

Summary
Ici, nous décrivons une stratégie simple et accessible pour visualiser, quantifier, et cartographier les cellules immunitaires dans les sections formelles de tissu tumoral paraffin-intégré. Cette méthodologie combine les techniques d’imagerie et d’analyse numérique existantes dans le but d’élargir la capacité de multiplexage et l’analyse multiparameter des essais d’imagerie.
Abstract
Le paysage immunitaire du microenvironnement de tumeur (TME) est un facteur déterminant dans la progression et la réponse de cancer à la thérapie. Plus précisément, la densité et l’emplacement des cellules immunitaires dans le TME ont des valeurs diagnostiques et pronostiques importantes. Le profilage multiommique du TME a augmenté de façon exponentielle notre compréhension des nombreux réseaux cellulaires et moléculaires régulant l’initiation et la progression des tumeurs. Cependant, ces techniques ne fournissent pas d’informations sur l’organisation spatiale des cellules ou des interactions cellule-cellule. Des techniques de multiplexage abordables, accessibles et faciles à exécuter qui permettent la résolution spatiale des cellules immunitaires dans les sections tissulaires sont nécessaires pour compléter les technologies à haut débit à base d’une seule cellule. Ici, nous décrivons une stratégie qui intègre l’imagerie en série, l’étiquetage séquentiel et l’alignement d’images pour générer des diapositives virtuelles multiparameter de sections de tissus entiers. Les diapositives virtuelles sont ensuite analysées de façon automatisée à l’aide de protocoles définis par l’utilisateur qui permettent l’identification, la quantification et la cartographie des populations cellulaires d’intérêt. L’analyse d’image est faite, dans ce cas à l’aide des modules d’analyse Tissuealign, Auteur, et HISTOmap. Nous présentons un exemple où nous avons appliqué cette stratégie avec succès à un spécimen clinique, maximisant l’information qui peut être obtenue à partir d’échantillons de tissus limités et fournissant une vue impartiale du TME dans toute la section tissulaire.
Introduction
Le développement du cancer est le résultat d’un processus multistep impliquant des interactions réciproques entre les cellules malignes et le TME. Autre que les cellules tumorales, le TME est composé de cellules nonmalignantes, de cellules stromales, de populations de cellules immunitaires et de matrice extracellulaire (ECM)1. L’organisation spatiale des différents composants cellulaires et structurels du tissu tumoral et l’échange dynamique entre le cancer et les cellules non cancéreuses voisines modulent finalement la progression et la réponse de tumeur à la thérapie2,3,4. Il a été démontré que la réponse immunitaire dans le cancer est spatiotemporally réglementée5,6. Différentes populations de cellules immunitaires infiltrant la lésion néoplastique et le tissu adjacent présentent des modèles de distribution spatiale distinctifs et des états variés d’activation et de différenciation associés à différentes fonctions (p. ex., pro- contre antitumor). Ces différentes populations immunitaires et leurs paramètres coevolve heures supplémentaires avec la tumeur et les compartiments stromaux.
L’émergence de technologies permettant le profilage multiomique à cellules individuelles a augmenté de façon exponentielle notre compréhension des nombreux réseaux cellulaires et moléculaires régulant la carcinogenèse et la progression tumorale. Cependant, la plupart des outils analytiques à haut débit à base de cellules simples nécessitent une perturbation tissulaire et l’isolement cellulaire unique, ce qui entraîne une perte d’information sur l’organisation spatiale des cellules et les interactions cellule-cellule7. Étant donné que l’emplacement et l’arrangement de cellules immunitaires spécifiques dans le TME ont une valeur diagnostique et pronostique, les technologies permettant la résolution spatiale sont un complément essentiel des techniques de profilage immunitaire à base de cellules uniques.
Traditionnellement, les techniques d’imagerie comme l’immunohistochimie (IHC) et l’immunofluorescence multiplexe (MIF) ont été limitées à un petit nombre de biomarqueurs qui peuvent être visualisés simultanément. Cette limitation a entravé l’étude de la dynamique spatiotemporal des cellules immunitaires tumeur-infiltrement, qui sont typiquement définies par plusieurs marqueurs phénotypiques. Les progrès récents dans les outils d’imagerie et d’analyse ont élargi les possibilités du multiplexe. De nouvelles technologies d’étiquetage à base d’anticorps comme l’histo-cytométrie et la cytométrie de masse d’imagerie ont été utilisées pour séparer spatialement jusqu’à 12 et 32 biomarqueurs, respectivement8,9. L’imagerie par spectrométrie de masse, une technique qui ne nécessite pas d’étiquetage, a le potentiel d’imager simultanément des milliers de biomarqueurs dans une seule section tissulaire10,11. Bien que ces techniques aient déjà montré un grand potentiel pour disséquer le paysage immunitaire des tissus dans le cancer, elles utilisent des équipements et des logiciels très sophistiqués et coûteux et ne sont pas facilement accessibles à la majorité des chercheurs.
Alternativement, la capacité de multiplexage de l’IHC traditionnel et mIF a été élargie grâce à l’utilisation de l’imagerie en série, des cycles séquentiels d’étiquetage, et l’imagerie spectrale7,12,13,14,15,16. Ces techniques génèrent plusieurs images à partir de la même ou à partir de sections de tissus série qui peuvent être consolidées en diapositives multiparameter virtuelles à l’aide d’un logiciel d’analyse d’images. Par conséquent, le nombre de marqueurs pouvant être visualisés et analysés simultanément augmente.
Ici, nous proposons une stratégie pour la conception rationnelle des essais de multiplex tissulaire à l’aide de réactifs disponibles dans le commerce, d’équipements de microscopie abordables et de logiciels conviviaux(figure 1). Cette méthodologie intègre l’imagerie en série, l’étiquetage du multiplexe séquentiel, l’imagerie des tissus entiers et l’alignement des tissus pour générer des diapositives virtuelles multiparameter qui peuvent être utilisées pour la quantification automatisée et la cartographie des cellules immunitaires dans les sections tissulaires. Grâce à cette stratégie, nous avons créé une diapositive virtuelle comprenant 11 biomarqueurs plus deux taches histologiques fréquemment utilisées : l’hématoxyline et l’éosine (H et E) et le picrosirius rouge (PSR). De multiples populations de cellules immunitaires ont été identifiées, localisées et quantifiées dans différents compartiments de tissu et leur distribution spatiale résolue utilisant des maçons de tissu. Cette stratégie maximise l’information qui peut être obtenue à partir de spécimens cliniques limités et s’applique aux échantillons de tissus archivés par la paraffine (FFPE) formalin-fixe, y compris les tissus entiers, les biopsies d’aiguilles de base et les microrésabres de tissu. Nous proposons cette méthodologie comme un guide utile pour concevoir des essais personnalisés pour l’identification, la quantification et la cartographie des populations de cellules immunitaires dans le TME.
Protocol
Trois sections ffPE en série du virus hpatocellulaire humain réséqué (HBV) ont été obtenues auprès du Centre hospitalier de l’Université de Montréal (CHUM) Hepatopancreatobiliary Cancer Clinical Database and Biological Specimen Dépôt (HBP Biobank). Les patients participant à cette banque de tissus ont donné leur consentement éclairé. Cette étude a été approuvée par le comité d’éthique institutionnel (Protocole numéro 09.237) et réalisée conformément à la Déclaration d’Helsinki.
1. Protocole de coloration de l’hématoxyline et de l’éosine (H et E)
REMARQUE : La coloration de la H-E a été réalisée par l’installation de base de pathologie moléculaire du Centre d’études du Centre hospitalier de l’Université de Montréal (CRCHUM) à l’aide du colorant à glissière robotique multiprogramme Shandon à l’aide du programme suivant.
- Pour la désaffinisation, immerger les toboggans 3x pendant 2,5 min chacun en substitut de xylène.
CAUTION : Les substituts de Xylène sont inflammables, irritants pour la peau, et nocifs s’ils sont inhalés. - Pour la réhydratation, immerger les diapositives dans 100% éthanol 3x pendant 2,5 minutes chacun. Laver pendant 1 min dans de l’eau distillée double (ddH2O) pour réhydrater.
- Incuber pendant 1 min en hématoxyline. Laver 3x pendant 1 min chacun en ddH2O.
- Incuber pour 5 s avec eosin. Laver 30 s avec 95% d’éthanol. Laver 2x pendant 1 min avec 100% d’éthanol.
AVERTISSEMENT : L’éthanol est inflammable et irritant pour les yeux. Eosin est un irritant pour les yeux. - Pour la déshydratation, immerger 3x pendant 1,5 min chacun dans le substitut de xylène. Mount glisse manuellement.
REMARQUE : Le délai estimé pour l’exécution de cette partie du protocole est de 30 min.
2. Protocole de coloration d’immunofluorescence multiplexe pour les sections FFPE
REMARQUE : Ce protocole a été adapté de Robertson et coll.17.
- Déparaffinisation et réhydratation
REMARQUE : Avant l’étiquetage des sections FFPE par l’IHC ou le MIF, la paraffine doit être supprimée. Le défaut d’enlever efficacement la paraffine entraîne une coloration sous-optimale.- Placez la section tissulaire FFPE de 4 m dans des porte-diapositives en verre. Sous le capot de fumée, immerger les toboggans dans un bocal Coplin contenant du xylène pré-réchauffer de 37 oC pendant 10 min.
CAUTION: Xylene est inflammable, un irritant de la peau, et nocif si inhalé. - Agiter manuellement les lames pendant 10 s toutes les 2 minutes. Répéter 1x dans le xylène frais pendant encore 5 minutes.
- Dans le capot chimique, immerger les diapositives séquentiellement pendant 5 min dans chacune des solutions suivantes : 1) xylène : éthanol (1:1 v/v); 2) 100% d’éthanol; 3) 70% d’éthanol; 4) 50% d’éthanol; 5) 30% d’éthanol; 6) saline tamponnée par le phosphate (PBS).
REMARQUE : Conservez les diapositives dans PBS jusqu’à ce qu’elles soient prêtes à effectuer la récupération de l’antigène. Gardez les sections décontrées hydratées en tout temps. Le séchage causera la liaison non spécifique d’anticorps et donc la coloration élevée de fond.
- Placez la section tissulaire FFPE de 4 m dans des porte-diapositives en verre. Sous le capot de fumée, immerger les toboggans dans un bocal Coplin contenant du xylène pré-réchauffer de 37 oC pendant 10 min.
- Récupération d’antigène induite par la chaleur
REMARQUE : Les antigènes peuvent être masqués sur la fixation formelle, empêchant la liaison d’anticorps et par conséquent la visualisation. L’utilisation de tampons et de procédures de démasquage d’antigène rétablissent partiellement la conformation indigène des épitopes et restaure ainsi la reconnaissance des anticorps. Le type de tampon et de durée de récupération d’antigènes devrait être optimisé pour les conditions spécifiques d’analyse (p. ex., cible, anticorps, tissus, etc.).- Plongez des diapositives déconsées dans un bocal Coplin contenant la solution de récupération d’antigènes (recette dans la Table des Matériaux).
- Placer le bocal Coplin fermé dans une cocotte-minute électrique avec de l’eau du robinet. Le niveau de l’eau ne doit pas dépasser la moitié de la hauteur du pot afin que l’eau ne se mélange pas avec la solution de récupération d’antigène.
- Fermer le couvercle et la soupape de pression de la cuisinière. Sélectionnez haute pression pendant 10 min et commencez. Une fois fait, débrancher la cuisinière, relâcher la pression, ouvrir le couvercle, et garder le pot à l’intérieur de la cuisinière pendant 30 minutes, permettant aux lames de refroidir.
- Blocage de la liaison non spécifique
- Transférer la grille avec les diapositives dans un bocal Coplin rempli de PBS. Rincer le tampon de récupération d’antigènes avec PBS 2x pendant 5 minutes chacun.
- Encerclez les sections tissulaires avec un stylo PAP pour créer une barrière hydrophobe. Immerger les diapositives dans un bocal Coplin contenant 0,1 M glycine dans PBS. Incuber pendant 15 minutes à température ambiante (RT).
REMARQUE : La glycine sature les groupes d’aldéhyde générés pendant la récupération d’antigène. Ces groupes pourraient lier les anticorps primaires et secondaires de façon non spécifique. - Rincer la solution de glycine en lavant 2x avec PBS pendant 5 min. Placez les diapositives dans une chambre d’humidité et ajoutez assez de solution de blocage pour couvrir toutes les sections tissulaires. Évitez de déborder la barrière hydrophobe. Incubate pendant 30 min à RT.
REMARQUE: La recette de la solution de blocage peut être trouvée dans la Table des Matériaux. La solution de blocage devrait contenir une protéine (p. ex., BSA) pour bloquer les sites de liaison non spécifiques. Il peut également incorporer des détergents comme Triton X-100 ou Tween 20 qui réduisent les interactions hydrophobes entre les anticorps et les cibles tissulaires, ce qui rend la reconnaissance des antigènes plus sélective. L’ajout de sérum total de 10 % provenant de l’espèce d’où provient le tissu bloquerait les récepteurs Fc et réduirait ainsi la liaison des anticorps non spécifiques. Enfin, l’ajout de 10 % du sérum provenant de l’espèce dans laquelle les anticorps secondaires ont été élevés réduirait au minimum l’attachement direct non spécifique des anticorps secondaires à la section tissulaire.
- Étiquetage de l’immunofluorescence
- Rincer avec PBS-Tween (0,1% v/v) 2x pendant 5 minutes chacun et placer les diapositives dans la chambre d’humidité.
- Ajouter le cocktail d’anticorps primaires réutilisés dans la solution de blocage. Incuber pendant la nuit à 4 oC. Les anticorps primaires et secondaires utilisés pour cette étude sont répertoriés dans le Tableau des matériaux.
REMARQUE : Le cocktail d’anticorps primaires devrait contenir soit des anticorps élevés chez différentes espèces, soit d’une même espèce, mais d’isotypes différents. Pour une liste des paires d’anticorps primaires-secondaires utilisées dans cette étude, consultez le tableau 2. Les détails de tous les anticorps utilisés sont dans le tableau des matériaux et le tableau 2. - Rincer avec PBS-Tween (0,1% v/v) 3x pendant 5 min et remettre les diapositives dans la chambre d’humidité. Dans l’obscurité, ajouter le cocktail d’anticorps secondaires et incuber pendant 1 h à RT.
REMARQUE : Lorsque les anticorps primaires proviennent de différentes espèces, les anticorps secondaires doivent être sélectionnés de façon à ce que chacun d’eux ne se lie qu’à l’un des anticorps primaires et non aux autres. Ceci est généralement réalisé en utilisant des anticorps secondaires tous élevés dans la même espèce tant que cette espèce diffère de l’espèce où les anticorps primaires ont été générés. Dans les cas où les anticorps primaires ont été élevés chez la même espèce mais ont des isotypes différents, des anticorps secondaires spécifiques à l’isotype devraient être utilisés. - Rincer avec PBS-Tween (0,1% v/v) 3x pour 5 min chacun. Rincer avec ddH2O. Enlever l’excès de liquide et monter dans les supports de montage avec DAPI. Le volume utilisé dépend de la taille de la section. Habituellement, 40 L est suffisant pour couvrir la surface d’une glissière de microscopie régulière.
- Placez la glissière de couverture sur la section et pressez doucement le support de montage excessif en évitant la formation de bulles. Laisser sécher les toboggans pendant 20 min à RT dans l’obscurité et conserver à 4 oC jusqu’à ce qu’ils soient prêts à être acquis.
- Acquérir des images pour tous les canaux à l’aide de l’ensemble du scanner de diapositives (voir Tableau des matériaux).
REMARQUE : Les anticorps ont été validés utilisant le tissu hpatocellulaire humain de carcinome comme contrôle positif. Pour chaque anticorps primaire, trois sections en série ont été tachées d’anticorps primaires, de contrôle d’isotype ou seulement de solution de blocage respectivement sans variation dans le reste du protocole de coloration. Les images acquises ont été comparées pour établir la spécificité de la coloration. La coloration a été considérée comme spécifique lorsque le signal de la section incubée avec l’anticorps primaire avait le modèle prévu et se distinguait facilement de l’arrière-plan. Les anticorps primaires donnant un signal de fond élevé ou des composants de tissu d’étiquetage dans l’isotype et aucune section primaire d’anticorps n’ont été considérées non spécifiques. Le délai estimé pour terminer cette partie du protocole est de 2 jours. Les contrôles requis comprennent : (1) Le contrôle de l’isotype pour établir la contribution de la liaison non spécifique de l’anticorps primaire au signal de fond. Une section est tachée de la même manière que les autres tissus de l’échantillon, sauf qu’elle est incubée avec un anticorps avec le même isotype et l’origine de l’anticorps primaire, mais spécifique pour une cible qui est absente dans la section tissulaire. Si l’anticorps de contrôle d’isotype approprié n’est pas disponible, il peut être remplacé par l’IgG total de la même espèce où l’anticorps primaire a été élevé dedans ; (2) Aucun contrôle primaire des anticorps (c.-à-d. le contrôle négatif) n’a pour établir la spécificité de la coloration et estimer la contribution de la liaison non spécifique des anticorps secondaires au signal de fond. Dans ce cas, la section de contrôle est tachée de la même manière que les autres sections, sauf qu’aucun anticorps primaire n’est ajouté; (3) Contrôle positif pour établir que la coloration fonctionne. Dans ce cas, la coloration est effectuée sur une section de tissu qui est connue pour exprimer le marqueur identifié par l’anticorps primaire.
3. Protocole de coloration rouge Picro-sirius (PSR)/vert rapide
REMARQUE : Le but de cette coloration est de visualiser les collagènes fibrillar I et III dans les sections tissulaires de la FFPE. Ce protocole a été adapté de Segnani et coll.18. Toutes les étapes sont effectuées dans une hotte chimique.
- Effectuez la déparaffinisation et la réhydratation des sections tissulaires semblables au protocole de coloration de l’immunofluorescence multiplexe pour les sections FFPE (article 2.1).
REMARQUE : Si la section à tacher a déjà été utilisée pour l’étiquetage de l’immunofluorescence et que la paraffine a déjà été enlevée, les étapes de déparaffinisation-réhydratation sont utiles pour enlever le support de montage. DAPI n’est pas supprimé en utilisant cette procédure, mais il ne perçage pas interférer avec la coloration PSR. - Plongez les diapositives dans un bocal contenant la solution picro-sirius rouge/vert vif (recette dans la Table des Matériaux) et incuber pendant 30 min à RT (plus de 30 min se traduit par une coloration non spécifique des noyaux des hépatocytes).
- Laver rapidement les diapositives en ddH2O (5 trempettes). Ensuite, lavez-vous rapidement à l’éthanol à 100% (5 trempettes). Laver pour 30 s en xylène-100% éthanol (1:1 v/v). Laver pour 30 s en xylène. Mont avec des supports de montage (voir Tableau des matériaux) avant que le xylène ne se soit totalement évaporé (ce qui aide au montage).
REMARQUE : Le temps estimé pour l’exécution de cette partie du protocole est de 1 h.
4. Elution d’anticorps provenant de sections tissulaires
REMARQUE : Afin de réutiliser les sections de tissu dans les essais d’étiquetage séquentiel, l’élimination complète des anticorps primaires et secondaires est exigée. Les anticorps liés ont été dépouillés comme décrit précédemment13.
Préchauffer un bain d’eau à 56 oC. Placez les sections à l’intérieur d’un bocal contenant un tampon de décapage (recette dans la Table des Matériaux),fermez le couvercle et scellez-le avec du ruban adhésif pour éviter les fuites pendant les secousses.
- Mettre le pot à l’intérieur du bain d’eau et incuber pendant 30 min avec agitation.
- Laver 4x pendant 15 min chacun en ddH2O à RT. Rinse avec PBS-Tween (0,1% v/v).
- Gardez les sections hydratées dans PBS-Tween ou en eau jusqu’à ce qu’elles soient prêtes à reprobe la section avec la deuxième série d’anticorps primaires.
REMARQUE : Le temps estimé pour l’exécution de cette partie du protocole est de 2 h. - Vérifier l’efficacité de la procédure d’elution d’anticorps.
REMARQUE : Avant d’utiliser le protocole pour l’elution d’anticorps dans un essai d’étiquetage séquentiel, l’efficacité de l’élimination des anticorps primaires et secondaires doit être vérifiée.- Effectuez l’acquisition de teinture et d’image d’une section avec une paire d’anticorps primaire-secondaire donnée, comme indiqué dans le protocole de coloration de l’immunofluorescence multiplexe pour les sections FFPE (sections 2.1-2.4.6).
- Lors de l’acquisition d’une image, effectuer l’écoulement des complexes d’anticorps primaires-secondaires liés aux tissus, comme indiqué dans les sections 4.1-4.3.
- Incuber la section avec le même anticorps secondaire et les mêmes conditions utilisées dans l’étape 2.4.3.
- Effectuez des étapes de lavage, de montage et d’acquisition d’images comme indiqué dans 2.4.4-2.4.6.
- Comparez côte à côte les images acquises avant et après le décapage afin d’établir si le signal spécifique a disparu ou non.
REMARQUE : La comparaison des images avant et après l’enlèvement des anticorps validera l’efficacité de la procédure d’elution. Cependant, il est normal de voir une augmentation du signal de fond dans tous les canaux, ainsi que la diffusion de DAPI. Cela limite le nombre de tours de décapage qui peuvent être exécutés sur la même section tissulaire. Trois tours de décapage semblent être le maximum.
5. Acquisition d’images
- Générer des images à l’aide d’un scanner à glissière entier.
- Utilisez un objectif 20x 0.75NA et une résolution de 0.3225 'm/pixel.
6. Analyse d’image
REMARQUE : La méthode décrite ici se réfère à l’exemple actuel. Veuillez consulter le tableau 1 et le texte pour s’adapter à d’autres échantillons spécifiques.
- Effectuez l’alignement tissulaire à l’aide du module Tissualign du logiciel d’analyse d’images (VIS dans ce protocole, voir Tableau des matériaux).
- Ouvrez le logiciel d’analyse d’image et cliquez sur l’onglet Tissuealign.
- Importer les images à aligner dans le plateau de diapositives en allant au fichier . Base de données et sélectionnez la première image à être alignée. Retournez à l’onglet Tissuealign et chargez l’image en cliquant sur le bouton Charge dans le plateau de diapositives. L’image apparaîtra dans le plateau de diapositives et dans l’espace de travail.
REMARQUE : Seule la pile d’intérêt doit être chargée dans le plateau de diapositives. - Répétez l’étape 6.1.2 pour toutes les images afin d’être alignées, en les chargeant une par une. Une fois que toutes les images d’intérêt sont chargées sur le plateau de diapositives procéder à lier les images en appuyant sur Next dans les étapes de flux de travail dans le ruban.
- Ensuite, faites glisser et laissez tomber la deuxième image sur le dessus de la première image. Les premières et deuxièmes images sont désormais liées. Répétez cette étape pour que les autres images soient alignées, une par une, de façon ordonnée. Le nom de la première image changera, indiquant qu’elle a été liée aux autres images. Simultanément, les images liées seront affichées dans l’espace de travail à droite du plateau de diapositives.
- À ce stade, alignez les images à l’aide d’un alignement automatique, d’un alignement semi-automatique ou d’un alignement manuel. Il est toujours préférable d’essayer l’alignement automatique d’abord. Pour l’alignement automatique appuyez sur le bouton Suivant dans les étapes de flux de travail (étape 3) dans le ruban.
- Examinez l’alignement automatique en naviguant à différents endroits du tissu et en vérifiant visuellement que les structures correspondantes dans différentes images sont disposées de la même manière dans les deux dimensions de l’image.
- Si le résultat de l’alignement automatique n’est pas satisfaisant, améliorez-le à l’aide de broches (utiliser un minimum de trois broches par image) indiquant des caractéristiques de tissu homologue dans les images liées. Une fois que les broches sont placées à des endroits homologues dans les images liées, l’utilisateur a deux choix : l’alignement semi-automatique ou l’alignement manuel. Pour l’alignement semi-automatique, cliquez sur le bouton Auto-align en fonction des points de repère actuels dans le ruban. Pour l’alignement manuel, cliquez sur le bouton Appliquer les épingles sur le ruban.
- Lorsqu’il est satisfait de l’alignement cliquez sur le bouton Suivant dans les étapes de flux de travail et enregistrer l’image composite dans la base de données.
REMARQUE : L’alignement de six diapositives couvrant 11 marqueurs plus les images H et E et PSR a pris 15 minutes dans l’analyse présentée.
- Effectuez la détection des tissus à l’aide du protocole défini par l’utilisateur Protocole d’analyse Package 1 (APP 1, tableau 1).
- Ouvrez le module d’analyse d’image du logiciel en cliquant sur l’onglet Analyse d’image dans le ruban.
- Importer l’image composite (alignée) en allant au fichier ( Base de données et sélection de l’image d’intérêt et en cliquant sur l’onglet Analyse d’image.
- Ouvrez le dialogue de sélection APP en cliquant sur l’icône Open APP et sélectionnez le paquet de protocole d’analyse (APP) à utiliser. Dans ce cas, sélectionnez APP 1 pour la détection des tissus.
- Une fois qu’APP 1 est ouvert, confirmez que APP1 fonctionne correctement en se rendant à un emplacement de tissu sélectionné et en cliquant sur le bouton Preview. Si les résultats sont satisfaisants, passez à l’étape suivante.
- Cliquez pour exécuter APP 1 et traiter l’image à l’aide de l’APP sélectionnée.
- Exporter les données (p. ex., images, mesures, etc.) lorsque l’analyse est effectuée en cliquant sur Fichier/Exportation.
REMARQUE : L’APP 1 crée une région d’intérêt (ROI) délimitant le tissu (tissu ROI) et calcule la zone du tissu. - Enregistrer l’image modifiée avec le retour sur investissement nouvellement créé en allant au fichier . Économisez.
REMARQUE : La détection du tissu et la création d’un retour sur investissement avec APP 1 dans l’exemple fourni ont pris 5 min dans la station d’analyse d’images décrite. La zone du tissu traité était de 3,2 cm2.
- Effectuer la segmentation des tissus dans Stroma et Parenchyma à l’aide de l’APP 2 (tableau 1).
REMARQUE : APP 2 fonctionne sur le tissu de roi prédéfini. APP 2 segmente le tissu dans les ROIs Stroma et Parenchyma.- Ouvrez le module d’analyse d’image en cliquant sur l’onglet Analyse d’image dans le ruban.
- Importer l’image contenant le tissu de retour sur investissement en allant au fichier . Base de données et sélection de l’image enregistrée dans l’étape 6.2.7. Retournez à l’onglet Analyse d’image et chargez l’image en cliquant sur le bouton Charge dans le plateau de diapositives. L’image apparaîtra dans le plateau de diapositives et dans l’espace de travail.
- Ouvrez APP 2 à l’aide du dialogue de sélection APP comme dans 6.2.3.
- Aperçu de l’APP 2 en traitant dans un champ de vision sélectionné. Si les résultats sont satisfaisants, exécutez APP 2 sur l’image complète en cliquant sur le bouton Run. Comme la sortie de l’APP 2, le tissu ROI est segmenté dans les ROIs Stroma et Parenchyma et leurs zones respectives déterminées. Résultats à l’exportation comme dans 6.2.6. Enregistrer l’image modifiée comme dans 6.2.7.
REMARQUE : Segmenter le tissu à Stroma et Parenchyma à l’aide d’APP 2 a pris 4 h dans la station d’analyse présentée. La zone du tissu traité était de 3,2 cm2.
- Identifier et quantifier les cellules CD4MD de FoxP3salutà l’aide du protocole défini par l’utilisateur APP 3(tableau 1).
REMARQUE : APP 3 travaille sur les ROIs Stroma et Parenchyma prédéfinis.- Ouvrez le module d’analyse d’images et importez l’image contenant les ROIs Stroma et Parenchyma comme en 6.3.1 et 6.3.2. Ouvrez APP 3 à l’aide du dialogue de sélection APP comme dans 6.2.3.
- Aperçu app 3 traitement dans un champ de vision sélectionné enrichi en FoxP3salutCD4 cellules. Si les résultats sont satisfaisants, exécutez APP 3 sur l’image complète. Comme la sortie de l’APP 3, tous les objets CD4MD FoxP3salutseront étiquetés et leurs coordonnées tissulaires stockées. Les densités d’objets FoxP3salutCD4MD dans les ROIs Stroma et Parenchyma seront déterminées. Exporter les résultats comme dans 6.2.6.
- Effectuez la thermomapping tissulaire des objets étiquetés FoxP3salutCD4MD.
- Ouvrez le protocole défini par l’utilisateur FoxP3salutCD4MD MAP à l’aide du dialogue de sélection APP comme dans 6.2.3.
REMARQUE : FoxP3salutCD4MD MAP utilise les coordonnées des objets étiquetés FoxP3salutCD4MD pour générer des maçons à chaleur de densité. Identifier et compter les objets étiquetés CD4MD de FoxP3enutilisant l’APP 3 a pris 25 minutes dans la station d’analyse d’images décrite. La zone du tissu traité était de 3,2 cm2. - Exécutez FoxP3salutCD4 MAP en appuyant sur le bouton Run. Exporter la carte thermique des tissus en cliquant sur le fichier . Exportations Zone de travail.
REMARQUE : Cartographier les objets étiquetés FoxP3salutCD4MD à l’aide de FoxP3salutCD4 MAP a pris 5 minutes dans la station d’analyse d’images décrite.
- Ouvrez le protocole défini par l’utilisateur FoxP3salutCD4MD MAP à l’aide du dialogue de sélection APP comme dans 6.2.3.
- Identifier et quantifier les objets CD8MD, CD68MD, MPOMD, 'SMA et CD34 ' à l’aide des protocoles définis par l’utilisateur APP 4, APP5, APP6, APP7 et APP 8, respectivement(tableau 1)comme le font la section 6.4 à 6.4.3.2 chargeant l’APP d’intérêt dans chaque cas.
REMARQUE : Les PPA 4 à 8 travaillent sur les ROIs Stroma et Parenchyma prédéfinis.
Representative Results
Aperçu de la stratégie de visualisation, de quantification et de cartographie des populations cellulaires d’intérêt pour le TME
Afin de quantifier les populations d’intérêts cellulaires (ICO) dans différents compartiments tissulaires (CTC) et de caractériser leur organisation spatiale, nous avons conçu un flux de travail qui intègre des techniques abordables et faciles à utiliser et maximise l’information de position qui peut être obtenue à partir de précieux spécimens cliniques de la FFPE(figure 1). Premièrement, des sections FFPE de tissus entiers en série ont été tachées pour la visualisation des ICO (p. ex., des cellules immunitaires) et des TC (p. ex., strome versus parenchyma)(figure 1, étape 1). Le nombre de sections consécutives à tacher devrait être réduit au minimum qui permet de visualiser les cellules d’intérêt ou les caractéristiques tissulaires nécessaires pour répondre à la question de la recherche. Plus le nombre de sections de série est faible, plus la ressemblance et la concordance de l’architecture tissulaire à travers les sections contigus sont faibles. En outre, la capacité de multiplexage peut être élargie par la réutilisation de sections tachées fluorescentes par des techniques de décapage et de réprobation19.
Une fois les étapes de coloration terminées, un scanner de diapositives entier a été utilisé pour numériser les images. Les images acquises à partir de sections en série ont été alignées et regroupées en une diapositive multiplexe virtuelle d’une manière automatisée(figure 1, section 2). Ensuite, un retour sur investissement pour le tissu a été délimité avec un protocole défini par l’utilisateur qui a identifié les pixels associés aux tissus (PTI) (figure 1, étape 3). Par la suite, le tissu de retour sur investissement a été segmenté en TCs défini comme roIs additionnel. (Figure 1, étape 4). Ensuite, les protocoles définis par l’utilisateur ont détecté et quantifié les ICO dans différents TC(figure 1, étape 5). Enfin, les maâces tissulaires des ICO ont été générées en fonction de leurs densités et de leurs coordonnées tissulaires(figure 1,étape 6).
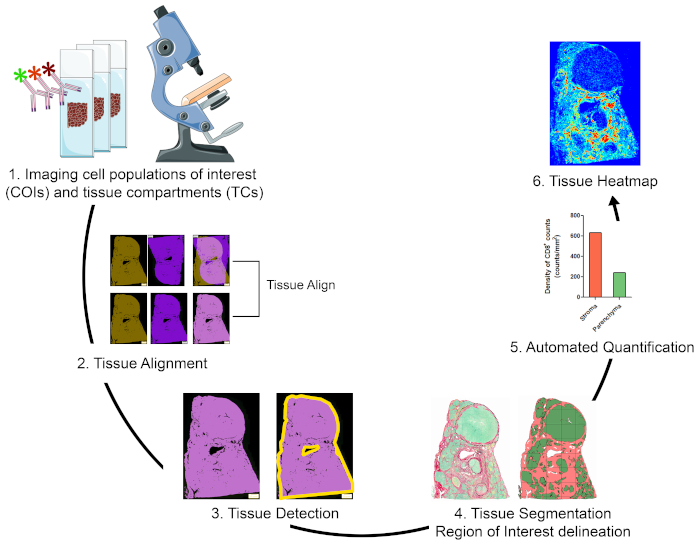
Figure 1 : Représentation schématique de la stratégie de visualisation, de quantification et de cartographie des cellules immunitaires dans le TME. (1) Des sections de tissus entiers en série ont été tachées pour l’étiquetage des COI et des TC. Les sections de tissus entiers teintées ont été numérisées à l’aide d’un scanner à glissière entier. (2) Les images acquises à partir de sections en série ont été liées, alignées et coregistered d’une manière automatisée à l’aide d’un module d’analyse Tissuealign. Une image composite a été générée à partir de l’alignement de haute précision des images individuelles. (3) Un protocole défini par l’utilisateur a été utilisé pour la détection automatisée des pixels associés aux tissus (PTP) dans l’image composite. (4) Le tissu a été segmenté en TC (p. ex., stroma et parenchyme) définis comme ROIs. (5) Des protocoles définis par l’utilisateur ont été utilisés pour la détection et la quantification automatisées des COI dans différents TC. (6) Des maques de tissu des COI ont été générés. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
CoIs et TCs d’imagerie
Trois sections de tissu entier série FFPE de tumeur résectée d’un sujet avec le carcinome hépatocellulaire HBV-associé ont été tachés dans un ou plusieurs tours de coloration comme dans la figure 2A. Section I a été tachée de H et E pour montrer l’architecture tissulaire, la morphologie cellulaire, et pour déterminer les paramètres cliniquement pertinents tels que le type de malignité, la catégorie de tumeur, et l’évaluation globale de l’infiltration immunitaire(figure 2C). Dans la section II contigue, deux séries de MIF ont été utilisées pour étiqueter les cellules parenchymales et non parenchymales du foie(figure 2A). Dans le premier tour, les vaisseaux normaux et de tumeur ont été visualisés utilisant la coloration de CD34 des cellules endothéliales. En outre, des cellules épithéliales (hépatocytes et cholangiocytes) ont été identifiées à l’aide de cytokeratine 8/18, et les cellules épipatiques hépatiques activées fibrogéniques ont été identifiées comme des cellules alpha lisses d’actine de muscle ('SMA')(figure 2C). Après l’acquisition d’images, les sections tissulaires ont été dépouillées et réprouvées avec des anticorps contre les macrophages (CD68), et les myofibroblastes (desmin). Pour mieux caractériser l’infiltration immunitaire de tumeur, la section de série adjacente III a été tachée utilisant deux tours de mIF pour les marqueurs cellulaires CD3, CD4, CD8, boîte à tête fourche P3 (FoxP3), et myeloperoxidase (MPO). Dans tous les cas, le DAPI a été utilisé comme comptoir nucléaire. Enfin, la section III a été tachée de tache de RSP et contre-attachée avec le vert rapide pour visualiser le collagène fibrillaire et segmenter le tissu en stroma et parenchyme (figure 2C).
Un scanner de diapositives entier équipé d’une lentille objective 20X a été utilisé pour numériser les sections tachées et pour créer des diapositives virtuelles. Six images ont été acquises à partir des trois sections en série(figure 2B) et les diapositives virtuelles analysées par la suite à l’aide du logiciel VIS selon la représentation schématique de la figure 1.
Analyse d’image
L’analyse d’image comprenait cinq étapes : 1) l’alignement tissulaire ; 2) détection des tissus; 3) segmentation tissulaire; 4) la quantification automatisée des ICO; et 5) cartographie de la chaleur tissulaire. Tous les protocoles d’analyse d’image ont été développés à l’aide du module Auteur du logiciel d’analyse d’images et sont désignés dans le texte sous le nom d’APP.
Alignement de tissu
Six diapositives virtuelles de trois sections en série, couvrant 11 marqueurs plus des taches H’amp;E et PSR, ont été chargées dans le module Tissualign du logiciel d’analyse d’images. Ensuite, les images ont été reliées, alignées et coregistered d’une manière automatisée, générant un 11-plex plus H 'amp; E et PSR image composite virtuelle, contenant toutes les couches des images individuelles (Figures 2A-C). L’alignement était précis dans le cas des images provenant de sections de série adjacentes, montrant les structures tissulaires correspondantes positionnées et disposées de façon homologue sur l’alignement(figure 2C et figure S1A). De plus, l’alignement était précis au niveau de la cellule individuelle pour les images provenant de la même section(figure S1B). Le temps d’alignement automatique dépend du nombre, de la taille, de la complexité et de la similitude des images à aligner. L’alignement des six diapositives virtuelles mentionnées ci-dessus a pris 15 minutes dans notre station VIS.

Figure 2 : Coloration des sections de tissus en série et alignement d’images. (A) Résumé des colorants effectués sur trois sections en série pour la visualisation des COI et des TC. Les nombres entre parenthèses indiquent la désignation d’image. Pour les sections II et III, les tissus ont été dépouillés et reprobés d’un deuxième cocktail d’anticorps. (B) Aperçu de six images individuelles de tissu entier avant et après l’alignement de tissu (gauche et droite, respectivement). Barre d’échelle de 3 500 m (C) Vue zoomée d’images alignées. Barre d’échelle de 80 m. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Détection des tissus
Une fois que les images ont été liées et alignées, nous avons cherché à identifier les PPP(figure 3A). Pour concevoir un APP pour la détection automatisée des PPP (APP 1, tableau 1), nous avons profité de deux propriétés qui différencient les PTP des pixels non associés aux tissus. Tout d’abord, le signal DAPI (bande bleue) est limité aux noyaux, qui sont situés exclusivement dans le tissu, ce qui signifie que tous les pixels DAPI sont un sous-ensemble de PTP. Deuxièmement, les PTP ont un signal d’autofluorescence plus élevé dans les bandes vertes et jaunes par rapport aux pixels non associés au tissu. Par conséquent, nous avons développé APP 1 pour la détection des tissus(tableau 1), qui détecte les PTP en fonction du signal de base dans ces canaux à l’aide de techniques de seuil simple. Des seuils pour les bandes bleues, vertes et jaunes ont été fixés de sorte que les PTP avaient des valeurs d’intensité de fond au-dessus des seuils, tandis que les pixels non associés au tissu avaient des valeurs inférieures. APP 1 pour la détection des tissus a été appliqué à l’image IIA, qui contient des couches dans les canaux bleu, vert et jaune(figure 3A). Comme sorties de l’APP 1, un masque vert vif a été posé sur le dessus des PPE, et un retour sur investissement appelé «Tissu» a été délimité (sortie, figure 3A). En outre, la zone du tissu a été déterminée comme variable de sortie quantitative. Étant donné que l’APP 1 n’intègre pas les pixels qui ne sont pas associés au tissu dans le tissu roi, ils ont été exclus de l’analyse ultérieure basée sur ce retour sur investissement(figure 3A). La précision de l’APP 1 à l’identification des PPP est indiquée dans la figure 3A.
Segmentation et délimitation des tissus des ROI pour les TC
Ensuite, nous avons procédé à définir différents compartiments à l’intérieur du tissu roi en segmentant le tissu en strome par rapport au parenchyme. Nous avons utilisé l’image tachée de LAR (IIIC, figure 2C), où le stroma peut être défini comme la zone associée au dépôt de collagènes fibrillar (bande rouge), le parenchyme comme zone où les collagènes fibrillar sont absents, et le colorant vert rapide de resténue prévaut (bande verte) (figure 3B). Nous avons créé APP 2 (tableau 1) pour délimiter numériquement les TC Stroma et Parenchyma. Cette APP travaille sur le tissu de roi prédéfini (sortie, figure 3A) et utilise des zones représentatives de stroma et de parenchyme pour la formation de l’outil Classifier intégré dans le module d’analyse d’images. Le Classificateur qualifié attribue les pixels à un stroma ou à une étiquette parenchyma (saumon et vert, respectivement, figure 3B). Lors de la classification des pixels, l’APP 2 a exécuté des opérations morphologiques visant à définir les ROIs Stroma et Parenchyma(figure 3B et tableau 1). Les performances de l’APP 2 à classer les pixels et à générer les IPP respectifs sont indiquées dans la figure 3B. En outre, APP 2 quantifie la zone du stroma et du parenchyme. Enfin, même si la segmentation se fait à l’aide de la section tachée de la RSP, les régions de stroma et de parenchyme décrites peuvent être transférées à n’importe quelle image alignée sur l’image de PSR.

Figure 3 : Détection/segmentation automatisée des tissus et génération d’IRM respectives. (A) Image IIA a été utilisé pour identifier les PTP (image gauche, barre d’échelle - 6.000 'm). Un masque vert vif a été attribué aux PPE à l’aide de l’APP 1 (tableau 1) générant un retour sur investissement appelé Tissu (sortie 1). Droite, l’encart affiche une vue zoomée démontrant la précision de l’APP 1 à détecter les PPP. Barre d’échelle de 350 m (B) Le tissu ROI (sortie 1) est segmenté en stroma et parenchyme à l’aide de l’APP 2. L’image de gauche montre une vue du tissu roi segmenté en stroma de roi (saumon) et parenchyme de ROI (vert). Barre d’échelle 4 500 m. Sur la droite, des vues zoomées de l’encart pour le tissu DE ROI, la coloration psR originale (image IIIC), et le stroma roIs et le parenchyme. Barre d’échelle à 250 m. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
Quantification automatisée des ICO
Ensuite, nous avons procédé à l’identification, la localisation et la quantification des COI dans les ROIs Stroma et Parenchyma. Des AAP 3 à 8(tableau 1) ont été créés pour localiser et compter les COI suivants : CD4-FoxP3MD, CD8MD, CD68MD, MPOMD, cellules DE LSMAMD et CD34MD, respectivement. APP 3 a été conçu pour localiser et compter les cellules CD4-FoxP3MD (image IIIA, figure 2C) comme marqueurs de substitution des lymphocytes T régulateurs (Tregs). Ce protocole détecte la colocalisation du signal à partir du facteur de transcription nucléaire FoxP3 (bande rouge) et de la teinture d’adn DAPI (bande bleue). Étant donné que les lymphocytes T récemment activés d’augmenter FoxP3, pour enrichir pour Tregs nous avons fixé des seuils pour présélectionner uniquement les cellules brillantes FoxP3 (FoxP3salut). Ensuite, parmi toutes les cellules préélectrées DAPI-FoxP3salut, seules celles qui étaient entourées de signaux CD4 en forme d’anneau lumineux (bande verte) ont été étiquetés et comptés comme FoxP3salutCD4 (étiquette rose, figure 4A). La densité des cellules FoxP3salutCD4MD dans les ROIs Stroma et Parenchyma ont été déterminées comme variables quantitatives de sortie de l’APP 3(figure 4A).
De même, les APP 4 à 6 ont été conçus pour la détection des cellules CD8MD, CD68MD et MPOMD. Ces APE partagent la même conception de base pour détecter et quantifier les ICO. Plus précisément, les ICO sont identifiés en fonction de l’intensité du signal du biomarqueur spécifique de la population cellulaire, puis plusieurs étapes morphologiques de posttraitement sont exécutées pour délimiter les cellules individuelles(tableau 1). Les cellules individuelles ou les ICO sont étiquetées, comptées et leurs coordonnées tissulaires sont enregistrées. Les APP 4 à 6 déterminent également la densité des ICO dans les ROIs Stroma et Parenchyma(figure 4B-D).
La qualité de notre coloration DAPI n’était pas assez bonne pour intégrer la segmentation des noyaux dans les APP 3 à 6, de sorte que nous ne pouvons pas nous assurer que tous les objets étiquetés individuellement sont des cellules individuelles. Pour cette raison, nous avons exprimé la densité des cellules dans les comptes d’objets étiquetés/mm2 (figure 4). Cependant, les agrégats cellulaires ont été séparés avec succès en cellules individuelles dans les étapes de post-traitement intégrées dans les APP 3 à 6, et l’inspection visuelle étendue a montré que la plupart des objets étiquetés correspondaient à des cellules simples.
Pour la détection de la zone 'SMA' et CD34', nous avons développé respectivement les APP 7 et 8(tableau 1). Les deux APP détectent le signal spécifique en fonction des seuils et déterminent le pourcentage de zone positive dans les ROIs Stroma et Parenchyma(figure 4E-F).
L’une des possibilités les plus intéressantes de générer des diapositives multiplex virtuelles est l’analyse de l’expression de colocalisation. Nous avons généré APP 10 pour détecter la colocalisation entre la SMA et le desmin, deux marqueurs co-exprimés par les myofibroblastes dans le foie. APP 10 utilise des seuils pour trouver des pixels positifs pour la SMA, le desmin et l’AMS plus desmin(tableau 1). En tant que variables quantitatives de sortie, l’APP 10 détermine la zone de l’AMD, la zone desminMD et la zone d’expression colocalisée de ces deux marqueurs(figure S3).

Figure 4 : Identification et quantification des COI dans les TC stroma et parenchyma. (A-F) Détection et quantification automatisées des CD4-FoxP3MD, CD8, CD68, MPOMD, 'SMA' et CD34MD COI dans les ROIs Stroma et Parenchyma à l’aide de protocoles 3, 4, 5, 6, 7 et 8, respectivement(tableau 1). Sur la gauche sont les images originales, au milieu les images traitées, et sur la droite les quantifications. Pour les figures 4A-D, barre d’échelle à 40 m. Pour les figures 4E et F, barre à l’échelle de 350 m. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Comme alternative à la quantification des COI dans les TC Stroma et Parenchyma, nous avons déterminé la densité des cellules immunitaires dans les différents nodules malins nommés 1 à 4 (figure 5A, H, et je). Le retour sur investissement pour chaque nodule a été délimité manuellement, comme indiqué dans la figure 5A. Les signatures immunitaires distinctives de tissu ont caractérisé chaque nodule, révélant davantage l’hétérogénéité intrinsèque du TME.
Les maçons de tissu
Comme mentionné ci-dessus, les PPP 3 à 8 stockent les coordonnées tissulaires de chaque objet étiqueté individuellement. Cette fonctionnalité permet la génération automatisée de cartes tissulaires où les régions de haute densité d’une population cellulaire donnée sont affichées comme points chauds (rouge), et les régions avec une densité relativement faible comme points froids (bleu foncé). Les valeurs de densité intermédiaire sont attribuées aux couleurs selon l’échelle de couleur indiquée dans la figure 5. Des maçons de tissu ont été générés par des APP qui ont divisé les images en cercles de 50 m de diamètre et ont attribué une couleur en fonction de la densité relative d’une COI donnée à l’intérieur du cercle. Comme l’indiqué la figure 5B-G, les modèles de positionnement et la répartition de l’intensité des différents ICO du TME étaient très variés. De plus, au niveau des nodules individuels, l’organisation de différentes populations dans la région tissulaire était unique(figure S2A-C). Pour donner un exemple de la puissance de cette technique et pour visualiser l’organisation spatiale des points chauds de différentes populations dans la même nodule, les points chauds de différents types de cellules ont été extraits manuellement et cartographiés ensemble sur le contour de nodule 2(figure S2, figure D, et figure E).

Figure 5 : Les maçons de tissu des COI dans le TME. (A) Picrosirius Rouge coloration montrant l’emplacement des nodules 1, 2, 3 et 4. (B-G) Les maçons de soie pour CD4-FoxP3MD, CD8, CD68, MPOMD, CD34MD et COIs de LSMAMD, respectivement. Le bleu foncé indique une densité relativement faible, et le rouge indique une densité relativement élevée. Les valeurs de densité intermédiaire sont attribuées aux couleurs en fonction de l’échelle de couleur montrée. (H et I) Quantification des COI dans les nodules 1, 2 et 3 à 4 organisés par type de cellule et par nodule, respectivement. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure supplémentaire S1 : Validation de l’alignement des tissus. (A) la coloration CD34 (en rouge) effectuée à la section II (entrée 1) est utilisée pour générer un masque CD34 en vert (sortie 1). Le masque vert (sortie 1) est superposé sur l’image H et E de la section de série alignée I (entrée 2). L’image de fusion montre la correspondance parfaite des structures vasculaires. Barre d’échelle à 50 m. (B) Image IIIA montrant la fusion de DAPI, CD4, et FoxP3 (entrée 1) a été utilisé pour générer une étiquette pour les cellules CD4-FoxP3 (sortie 1 en magenta). L’étiquette de sortie 1 a été transférée sur l’image alignée IIIB (entrée 2) et montre une correspondance parfaite entre les paires FoxP3/DAPI et CD4/CD3 dans l’image de fusion. Barre d’échelle de 15 m. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure supplémentaire S2 : Vue zoomée des maçons de tissu. (A-C) Les maçons de tissu pour les cellules CD4-FoxP3MD, CD8MD, CD68MD et MPOMD dans les nodules 1-4. Les barres d’échelle dans les nodules 1, 2 et 3 à 4 représentent respectivement 1 500 m, 700 m et 500 m. (D) Contour de nodule 2 avec ligne solide noire. (E) Les points chauds des cellules CD4-FoxP3MD, CD8MD, CD68MD et MPOMD en nodule 2 ont été extraits et cartographiés ensemble sur le contour nodule 2 défini dans D. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure supplémentaire S3 : Analyse de colocalisation. (A) Sur la gauche et le milieu sont des images de l’étiquette de LMA en vert et desmin étiquette en rouge respectivement. Sur la droite se trouve une zone doublement positive de la SMA/desmin en jaune. (B) Quantification de la zone de la SMA, de la zone de desmin et de la zone double positive de la SMA/desmin. Barre d’échelle à 150 m. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
| Application | But | Classification | Classification | Étapes post-traitement | Variables de sortie |
| Méthode | fonctionnalités | ||||
| (valeur pixel) | |||||
| 1 | Détection des tissus | Seuil | Canal DAPI (150) | o Objets d’étiquette avec des valeurs colocalisées au-dessus du seuil pour les 3 canaux | o Tissu ROI |
| Canal FITC/A488 (120) | o Fermer l’objet positif 5 pixels | o Zone tissulaire | |||
| Canal TRITC/A568 (40) | o Créer des tissus ROI | ||||
| 2 | Segmentation des tissus | Forêt de décision | RGB-R médiane | o Trous de remplissage | o ROI Stroma |
| RGB-G médiane | o Créer le ROI Stroma | o Région de Stroma | |||
| RGB-B médiane | o Créer le ROI Parenchyma | o ROI Parenchyma | |||
| Médiane IHS-S | o Région de Parenchyma | ||||
| Médiane de H et E Eosin | |||||
| 3 | Pour localiser et quantifier les cellules CD4MD FoxP3MD | Seuil | Canal DAPI (-gt;600) | o Objets d’étiquette avec colocalisation de DAPI et Cy5/A647, entourés par le signal FITC/A488 | o Comptes et densité des cellules CD4-FoxP3MD dans ROIs Stroma et Parenchyma |
| Canal FITC/A488 poly lissage (gt;850) | o Objets clairs de moins de 7 m2 | o Coordonnées des cellules CD4-FoxP3MD individuelles | |||
| Canal Cy5/A647 (-gt;800) | |||||
| 4 | Localiser et quantifier les cellules CD8MD | Seuil | Canal DAPI (lt;1200) | o Objets positifs clairs de moins de 15 m2 | o Comptes et densité des cellules CD8MD dans ROIs Stroma et Parenchyma |
| Canal Cy5/A647 médiane (-gt;80) | o Fermer les objets positifs 2 pixels | o Coordonnées des cellules individuelles | |||
| o Objets séparés | |||||
| 5 | Localiser et quantifier les cellules CD68MD | Seuil | Canal FITC/A488 (-gt;200) | o Objets positifs clairs de moins de 20 m2 | o Comptes et densité des cellules CD68MD dans ROIs Stroma et Parenchyma |
| o Dilate objets positifs 3 pixels | o Coordonnées des cellules CD68MD individuelles | ||||
| o Objets séparés | |||||
| 6 | Localiser et quantifier les cellules MPOMD | Seuil | Canal DAPI (-400) | o Objets clairs de moins de 5 m2 | o Nombres et densité des cellules MPOMD dans roIs Stroma et Parenchyma. |
| Canal TRITC/A568 (900-4000) | o Dilate 3 pixels objets positifs | o Coordonnées des cellules MPOMD individuelles. | |||
| o Objets séparés | |||||
| 7 | Localiser et quantifier la zone de la zone de l’ESMA | Seuil | Canal TRITC/CF568 (-gt;1050) | o Objets positifs clairs de moins de 25 m2 | o Comptes et densité de la zone de l’ISMD dans les ROIs Stroma et Parenchyma |
| o Dilate 3 pixels objets positifs | o Coordonnées des pixels de LSMA | ||||
| 8 | Localiser et quantifier la zone CD34MD | Seuil | Canal DAPI (lt;5000) | o Objets positifs clairs de moins de 25 m2 | o Comptes et densité de la zone CD34MD dans roIs Stroma et Parenchyma |
| Canal Cy5/A647 médiane (-gt;120) | o Dilate 3 pixels objets positifs | o Coordonnées des pixels CD34MD | |||
| 9 | Créer des maçons de tissu pour une population cellulaire donnée | Carte thermique de l’objet | Carte thermique de l’objet | o Carte chauffante | |
| Rayon de dessin 50 m | --- | ||||
| 10 | Quantifier la colocalisation entre la SMA et Lesmin | Seuil | Channel TRITC (CF568) (-gt;1050) | o Objets d’étiquette avec des valeurs supérieures au seuil pour TRITC (CF568) | o Quantifier l’expression colocalisée de l’ESMA et des Desmin |
| Canal Cy5 (A647) (gt;1000) | o Objets d’étiquette avec des valeurs supérieures au seuil pour Cy5 (A647) | ||||
| o Objets d’étiquette avec colocalisation de valeurs supérieures au seuil pour TRITC (CF568) et Cy5 (A647) | |||||
| o Objets positifs clairs de moins de 25 m2 |
Tableau 1 : Paramètres généraux utilisés pour la conception des PPA utilisés pour l’analyse d’images. Les paramètres spécifiés dans ce tableau sont ajustés aux caractéristiques uniques des images utilisées dans cette analyse (p. ex., fond, artefacts, etc.) et peuvent ne pas s’appliquer à d’autres images. Étant donné que les étapes post-traitement mentionnées ont été définies pour les images spécifiques analysées dans cette étude, elles ne sont pas intentionnellement détaillées. L’utilisateur doit personnaliser les APP sur les images à analyser.
| Section/Staining | Anticorps primaires | Anticorps secondaires |
| Section II/1st Staining | Souris IgG2a anti-humain 'SMA Souris IgG1 anti-humain CD34 Lapin anti-humain Cytokeratin 8/18 |
Chèvre anti-souris IgG2a CF568 Rat anti-souris IgG1 A647 Âne anti-lapin A488 |
| Section II/2nd Staining | Lapin anti-humain Desmin Souris anti-humaine CD68 |
Âne anti-lapin A647 Âne anti-souris DyLight 755 |
| Section III/1st Staining | Souris anti-humaine CD4 Lapin anti-humain FoxP3 Chèvre anti-humain MPO |
Âne anti-souris A488 Âne anti-lapin A647 Âne anti-chèvre A568 |
| Section III/2nd Staining | Lapin anti-humain CD3 Souris anti-humaine CD8 |
Âne anti-souris DyLight 755 Âne anti-lapin A647 |
Tableau 2 : Paires d’anticorps primaires-secondaires pour le MIF.
Discussion
Des techniques simples, accessibles et faciles à exécuter qui permettent la résolution spatiale des cellules immunitaires dans les sections tissulaires sont nécessaires pour cartographier le paysage immunitaire dans le cancer et d’autres troubles immunologiques. Ici, nous décrivons une stratégie qui intègre des techniques d’étiquetage et d’analyse numérique largement disponibles pour élargir la capacité de multiplexage et l’évaluation multidimensionnelle des essais d’imagerie12,13,17,19. La coloration de trois sections en série pour différents marqueurs, et la réutilisation des sections par des techniques de décapage et de réprobation, nous ont permis de visualiser 11 paramètres en plus des taches de H et E et de PSR. Six images de ces sections ont été alignées de façon automatisée à l’aide du module d’alignement des tissus. L’alignement était précis au niveau de la cellule individuelle pour les images provenant de la même section et très concordant pour les images provenant de sections voisines. Le multiplexe virtuel nous a permis de déterminer comment les marqueurs visualisés dans une section se rapportent spatialement à des marqueurs visualisés dans une autre section contigu. Alors que certaines des colorations étiquetées COIs, d’autres étiquetés TCs, nous permettant de quantifier coIs dans les différents TC. L’utilisation d’outils logiciels pour la quantification automatisée des ICO a grandement simplifié et accéléré le traitement des images. En outre, l’analyse numérique a été appliquée à des sections de tissus entiers au lieu de certains champs de vision, ce qui a donné lieu à une représentation impartiale du TME. En outre, parce que les coordonnées tissulaires des COI ont été enregistrées, il était possible de générer des thermomaps tissulaires.
Il y a plusieurs secteurs dans ce protocole où le dépannage peut être nécessaire. Premièrement, une mauvaise récupération d’antigène peut affecter la qualité du mIF, de sorte que le type de tampon et de durée de récupération d’antigènes devrait être optimisé pour les conditions spécifiques d’essai/biomarqueur utilisées. Deuxièmement, le type de solution de blocage utilisée devrait être adapté aux tissus/antigènes/espèces d’anticorps primaires et secondaires. Dans nos mains, l’ajout de sérum total de 10% de l’espèce où le tissu provient des récepteurs bloqués Fc, et donc réduit la liaison des anticorps non spécifiques. L’ajout de 10 % du sérum provenant de l’espèce dans laquelle les anticorps secondaires ont été élevés réduirait au minimum l’attachement direct non spécifique des anticorps secondaires à la section tissulaire. Troisièmement, la validation de la spécificité des anticorps primaires et secondaires à l’aide des contrôles positifs et négatifs appropriés est essentielle. Quatrièmement, l’augmentation de l’autofluorescence dans certains canaux et la diffusion du DAPI lors du décapage primaire des anticorps sont également courantes. Pour répondre à l’autofluorescence améliorée, nous avons utilisé des paires d’anticorps primaires/secondaires où le signal spécifique avait des valeurs d’intensité au moins 5x celle de l’arrière-plan. Enfin, certains anticorps d’affinité élevée ne peuvent pas être écaillés avec des procédures régulières de décapage. Dans ce cas, nous recommandons d’utiliser de tels anticorps dans la dernière série d’étiquetage. L’utilisateur peut avoir à essayer différentes séquences de coloration pour trouver la configuration optimale pour les anticorps d’intérêt. L’efficacité du décapage doit être confirmée avant de passer à une deuxième ou troisième série d’étiquetage.
La principale limitation et le défi de cette stratégie est de trouver les bonnes combinaisons d’anticorps fluorescents primaires et secondaires pour les marqueurs d’intérêt. Trouver des anticorps primaires élevés dans différentes espèces ou avec des isotypes différents qui pourraient être utilisés simultanément est limité par ce qui est disponible dans le commerce. La plupart des scanners à glissière entiers sont équipés de lampes et de filtres qui permettent l’imagerie d’un maximum de cinq canaux, et les anticorps secondaires dans les bonnes espèces et le fluorophore droit ne sont pas toujours disponibles. Nous avons partiellement surmonté ces limitations à l’aide de colorants en série et d’étiquetage séquentiel. Plusieurs combinaisons d’anticorps peuvent devoir être testées pour arriver à la meilleure combinaison pour les marqueurs d’intérêt. Une autre limitation est la qualité de la coloration DAPI, parce que le décapage et la réprobation ne permettent pas toujours d’effectuer la segmentation des noyaux.
Le module d’alignement des tissus nécessite une formation minimale et aucune compétence de programmation de la part des utilisateurs. Le logiciel permet théoriquement l’alignement d’un nombre illimité d’images. Cependant, l’alignement précis dépend de la relation des sections, où des sections plus proches qui sont plus histologically concordant sont plus précisément alignées. Nous avons utilisé le module Auteur de VIS pour générer les APP. Une connaissance de base de l’analyse d’image est nécessaire pour créer des APP, mais c’est également le cas lors de l’utilisation de tout autre logiciel d’analyse d’images. Les avantages uniques de VIS par rapport à d’autres logiciels d’analyse d’images comprennent l’alignement automatisé des images à partir de sections préparées à l’aide de différentes méthodes (p. ex., IF, histochemistry, IHC). Cela permet des études de colocalisation de plusieurs marqueurs d’intérêt à l’aide de multiplexe virtuel. En outre, la conception flexible et conviviale des PPP permet une personnalisation spécifique à l’utilisateur. La quantification et la cartographie automatisées, ainsi que la possibilité de traiter des sections de tissus entiers, permet d’économiser du temps et de réduire les biais par rapport au comptage manuel par inspection visuelle.
Cette stratégie est un outil de recherche très utile pour l’immunologie tissulaire dans le contexte du cancer et de l’autoimmunité, mais elle n’est pas en mesure d’être utilisée cliniquement. Avec une normalisation et une validation supplémentaires, il peut être utilisé à l’avenir pour de multiples applications (p. ex., cartographier le paysage immunitaire du cancer afin de prédire et de surveiller la réponse aux agents immunothérapeutiques). Il peut également être adapté à différentes conditions inflammatoires (p. ex., maladie inflammatoire de l’intestin) pour combiner l’évaluation pathologique avec les biomarqueurs pronostiques.
Les principales étapes critiques de ce protocole sont l’efficacité/spécificité de l’étiquetage et la robustesse des APP conçus pour l’utilisation ou le biomarqueur prévu. Par conséquent, une validation régulière par inspection visuelle, en particulier lors de la conception d’une nouvelle APP, est essentielle. L’utilisation efficace de plusieurs séries de décapage et de réprobation ou différents types de taches sur la même section sont des composants critiques et peuvent être spécifiques aux tissus ou à la section. Il est essentiel de vérifier l’efficacité de ces processus avant de procéder à une analyse de lots volumines.
En résumé, nous fournissons une stratégie qui maximise l’information quantitative et spatiale qui peut être obtenue à partir d’échantillons de tissus cliniques précieux. Les ressources, l’équipement et les connaissances nécessaires à la mise en œuvre de cette méthodologie sont largement accessibles. Nous proposons cette méthodologie comme un guide utile pour la planification des essais visant à identifier, quantifier et cartographier les populations de cellules immunitaires dans le TME.
Disclosures
Les auteurs ne déclarent aucun conflit d’intérêts.
Acknowledgments
Nous remercions le participant à l’étude. Nous remercions Louise Rousseau, coordonnatrice de la biobanque HBP pour la récupération des échantillons de tissus et de toutes les informations cliniques associées. Nous reconnaissons la pathologie moléculaire et les installations de base d’imagerie cellulaire au CRCHUM et Michael Persch de Visiopharm pour une excellente assistance technique. Financement : Cette étude a été appuyée par des subventions de la Fondation canadienne du foie, du Réseau sida et des maladies infectieuses du Fonds de recherche du Québec-Santé (FRQS) et du Réseau SIDA-MI et du Réseau canadien contre l’hépatite C (CanHepC). CanHepC est financé par une initiative conjointe des Instituts de recherche en santé du Canada (IRSC) (NHC-142832) et de l’Agence de la santé publique du Canada. M.F.M. a reçu des bourses de l’Université de Montréal, de la Bourse Gabriel Marquis et de la FRQS. T.F. a reçu des bourses doctorales des IRSC et de CanHepC. S.T. est titulaire de la Chaire Roger-Des-Groseillers en chirurgie hénotobiliaire et pancréatique oncologique, Université de Montréal.
Contributions d’auteurs : M.F.M. a conçu, effectué des expériences et analysé des données. T.F. a conçu des expériences. A.C-B. fourni des conseils techniques. G.S. a effectué toute l’évaluation pathologique du sujet de l’étude et a fourni des commentaires sur tous les aspects pathologiques. L.M. a effectué des colorants, optimisé et effectué l’acquisition d’images. M.N.A. a effectué la tache de RFP et a fourni une contribution technique précieuse. Le N.-B. a contribué à l’analyse de l’image. S.T. est le chercheur principal de la biobanque HBP et est responsable de superviser l’exploitation globale de la biobanque. Il a également fourni des commentaires inestimables sur tous les aspects du projet et ses implications cliniques. M.F.M, T.F., et N.H.S. ont conceptualisé et conçu l’étude. N.H.S. a supervisé le travail et obtenu du financement. M.F.M., T.F., A.C-B et N.H.S. ont écrit le manuscrit. Tous les auteurs ont examiné et approuvé le manuscrit.
Materials
| Name | Company | Catalog Number | Comments |
| Antigen Retrieval Solution: Sodium Citrate Buffer (10 mM Sodium Citrate, 0.05% v/v Tween 20, pH 6.0) | |||
| Blocking Solution: 1 % BSA, 10 % filtered human serum, 10 % filtered donkey serum, 0.1 % Tween 20, and 0.3% Triton in PBS | |||
| Bovine serum albumin (BSA) | Multicell | 800-095-EG | |
| Coplin jars (EASYDIP SLIDE STAINING SYSTEM) | Newcomersupply | 5300KIT | |
| Cover slides | Fisherbrand | 12-545E 22*50 | |
| Direct Red 80 | Sigma Aldrich | 365548 | |
| Donkey Serum | Sigma Aldrich | D9663 | |
| Ethanol 100% | |||
| Electric pressure cooker | Salton | ||
| Eosin | Leica Biosystems | 3801600 | CAUTION, eye irritation |
| Fast Green FCF | Sigma Aldrich | F7252 | CAUTION, harmful by inhalation, ingestion and skin absortion |
| FFPE section (4μm) slides | |||
| Glycine 0,1 M in PBS | |||
| Hematoxylin Stain Solution, Gil 1. Formulation, Regular Strength | Ricca Chemical Company | 3535-32 | |
| Holder (EasyDip Staining Jar Holder) | Newcomersupply | 5300RK | |
| Human Serum | Gemini | 22210 | |
| Humidity chamber | Millipore Sigma | Z670138-1EA | |
| Pap pen | abcam | ab2601 | |
| PBS | |||
| PBS-Tween 20 (0.1% v/v) | |||
| Permount Mounting Media | Fisher Chemical | SP15-500 | |
| Picric Acid 1.3 % | Sigma Aldrich | P6744 | CAUTION, skin and eye irritation |
| Picro-Sirius Red/Fast Green solution: Fast Green 0.1 % w/v + Sirius Red 0.2 % w/v in 1,3 % picric acid solution | |||
| Primary Antibody Anti-αSMA | Mouse IgG2a 1A4 | Sigma A2547 | Dilution 1/100 |
| Primary Antibody Anti-CD34 | Mouse IgG1 HPCA1/763 | Novus Biologicals NBP2-44568 | Dilution 1/250 |
| Primary Antibody Anti-Cytokeratin 8/18 | Rabbit EP17/EP30 | Agilent IR09461-2 | Ready to use |
| Primary Antibody Anti-CD68 | Mouse KP1 | Abcam ab955 | Dilution 1/200 |
| Primary Antibody Anti-Desmin | Rabbit Polyclonal | Invitrogen PA5-16705 | Dilution 1/200 |
| Primary Antibody Anti-CD4 | Mouse N1UG0 | Affymetrix 14-2444 | Dilution 1/250 |
| Primary Antibody Anti-FoxP3 | Rabbit 1054C | R & D MAB8214 | Dilution 1/100 |
| Primary Antibody Anti-MPO | Goat Polyclonal | R & D Systems AF3667 | Dilution 1/250 |
| Primary Antibody Anti-CD3 | Rabbit SP7 | Abcam ab16669 | Dilution 1/200 |
| Primary Antibody Anti-CD8 | Mouse C8/144B | Invitrogen 14-0085-80 | Dilution 1/200 |
| Secondary Antibody Donkey anti-mouse A488 | Polyclonal | Invitrogen A-21202 | Dilution 1/500 |
| Secondary Antibody Donkey anti-Rabbit A488 | Polyclonal | Invitrogen A-21206 | Dilution 1/500 |
| Secondary Antibody Donkey anti-goat A568 | Polyclonal | Invitrogen A-11057 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit A647 | Polyclonal | Invitrogen A-31573 | Dilution 1/500 |
| Secondary Antibody Rat anti-mouse IgG1 A647 | RMG1-1 | Biolegend 406618 | Dilution 1/500 |
| Secondary Antibody Goat anti-mouse IgG2a CF568 | Polyclonal | Sigma Aldrich SAB4600315 | Dilution 1/500 |
| Secondary Antibody Donkey anti-mouse DyLight 755 | Polyclonal | Invitrogen SA5-10171 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit DyLight 755 | Polyclonal | Invitrogen SA5-10043 | Dilution 1/500 |
| SDS | BioShop | SDS001,500 | CAUTION, oral skin and eye toxicity |
| Shandon multi-program robotic slide stainer | LabX | 11384903 | |
| Shandon Xylene Substitute, | Thermo Fisher Scientific | CA89413-336 | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Shaking water bath | |||
| SlowFade Gold antifade reagent with DAPI | Invitrogen | S36938 | |
| Sodium Citrate Dihydrate | Millipore Sigma | 1545801 | CAUTION, eye irritation |
| Stripping Buffer: mix 20 ml 10% w/v SDS with 12.5 ml 0.5 M Tris-HCl (pH 6.8), and 67.5 ml ultra-pure water. Under a fume hood, add 800 uL of 2-mercapto ethanol (114,4 mM final concentration) | |||
| Triton X-100 | Sigma Aldrich | T8787-50ML | |
| Tris-HCl | BioShop | 77-86-1 | |
| Tween 20 | Fisher Scientific | BP337-500 | |
| VIS Software | Visiopharm | ||
| Whole slide scanner Olympus BX61VS | Olympus | Microscope: Olympus Slide Scanner BX61VS, 5 slides scanner, motorized stage, autofocus. Camera: Lightsource: Xcite-120. Filters: BrightLine® Sedat filter set (# LED-DA/FI/TR/Cy5-4X4M-B-000, Semrock) | |
| Xylene | Sigma Aldrich | 214736-4L | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Xylene : Ethanol solution (1:1 v/v) | |||
| 2-mercaptoethanol | Sigma | M6250 | CAUTION, harmful by ingestion, inhalation, fatal if sking absortion. Eye irritation. Use fume hood |
References
- Greten, F. R., Grivennikov, S. I. Inflammation and Cancer:Triggers, Mechanisms, and Consequences. Immunity. 51 (1), 27-41 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Binnewies, M., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine. 24 (5), 541-550 (2018).
- Taube, J. M., et al. Implications of the tumor immune microenvironment for staging and therapeutics. Modern Pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 31 (2), 214-234 (2018).
- Bindea, G., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 39 (4), 782-795 (2013).
- Galon, J., et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. The Journal of Pathology. 232 (2), 199-209 (2014).
- Finotello, F., Eduati, F. Multi-Omics Profiling of the Tumor Microenvironment: Paving the Way to Precision Immuno-Oncology. Frontiers in Oncology. 8, 430 (2018).
- Gerner, M. Y., Kastenmuller, W., Ifrim, I., Kabat, J., Germain, R. N. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 37 (2), 364-376 (2012).
- Giesen, C., et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nature Methods. 11 (4), 417-422 (2014).
- Porta Siegel, T., et al. Mass Spectrometry Imaging and Integration with Other Imaging Modalities for Greater Molecular Understanding of Biological Tissues. Molecular Imaging and Biology : MIB: the official publication of the Academy of Molecular Imaging. 20 (6), 888-901 (2018).
- Buchberger, A. R., DeLaney, K., Johnson, J., Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Analytical Chemistry. 90 (1), 240-265 (2018).
- Pirici, D., et al. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. Journal of Histochemistry and Cytochemistry. 57 (6), 567-575 (2009).
- Gendusa, R., Scalia, C. R., Buscone, S., Cattoretti, G. Elution of High-affinity (>10-9 KD) Antibodies from Tissue Sections: Clues to the Molecular Mechanism and Use in Sequential Immunostaining. Journal of Histochemistry and Cytochemistry. 62 (7), 519-531 (2014).
- van der Loos, C. M. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. Journal of Histochemistry and Cytochemistry. 56 (4), 313-328 (2008).
- Stack, E. C., Wang, C., Roman, K. A., Hoyt, C. C. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 70 (1), 46-58 (2014).
- Toth, Z. E., Mezey, E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. Journal of Histochemistry and Cytochemistry. 55 (6), 545-554 (2007).
- Robertson, D., Savage, K., Reis-Filho, J. S., Isacke, C. M. Multiple immunofluorescence labeling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell Biology. 9, 13 (2008).
- Segnani, C., et al. Histochemical Detection of Collagen Fibers by Sirius Red/Fast Green Is More Sensitive than van Gieson or Sirius Red Alone in Normal and Inflamed Rat Colon. PloS One. 10 (12), 0144630 (2015).
- Bolognesi, M. M., et al. Multiplex Staining by Sequential Immunostaining and Antibody Removal on Routine Tissue Sections. Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
