fonds de recherche du Québec-Santé (FRQS) AIDS and Infectious Disease Network (Réseau SIDA-MI) y la Red Canadiense sobre Hepatitis C (CanHepC). CanHepC está financiado por una iniciativa conjunta de los Institutos Canadienses de Investigación Sanitaria (CIHR) (NHC-142832) y la Agencia de Salud Pública del Canadá. M.F.M. recibió becas de la Universidad de Montreal, Bourse Gabriel Marquis y FRQS. T.F. recibió becas de doctorado de CIHR y CanHepC. S.T. tiene la Cátedra Roger-Des-Groseillers en cirugía oncológica hepatobiliar y pancreática, Université de Montréal.
Contribuciones del autor: M.F.M. diseñó, realizó experimentos y analizó datos. T.F. diseñó experimentos. A.C-B. proporcionaron orientación técnica. G.S. realizó toda la evaluación patológica del sujeto del estudio y aportó información sobre todos los aspectos patológicos. L.M. realizó la tinción de H&E, optimizó y realizó la adquisición de imágenes. M.N.A. realizó la mancha de PSR y proporcionó valiosos aportes técnicos. N.B. contribuyó al análisis de imágenes. S.T. es el investigador principal del biobanco HBP y es responsable de supervisar el funcionamiento general del biobanco. También aportó inestimables aportaciones sobre todos los aspectos del proyecto y sus implicaciones clínicas. M.F.M, T.F. y N.H.S. conceptualizaron y diseñaron el estudio. N.H.S. supervisó el trabajo y obtuvo fondos. M.F.M., T.F., A.C-B y N.H.S. escribieron el manuscrito. Todos los autores revisaron y aprobaron el manuscrito.
Materials
| Name | Company | Catalog Number | Comments |
| Antigen Retrieval Solution: Sodium Citrate Buffer (10 mM Sodium Citrate, 0.05% v/v Tween 20, pH 6.0) | |||
| Blocking Solution: 1 % BSA, 10 % filtered human serum, 10 % filtered donkey serum, 0.1 % Tween 20, and 0.3% Triton in PBS | |||
| Bovine serum albumin (BSA) | Multicell | 800-095-EG | |
| Coplin jars (EASYDIP SLIDE STAINING SYSTEM) | Newcomersupply | 5300KIT | |
| Cover slides | Fisherbrand | 12-545E 22*50 | |
| Direct Red 80 | Sigma Aldrich | 365548 | |
| Donkey Serum | Sigma Aldrich | D9663 | |
| Ethanol 100% | |||
| Electric pressure cooker | Salton | ||
| Eosin | Leica Biosystems | 3801600 | CAUTION, eye irritation |
| Fast Green FCF | Sigma Aldrich | F7252 | CAUTION, harmful by inhalation, ingestion and skin absortion |
| FFPE section (4μm) slides | |||
| Glycine 0,1 M in PBS | |||
| Hematoxylin Stain Solution, Gil 1. Formulation, Regular Strength | Ricca Chemical Company | 3535-32 | |
| Holder (EasyDip Staining Jar Holder) | Newcomersupply | 5300RK | |
| Human Serum | Gemini | 22210 | |
| Humidity chamber | Millipore Sigma | Z670138-1EA | |
| Pap pen | abcam | ab2601 | |
| PBS | |||
| PBS-Tween 20 (0.1% v/v) | |||
| Permount Mounting Media | Fisher Chemical | SP15-500 | |
| Picric Acid 1.3 % | Sigma Aldrich | P6744 | CAUTION, skin and eye irritation |
| Picro-Sirius Red/Fast Green solution: Fast Green 0.1 % w/v + Sirius Red 0.2 % w/v in 1,3 % picric acid solution | |||
| Primary Antibody Anti-αSMA | Mouse IgG2a 1A4 | Sigma A2547 | Dilution 1/100 |
| Primary Antibody Anti-CD34 | Mouse IgG1 HPCA1/763 | Novus Biologicals NBP2-44568 | Dilution 1/250 |
| Primary Antibody Anti-Cytokeratin 8/18 | Rabbit EP17/EP30 | Agilent IR09461-2 | Ready to use |
| Primary Antibody Anti-CD68 | Mouse KP1 | Abcam ab955 | Dilution 1/200 |
| Primary Antibody Anti-Desmin | Rabbit Polyclonal | Invitrogen PA5-16705 | Dilution 1/200 |
| Primary Antibody Anti-CD4 | Mouse N1UG0 | Affymetrix 14-2444 | Dilution 1/250 |
| Primary Antibody Anti-FoxP3 | Rabbit 1054C | R & D MAB8214 | Dilution 1/100 |
| Primary Antibody Anti-MPO | Goat Polyclonal | R & D Systems AF3667 | Dilution 1/250 |
| Primary Antibody Anti-CD3 | Rabbit SP7 | Abcam ab16669 | Dilution 1/200 |
| Primary Antibody Anti-CD8 | Mouse C8/144B | Invitrogen 14-0085-80 | Dilution 1/200 |
| Secondary Antibody Donkey anti-mouse A488 | Polyclonal | Invitrogen A-21202 | Dilution 1/500 |
| Secondary Antibody Donkey anti-Rabbit A488 | Polyclonal | Invitrogen A-21206 | Dilution 1/500 |
| Secondary Antibody Donkey anti-goat A568 | Polyclonal | Invitrogen A-11057 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit A647 | Polyclonal | Invitrogen A-31573 | Dilution 1/500 |
| Secondary Antibody Rat anti-mouse IgG1 A647 | RMG1-1 | Biolegend 406618 | Dilution 1/500 |
| Secondary Antibody Goat anti-mouse IgG2a CF568 | Polyclonal | Sigma Aldrich SAB4600315 | Dilution 1/500 |
| Secondary Antibody Donkey anti-mouse DyLight 755 | Polyclonal | Invitrogen SA5-10171 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit DyLight 755 | Polyclonal | Invitrogen SA5-10043 | Dilution 1/500 |
| SDS | BioShop | SDS001,500 | CAUTION, oral skin and eye toxicity |
| Shandon multi-program robotic slide stainer | LabX | 11384903 | |
| Shandon Xylene Substitute, | Thermo Fisher Scientific | CA89413-336 | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Shaking water bath | |||
| SlowFade Gold antifade reagent with DAPI | Invitrogen | S36938 | |
| Sodium Citrate Dihydrate | Millipore Sigma | 1545801 | CAUTION, eye irritation |
| Stripping Buffer: mix 20 ml 10% w/v SDS with 12.5 ml 0.5 M Tris-HCl (pH 6.8), and 67.5 ml ultra-pure water. Under a fume hood, add 800 uL of 2-mercapto ethanol (114,4 mM final concentration) | |||
| Triton X-100 | Sigma Aldrich | T8787-50ML | |
| Tris-HCl | BioShop | 77-86-1 | |
| Tween 20 | Fisher Scientific | BP337-500 | |
| VIS Software | Visiopharm | ||
| Whole slide scanner Olympus BX61VS | Olympus | Microscope: Olympus Slide Scanner BX61VS, 5 slides scanner, motorized stage, autofocus. Camera: Lightsource: Xcite-120. Filters: BrightLine® Sedat filter set (# LED-DA/FI/TR/Cy5-4X4M-B-000, Semrock) | |
| Xylene | Sigma Aldrich | 214736-4L | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Xylene : Ethanol solution (1:1 v/v) | |||
| 2-mercaptoethanol | Sigma | M6250 | CAUTION, harmful by ingestion, inhalation, fatal if sking absortion. Eye irritation. Use fume hood |
References
- Greten, F. R., Grivennikov, S. I. Inflammation and Cancer:Triggers, Mechanisms, and Consequences. Immunity. 51 (1), 27-41 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Binnewies, M., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine. 24 (5), 541-550 (2018).
- Taube, J. M., et al. Implications of the tumor immune microenvironment for staging and therapeutics. Modern Pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 31 (2), 214-234 (2018).
- Bindea, G., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 39 (4), 782-795 (2013).
- Galon, J., et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. The Journal of Pathology. 232 (2), 199-209 (2014).
- Finotello, F., Eduati, F. Multi-Omics Profiling of the Tumor Microenvironment: Paving the Way to Precision Immuno-Oncology. Frontiers in Oncology. 8, 430 (2018).
- Gerner, M. Y., Kastenmuller, W., Ifrim, I., Kabat, J., Germain, R. N. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 37 (2), 364-376 (2012).
- Giesen, C., et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nature Methods. 11 (4), 417-422 (2014).
- Porta Siegel, T., et al. Mass Spectrometry Imaging and Integration with Other Imaging Modalities for Greater Molecular Understanding of Biological Tissues. Molecular Imaging and Biology : MIB: the official publication of the Academy of Molecular Imaging. 20 (6), 888-901 (2018).
- Buchberger, A. R., DeLaney, K., Johnson, J., Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Analytical Chemistry. 90 (1), 240-265 (2018).
- Pirici, D., et al. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. Journal of Histochemistry and Cytochemistry. 57 (6), 567-575 (2009).
- Gendusa, R., Scalia, C. R., Buscone, S., Cattoretti, G. Elution of High-affinity (>10-9 KD) Antibodies from Tissue Sections: Clues to the Molecular Mechanism and Use in Sequential Immunostaining. Journal of Histochemistry and Cytochemistry. 62 (7), 519-531 (2014).
- van der Loos, C. M. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. Journal of Histochemistry and Cytochemistry. 56 (4), 313-328 (2008).
- Stack, E. C., Wang, C., Roman, K. A., Hoyt, C. C. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 70 (1), 46-58 (2014).
- Toth, Z. E., Mezey, E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. Journal of Histochemistry and Cytochemistry. 55 (6), 545-554 (2007).
- Robertson, D., Savage, K., Reis-Filho, J. S., Isacke, C. M. Multiple immunofluorescence labeling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell Biology. 9, 13 (2008).
- Segnani, C., et al. Histochemical Detection of Collagen Fibers by Sirius Red/Fast Green Is More Sensitive than van Gieson or Sirius Red Alone in Normal and Inflamed Rat Colon. PloS One. 10 (12), 0144630 (2015).
- Bolognesi, M. M., et al. Multiplex Staining by Sequential Immunostaining and Antibody Removal on Routine Tissue Sections. Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
