

Summary
Her beskriver vi en enkel og tilgjengelig strategi for visualisering, kvantifisering og kartlegging av immunceller i formalin-faste parafin-innebygde tumorvevseksjoner. Denne metoden kombinerer eksisterende bilde- og digitale analyseteknikker med det formål å utvide multipleksjonsevnen og multiparameteranalysen av bildebehandlingsanalyser.
Abstract
Immunlandskapet i tumormikromiljøet (TME) er en avgjørende faktor i kreftprogresjon og respons på terapi. Spesielt har tettheten og plasseringen av immunceller i TME viktige diagnostiske og prognostiske verdier. Multiomic profilering av TME har eksponentielt økt vår forståelse av de mange cellulære og molekylære nettverk som regulerer tumorinitiering og progresjon. Disse teknikkene gir imidlertid ikke informasjon om den romlige organiseringen av celler eller cellecelleinteraksjoner. Rimelig, tilgjengelig og lett å utføre multipleksing teknikker som tillater romlig oppløsning av immunceller i vev seksjoner er nødvendig for å utfylle enkelt celle-baserte high-throughput teknologier. Her beskriver vi en strategi som integrerer seriebilde, sekvensiell merking og bildejustering for å generere virtuelle multiparameterlysbilder av hele vevsseksjoner. Virtuelle lysbilder analyseres deretter på en automatisert måte ved hjelp av brukerdefinerte protokoller som muliggjør identifisering, kvantifisering og kartlegging av cellepopulasjoner av interesse. Bildeanalysen er gjort, i dette tilfellet ved hjelp av analysemodulene Tissuealign, Author og HISTOmap. Vi presenterer et eksempel der vi brukte denne strategien med hell til en klinisk prøve, maksimere informasjonen som kan fås fra begrensede vevsprøver og gi et objektivt syn på TME i hele vevsdelen.
Introduction
Kreftutvikling er et resultat av en flertrinnsprosess som involverer gjensidige interaksjoner mellom ondartede celler og TME. Annet enn tumorceller består TME av ikke-ondartede celler, stromale celler, immuncellepopulasjoner og ekstracellulær matrise (ECM)1. Den romlige organiseringen av de forskjellige cellulære og strukturelle komponentene i tumorvevet og den dynamiske utvekslingen mellom kreft og nærliggende ikke-kreftceller modulerer til slutt tumorprogresjon og respons på terapi2,,3,4. Det har vist seg at immunresponsen hos kreft er spatiotempoally regulert5,6. Ulike immuncellepopulasjoner som infiltrerer neoplastisk lesjon og tilstøtende vev viser karakteristiske romlige distribusjonsmønstre og varierte aktiverings- og differensieringstilstander forbundet med forskjellige funksjoner (f.eks. pro- versus antitumor). Disse forskjellige immunpopulasjonene og deres parametere dreier seg om overtid med svulsten og stromale rom.
Fremveksten av teknologier som tillater encellede multiomics profilering har eksponentielt økt vår forståelse av de mange cellulære og molekylære nettverk som regulerer kreftfremkallende og tumorprogresjon. De fleste enkeltcellebaserte analytiske verktøy med høy gjennomstrømning krever imidlertid vevsforstyrrelser og encellede isolering, noe som resulterer i tap av informasjon om den romlige organiseringen av celler og cellecelleinteraksjoner7. Fordi plasseringen og plasseringen av spesifikke immunceller i TME har diagnostisk og prognostisk verdi, er teknologier som tillater romlig oppløsning et viktig komplement av enkeltcellebaserte immunprofileringsteknikker.
Tradisjonelt har bildeteknikker som immunohistochemistry (IHC) og multipleks immunofluorescens (mIF) vært begrenset til et lite antall biomarkører som kan visualiseres samtidig. Denne begrensningen har hindret studien av spatiotemporal dynamikken i tumor-infiltrerende immunceller, som vanligvis er definert av flere fenotypisk markører. Nylige fremskritt innen bildebehandling og analytiske verktøy har utvidet mulighetene for multipleksing. Nye antistoffbaserte merkingsteknologier som histo-cytometri og bildemassecytometri har blitt brukt til å skille opptil 12 og 32 biomarkører, henholdsvis8,9. Massespektrometri bildebehandling, en teknikk som ikke krever merking, har potensial til å bilde tusenvis av biomarkører samtidig i en enkelt vev seksjon10,11. Selv om disse teknikkene allerede har vist stort potensial for å dissekere vevimmunlandskapet i kreft, bruker de svært sofistikert og dyrt utstyr og programvare og er ikke lett tilgjengeligfor de fleste forskere.
Alternativt har multipleksjonsevnen til tradisjonell IHC og mIF blitt utvidet gjennom bruk av seriell bildebehandling, sekvensielle runder med merking og spektral bildebehandling7,,12,,13,,14,15,16. Disse teknikkene genererer flere bilder fra samme eller fra serievevseksjoner som kan konsolideres til virtuelle multiparameterlysbilder ved hjelp av bildeanalyseprogramvare. Som et resultat øker antall markører som kan visualiseres og analyseres samtidig.
Her foreslår vi en strategi for rasjonell utforming av vev multipleks analyser ved hjelp av kommersielt tilgjengelige reagenser, rimelig mikroskopi utstyr, og brukervennlig programvare (Figur 1). Denne metoden integrerer seriebilde, sekvensiell multipleksmerking, hele vevsavbildning og vevsjustering for å generere virtuelle multiparameterlysbilder som kan brukes til automatisert kvantifisering og kartlegging av immunceller i vevsseksjoner. Ved hjelp av denne strategien opprettet vi en virtuell lysbilde bestående av 11 biomarkører pluss to ofte brukte histologiske flekker: hematoksylin og eosin (H&E) og picrosirius rød (PSR). Flere immuncellepopulasjoner ble identifisert, lokalisert og kvantifisert i forskjellige vevsrom og deres romlige fordeling løst ved hjelp av vevsvarmekart. Denne strategien maksimerer informasjonen som kan oppnås fra begrensede kliniske prøver og gjelder for formalin-fast parafin-innebygd (FFPE) arkivertvevsprøver, inkludert hele vev, kjerne nål biopsier, og vev mikroarrays. Vi foreslår denne metodikken som en nyttig veiledning for utforming av tilpassede analyser for identifisering, kvantifisering og kartlegging av immuncellepopulasjoner i TME.
Protocol
Tre seriell FFPE seksjoner fra resected hepatitt B virus (HBV)-assosiert humant hepatocellulært karsinom ble hentet fra Centre hospitalier de l'Université de Montréal (CHUM) Hepatopancreatobiliary Cancer Clinical Database og Biological Specimen Depotet (HBP Biobank). Pasienter som deltar i denne vevsbanken ga informert samtykke. Denne studien ble godkjent av den institusjonelle etikkkomiteen (protokollnummer 09.237) og utført i samsvar med Helsingforserklæringen.
1. Hematoksylin og eosin (H&E) farging protokoll
MERK: H&E-fargingen ble utført av det molekylære patologikjerneanlegget til Centre de Recherches du Centre hospitalier de l'Université de Montréal (CRCHUM) ved hjelp av Shandon multiprogram robotskliefarperen ved hjelp av følgende program.
- For deparaffinisering, dypp lysbilder 3x for 2,5 min hver i xylen erstatning.
FORSIKTIG: Xylensubstitutter er brannfarlige, hudirriterende stoffer og skadelige ved innånding. - For rehydrering, dypp lysbilder i 100% etanol 3x i 2,5 min hver. Vask i 1 min i dobbelt destillert vann (ddH2O) for å rehydrere.
- Inkuber i 1 min i hematoksylin. Vask 3x i 1 min hver i ddH2O.
- Inkuber for 5 s med eosin. Vask 30 s med 95% etanol. Vask 2x i 1 min med 100% etanol.
FORSIKTIG: Etanol er brannfarlig og et øyeirriterende. Eosin er et øye irriterende. - For dehydrering, senk 3x i 1,5 min hver i xylenerstatningen. Montere lysbilder manuelt.
MERK: Beregnet tid for å utføre denne delen av protokollen er 30 min.
2. Multiplex immunofluorescens farging protokoll for FFPE seksjoner
MERK: Denne protokollen ble tilpasset fra Robertson et al.17.
- Deparaffinisering og rehydrering
MERK: Før antistoffmediert merking av FFPE-seksjoner av IHC eller mIF, skal parafinen fjernes. Hvis du ikke fjerner parafinen effektivt, kan det bli suboptimal farging.- Plasser 4 μm FFPE vevsdelen glir i glassglideholdere. Under røykhetten setter du ned skliene i en Coplin-krukke som inneholder 37 °C før varmet xylen i 10 min.
FORSIKTIG: Xylen er brannfarlig, en hudirriterende og skadelig ved innånding. - Rør skliene manuelt i 10 s hver 2 min. Gjenta 1x i frisk xylen i ytterligere 5 min.
- I den kjemiske hetten, senk lysbildene sekvensielt i 5 min i hver av følgende løsninger: 1) xylen : etanol (1:1 v / v); 2) 100% etanol; 3) 70% etanol; 4) 50% etanol; 5) 30% etanol; 6) fosfatbufret saltvann (PBS).
MERK: Oppbevar lysbildene i PBS til de er klare til å utføre antigengjenfinningen. Hold de devoksed delene hydrert til enhver tid. Uttørking vil føre til uspesifikk antistoffbinding og derfor høy bakgrunnsfarging.
- Plasser 4 μm FFPE vevsdelen glir i glassglideholdere. Under røykhetten setter du ned skliene i en Coplin-krukke som inneholder 37 °C før varmet xylen i 10 min.
- Varmeindusert antigenhenting
MERK: Antigener kan maskeres ved formalinfiksering, og forhindrer antistoffbinding og dermed visualisering. Bruken av antigenavsløringsbuffere og prosedyrer gjenoppretter delvis den innfødte konformasjonen av epitoper og gjenoppretter dermed antistoffgjenkjenning. Typen antigengjenfinningsbuffer og varighet bør optimaliseres for de spesifikke analyseforholdene (f.eks. mål, antistoff, vev osv.).- Dypp dewaxed lysbilder i en Coplin krukke som inneholder antigen gjenfinning løsning (oppskrift i Materialtabellen).
- Plasser lukket Coplin krukke i en elektrisk trykkoker med vann fra springen. Vannstanden bør ikke overstige halvparten av høyden på krukken slik at vannet ikke blandes med antigengjenfinningsløsningen.
- Lukk lokket og trykkventilen på komfyren. Velg høyt trykk i 10 min og start. Når du er ferdig, koble fra komfyren, slipp trykket, åpne lokket og hold krukken inne i komfyren i 30 minutter, slik at skliene avkjøles.
- Blokkering av uspesifikk binding
- Overfør stativet med lysbildene til en Coplin-krukke fylt med PBS. Skyll av antigengjenfinningsbufferen med PBS 2x i 5 min hver.
- Omkranser vevsseksjonene med en PAP-penn for å skape en hydrofob barriere. Dypp lysbildene i en Coplin-krukke som inneholder 0,1 M glycin i PBS. Inkuber i 15 min ved romtemperatur (RT).
MERK: Glycine metter aldehydgruppene som genereres under antigengjenfinning. Disse gruppene kan binde primære og sekundære antistoffer uspesifikt. - Skyll av glycinoppløsningen ved å vaske 2x med PBS i 5 min. Plasser lysbildene i et fuktighetskammer og legg til nok blokkeringsløsning for å dekke alle vevsseksjonene. Unngå å overfylte den hydrofobe barrieren. Inkuber i 30 min på RT.
MERK: Oppskriften på blokkeringsløsningen finnes i materialtabellen. Blokkeringsløsningen skal inneholde et protein (f.eks. BSA) for å blokkere ikke-spesifikke bindingssteder. Det kan også omfatte vaskemidler som Triton X-100 eller Tween 20 som reduserer hydrofobe interaksjoner mellom antistoffer og vevsmål, og dermed gjør antigengjenkjenning mer selektiv. Tillegg av 10% totalt serum fra arten der vevet kommer fra ville blokkere Fc reseptorer, og dermed redusere uspesifikk antistoff binding. Til slutt, tillegg av 10% av serum fra arten de sekundære antistoffene ble reist i ville minimere direkte uspesifikk vedlegg av sekundære antistoffer mot vevsdelen.
- Immunfluorescens merking
- Skyll med PBS-Tween (0,1 % v/v) 2x i 5 min hver og plasser lysbildene tilbake i fuktighetskammeret.
- Tilsett cocktail av primære antistoffer resuspendert i blokkeringsløsning. Inkuber over natten ved 4 °C. Primære og sekundære antistoffer som brukes til denne studien er oppført i Materialtabellen.
MERK: Cocktailen av primære antistoffer bør inneholde enten antistoffer oppvokst i forskjellige arter, eller fra samme art, men av forskjellige isotyper. For en liste over de primære sekundære antistoffparene som brukes i denne studien, se tabell 2. Detaljer om alle antistoffer som brukes er i materialtabellen og tabell 2. - Skyll med PBS-Tween (0,1 % v/v) 3x i 5 min og plasser lysbildene tilbake i fuktighetskammeret. I mørket, legg til cocktail av sekundære antistoffer og inkuber for 1 h på RT.
MERK: Når de primære antistoffene er fra forskjellige arter, bør de sekundære antistoffene velges slik at hver av dem bare binder seg til et av de primære antistoffene og ikke til hverandre. Dette oppnås vanligvis ved hjelp av sekundære antistoffer som alle er oppvokst i samme art så lenge denne arten er forskjellig fra arten der de primære antistoffene ble generert. I tilfeller der de primære antistoffene ble reist i samme art, men har forskjellige isotyper, bør isotypespesifikke sekundære antistoffer brukes. - Skyll med PBS-Tween (0,1 % v/v) 3x i 5 min hver. Skyll med ddH2O. Fjern overflødig væske og monter i monteringsmediet med DAPI. Volumet som brukes, avhenger av størrelsen på inndelingen. Vanligvis er 40 μL nok til å dekke overflaten av et vanlig mikroskopilysbilde.
- Plasser dekselet skyves på seksjonen og klem forsiktig ut overflødig monteringsmateriale unngå bobledannelse. La skliene tørke i 20 minutter ved RT i mørket og oppbevar ved 4 °C til de er klare for oppkjøp.
- Hent bilder for alle kanalene ved hjelp av hele lysbildeskanneren (se Materialarket).
MERK: Antistoffene ble validert ved hjelp av humant hepatocellulært karsinomvev som en positiv kontroll. For hvert primære antistoff ble tre seriedeler farget med enten primær antistoff, isotypekontroll eller bare blokkeringsløsning henholdsvis uten variasjon i resten av fargeprotokollen. De ervervede bildene ble sammenlignet med å fastslå farsifisiteten. Farging ble ansett som spesifikk når signalet i avsnittet inkubert med primær antistoff hadde forventet mønster og var lett skilles fra bakgrunnen. Primære antistoffer som gir et høyt bakgrunnssignal eller merking av vevskomponenter i isotypen, og ingen primære antistoffseksjoner ble ansett som uspesifikke. Beregnet tid for å fullføre denne delen av protokollen er 2 dager. Nødvendige kontroller inkluderer: (1) Isotype kontroll for å etablere bidrag et uspesifikt binding av det primære antistoffet til bakgrunnssignalet. En del er farget på samme måte som det andre prøvevevet bortsett fra at den inkuberes med et antistoff med samme isotype og opprinnelse av det primære antistoffet, men spesifikt for et mål som er fraværende i vevsdelen. Hvis riktig isotype kontroll antistoff ikke er tilgjengelig, kan det erstattes av total IgG fra samme art hvor det primære antistoffet ble reist i; (2) Ingen primær antistoffkontroll (dvs. negativ kontroll) for å etablere spesifisiteten til farsingen og å estimere bidraget fra uspesifikk binding av sekundære antistoffer mot bakgrunnssignalet. I dette tilfellet er kontrolldelen farget på samme måte som de andre seksjonene, bortsett fra at ingen primære antistoff er lagt til; (3) Positiv kontroll for å fastslå at faringen fungerer. I dette tilfellet utføres farging på en vevsseksjon som er kjent for å uttrykke markøren anerkjent av det primære antistoffet.
3. Picro-sirius rød (PSR)/rask grønn fargingsprotokoll
MERK: Målet med denne farging er å visualisere flimmer kollagen I og III i FFPE vev seksjoner. Denne protokollen ble tilpasset fra Segnani et al.18. Alle trinnene utføres i en kjemisk hette.
- Utfør deparaffinisering og rehydrering av vevsseksjoner som ligner på multipleks immunofluorescensfargingsprotokollen for FFPE-seksjonene (avsnitt 2.1).
MERK: Hvis delen som skal merkes tidligere har blitt brukt til immunfluorescensmerking og parafinen allerede er fjernet, er deparaffiniseringsrehydreringstrinnene nyttige for å fjerne monteringsmediet. DAPI fjernes ikke ved hjelp av denne prosedyren, men det forstyrrer ikke psr-farsfarging. - Dypp lysbildene i en krukke som inneholder den picro-sirius røde/raske grønne løsningen (oppskrift i materialtabellen)og inkuber i 30 min ved RT (mer enn 30 minutter resulterer i uspesifikk farging av kjernene i hepatocytter).
- Vask lysbilder raskt i ddH2O (5 dips). Vask deretter raskt i etanol 100% (5 dips). Vask i 30 s i xylen-100% etanol (1:1 v / v). Vask i 30 s i xylen. Monter med monteringsmidler (se materialtabellen) før xylen en helt har fordampet (dette hjelper med monteringen).
MERK: Beregnet tid for å utføre denne delen av protokollen er 1 t.
4. Elution av antistoffer fra vevseksjoner
MERK: For å gjenbruke vevsseksjoner i sekvensielle merkingsanalyser, er det nødvendig med fullstendig fjerning av primære og sekundære antistoffer. Bundne antistoffer ble strippet som tidligere beskrevet13.
Forvarm et vannbad til 56 °C. Sett seksjonene i en krukke som inneholder strippingbuffer (oppskrift i materialbordet), lukk lokket og forsegle det med parafinfilmtape for å forhindre lekkasje under risting.
- Sett krukken inne i vannbadet og inkuber i 30 min med agitasjon.
- Vask 4x i 15 min hver i ddH2O ved RT. Skyll med PBS-Tween (0,1% v/v).
- Hold seksjonene hydrert i PBS-Tween eller vann til de er klare til å reprobe seksjonen med den andre runden av primære antistoffer.
MERK: Beregnet tid for å utføre denne delen av protokollen er 2 h. - Kontroller effektiviteten av antistoffelutionprosedyren.
MERK: Før du bruker protokollen for antistoffelution i en sekvensiell merkingsanalyse, bør effektiviteten av fjerning av primære og sekundære antistoffer verifiseres.- Utfør farging og bildeoppkjøp av en seksjon med et gitt primært sekundært antistoffpar av interesse som angitt i multipleks immunofluorescensfargingsprotokollen for FFPE-seksjoner (avsnitt 2.1–2.4.6).
- Ved bildeoppkjøp utfører du elution av vevsbundne primære antistoffkomplekser som angitt i pkt. 4.1–4.3.
- Inkuber seksjonen med samme sekundære antistoff og samme tilstand som brukes i trinn 2.4.3.
- Utfør trinn for oppvasking, montering og bildesom angitt i 2.4.4–2.4.6.
- Sammenlign side ved side bilder ervervet før og etter stripping for å fastslå hvorvidt det spesifikke signalet har forsvunnet.
MERK: Sammenligning av bilder før og etter fjerning av antistoffer vil validere effektiviteten av elutionprosedyren. Det er imidlertid normalt å se en økning i bakgrunnssignalet i alle kanalene, samt diffusjon av DAPI. Dette begrenser antall runder med stripping som kan utføres på samme vev seksjon. Tre runder med stripping ser ut til å være det maksimale.
5. Bildeoppkjøp
- Generer bilder ved hjelp av en hel lysbildeskanner.
- Bruk et objektivobjektiv på 20 x 0,75NA og en oppløsning på 0,3225 μm/piksel.
6. Bildeanalyse
MERK: Metoden som er beskrevet her, refererer til gjeldende eksempel. Se tabell 1 og teksten for å tilpasse seg andre spesifikke eksempler.
- Utfør vevjustering ved hjelp av Tissualign-modulen for bildeanalyseprogramvaren (VIS i denne protokollen, se Materialtabellen).
- Åpne bildeanalyseprogramvaren og klikk på Vevsjustering-fanen.
- Importer bildene som skal justeres i lysbildeskuffen ved å gå til Fil | Database og velg det første bildet som skal justeres. Gå tilbake til kategorien Vevsjustering og last bildet ved å klikke Last inn-knappen i lysbildeskuffen. Bildet vises i lysbildeskuffen og i arbeidsområdet.
MERK: Bare stakken av interesse skal legges inn i lysbildeskuffen. - Gjenta trinn 6.1.2 for alle bildene i rekkefølgen som skal justeres, og last dem en etter en. Når alle bilder av interesse er lastet inn i lysbildeskuffen fortsette å koble bildene ved å trykke Neste i arbeidsflyttrinnene i båndet.
- Deretter drar og slipper du det andre bildet oppå det første bildet. De første og andre bildene er nå koblet sammen. Gjenta dette trinnet for de andre bildene som skal justeres, en etter en, på en ryddig måte. Navnet på det første bildet endres, noe som indikerer at det er koblet til de andre bildene. Samtidig vises de koblede bildene i arbeidsområdet til høyre for lysbildeskuffen.
- På dette tidspunktet justerer du bildene enten ved hjelp av automatisk justering, halvautomatisk justering eller manuell justering. Det er alltid å foretrekke å prøve automatisk justering først. For automatisk justering, trykk på Neste-knappen i arbeidsflyttrinnene (trinn 3) på båndet.
- Se gjennom den automatiske justeringen ved å navigere forskjellige plasseringer av vevet og visuelt bekrefte at de tilsvarende strukturene i forskjellige bilder er ordnet på samme måte i de to dimensjonene på bildet.
- Hvis resultatet av den automatiske justeringen ikke er tilfredsstillende, kan du forbedre den ved hjelp av pinner (bruk minst tre pinner per bilde) som indikerer homologvevsfunksjoner i de koblede bildene. Når pinnene er plassert på homologe steder i de koblede bildene, har brukeren to valg: halvautomatisk justering eller manuell justering. For halvautomatisk justering klikker du på knappen Automatisk justering basert på gjeldende pinpoints i båndet. For manuell justering klikker du på knappen Bruk pins på båndet.
- Når du er fornøyd med justeringen, klikker du på Neste-knappen i arbeidsflyttrinnene og lagrer det sammensatte bildet i databasen.
MERK: Justere seks lysbilder som strekker seg over 11 markører pluss H&E- og PSR-bildene tok 15 minutter i analysen som ble presentert.
- Utfør vevsdeteksjon ved hjelp av den brukerdefinerte protokollen Analysis Protocol Package 1 (APP 1, tabell 1).
- Åpne Bildeanalyse-modulen for programvaren ved å klikke kategorien Bildeanalyse på båndet.
- Importere det sammensatte (justerte) bildet ved å gå til Fil | Database og velge bildet av interesse og klikke tilbake kategorien Bildeanalyse.
- Åpne dialogboksen APP-valg ved å klikke på Åpne APP-ikonet og velge hvilken Analysis Protocol Package (APP) du vil bruke. I dette tilfellet velger du APP 1 for vevsdeteksjon.
- Når APP 1 er åpnet, må du bekrefte at APP1 fungerer Preview som den skal ved å gå til et valgt vevssted og klikke på Forhåndsvisning-knappen. Hvis resultatene er tilfredsstillende, går du til neste trinn.
- Klikk for å kjøre APP 1 og behandle bildet ved hjelp av den valgte APPEN.
- Eksporter dataene (f.eks. bilder, målinger osv.) når analysen utføres ved å klikke fil/eksport.
MERK: APP 1 skaper en region av interesse (ROI) delineating vevet (ROI Tissue) og beregner området av vevet. - Lagre det endrede bildet med det nyopprettede AVKASTNINGEN ved å gå til Fil | Lagre.
MERK: Oppdage vevet og opprette en avkastning med APP 1 i det angitte eksemplet tok 5 min i bildeanalysestasjonen som er beskrevet. Området av vevet behandlet var 3,2 cm2.
- Utfør vevssegmentering i Stroma og Parenchyma ved hjelp av APP 2 (Tabell 1).
MERK: APP 2 fungerer på det forhåndsdefinerte avkastningen vev. APP 2 segmenterer vevet i ROIs Stroma og Parenchyma.- Åpne Bildeanalyse-modulen ved å klikke kategorien Bildeanalyse på båndet.
- Importer bildet som inneholder ROI-vevet ved å gå til Fil | Database og velge bildet som er lagret i trinn 6.2.7. Gå tilbake til Bildeanalyse-fanen og last inn bildet ved å klikke Last inn-knappen i lysbildeskuffen. Bildet vises i lysbildeskuffen og i arbeidsområdet.
- Åpne APP 2 ved hjelp av dialogboksen APP-valg som i 6.2.3.
- Forhåndsvis APP 2 ved å behandle i et valgt synsfelt. Hvis resultatene er tilfredsstillende, kjører du APP 2 Run på hele bildet ved å klikke på Kjør-knappen. Som utgangen av APP 2 segmenterer roivevet i ROIs Stroma og Parenchyma og deres respektive områder bestemmes. Eksport resultater som i 6.2.6. Lagre det endrede bildet som i 6.2.7.
MERK: Segmentering av vevet i Stroma og Parenchyma ved hjelp av APP 2 tok 4 timer i analysestasjonen som ble presentert. Området av vevet behandlet var 3,2 cm2.
- Identifisere og kvantifisere FoxP3hiCD4+-celler ved hjelp av den brukerdefinerte protokollen APP 3 (Tabell 1).
MERK: APP 3 fungerer på de forhåndsdefinerte ROIs Stroma og Parenchyma.- Åpne Bildeanalyse-modulen og importer bildet som inneholder ROIs Stroma og Parenchyma som i 6.3.1 og 6.3.2. Åpne APP 3 ved hjelp av dialogboksen APP-valg som i 6.2.3.
- Forhåndsvis APP 3-behandling i et valgt synsfelt beriket i FoxP3-høy-CD4+-celler.hi Hvis resultatene er tilfredsstillende, kjører du APP 3 på hele bildet. Som utgangen av APP 3, alle de enkelte FoxP3hiCD4 + objekter vil bli merket og deres vev koordinater lagret. Tettheter av FoxP3hiCD4 + objekter i ROIs Stroma og Parenchyma vil bli bestemt. Eksporter resultatene som i 6.2.6.
- Utfør vevvarmetilordning av FoxP3hiCD4+ merkede objekter.
- Åpne den brukerdefinerte protokollen FoxP3hiCD4+ MAP ved hjelp av dialogboksen APP-valg som i 6.2.3.
MERK: FoxP3hiCD4+ MAP bruker koordinatene til FoxP3hiCD4+ merkede objekter for generering av varmekart for tetthet. Identifisere og telle FoxP3hiCD4 + merkede objekter ved hjelp av APP 3 tok 25 min i bildeanalysestasjonen beskrevet. Området av vevet behandlet var 3,2 cm2. - Kjør FoxP3hiCD4+ MAP ved å trykke på Kjør-knappen. Eksporter vevsvarmekartet ved å klikke fil | Eksport | Arbeidsområde.
MERK: Kartlegging FoxP3hiCD4 + merkede objekter ved hjelp av FoxP3hiCD4 + MAP tok 5 min i bildeanalysestasjonen beskrevet.
- Åpne den brukerdefinerte protokollen FoxP3hiCD4+ MAP ved hjelp av dialogboksen APP-valg som i 6.2.3.
- Identifiser og kvantifiser CD8+, CD68+, MPO+, αSMA og CD34 + objekter ved hjelp av de brukerdefinerte protokollene APP 4, APP5, APP6, APP7 og APP 8, henholdsvis (Tabell 1) som gjort i avsnitt 6.4 til 6.4.3.2 som laster INN APP av interesse i hvert tilfelle.
MERK: APPs 4 til 8 arbeider med de forhåndsdefinerte ROIs Stroma og Parenchyma.
Representative Results
Oversikt over strategien for visualisering, kvantifisering og kartlegging av cellepopulasjoner av interesse i TME
For å kvantifisere cellepopulasjoner av interesse (COIer) i forskjellige vevsrom (TCer) og for å karakterisere deres romlige organisasjon, designet vi en arbeidsflyt som integrerer rimelige og brukervennlige teknikker og maksimerer posisjonsinformasjonen som kan fås fra dyrebare FFPE kliniske prøver (figur 1). For det første ble seriell hele vev FFPE seksjoner farget for visualisering av COIer (f.eks immunceller) og TCer (f.eks stroma versus parenchyma) (Figur 1, trinn 1). Antall påfølgende seksjoner som skal lagres bør holdes til det minste som tillater visualisering av cellene av interesse eller vev funksjoner som trengs for å løse forskningsspørsmålet. Jo mindre antall seriedeler, desto høyere vevsarkitektur likhet og konkordans på tvers av sammenhengende seksjoner. I tillegg kan multipleksing ser ut til å utvides gjennom gjenbruk av fluorescerende fargede seksjoner gjennom stripping og reprobing teknikker19.
Når flekkertrinnene var gjort, ble en hel lysbildeskanner brukt til å digitalisere bildene. Bilder hentet fra seriedeler ble justert og konsolidert i et virtuelt multiplekslysbilde på en automatisert måte (Figur 1, seksjon 2). Deretter ble en avkastning for vevet avgrenset med en brukerdefinert protokoll som identifiserte vevstilknyttede piksler (TAPer) (Figur 1, trinn 3). Deretter ble avkastningsvevet segmentert i TCer definert som ekstra ROIer. (Figur 1, trinn 4). Deretter oppdaget og kvantifiserte cois i forskjellige TCer(figur 1, trinn 5). Til slutt ble vevsvarmekart av COIer generert basert på deres tettheter og deres vevskoordinater (Figur 1, trinn 6).
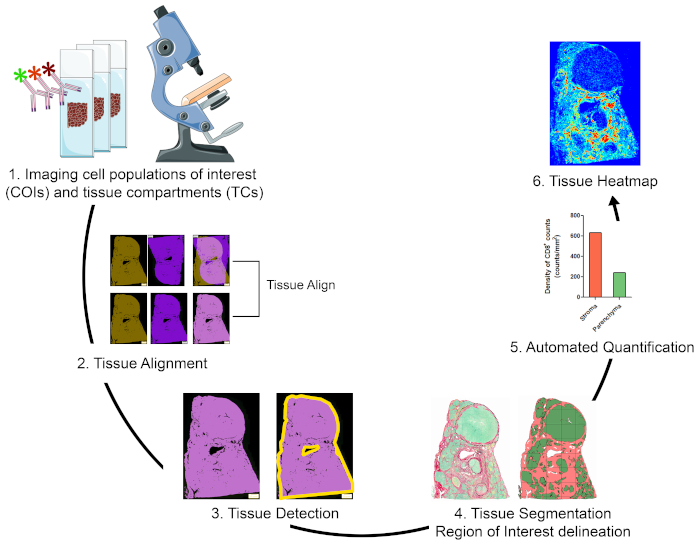
Figur 1: Skjematisk representasjon av strategien for visualisering, kvantifisering og kartlegging av immunceller i TME. (1) Seriell hele vev seksjoner ble farget for merking COIs og TCer. Farget hele vev seksjoner ble digitalisert ved hjelp av en hel lysbilde skanner. (2) Bilder hentet fra seriedeler ble koblet sammen, justert og coregistered på en automatisert måte ved hjelp av en Tissuealign analysemodul. Et sammensatt bilde ble generert fra høy presisjon justering av individuelle bilder. (3) En brukerdefinert protokoll ble brukt til automatisert påvisning av vevstilknyttede piksler (TAPer) i det sammensatte bildet. (4) Vevet ble segmentert i TCer (f.eks. stroma og parenchyma) definert som ROIer. (5) Brukerdefinerte protokoller ble brukt til automatisert deteksjon og kvantifisering av COI-er i forskjellige TCer. (6) Vevsvarmekart over COIer ble generert. Vennligst klikk her for å se en større versjon av denne figuren.
Bilde-COI-er og-tCer
Tre seriell FFPE hele vev deler av resected tumor fra et emne med HBV-assosiert hepatocellulært karsinom ble farget i en eller flere runder med farging som i figur 2A. Seksjon Jeg ble farget med H&E for å vise vevsarkitekturen, cellemorfologien, og for å bestemme klinisk relevante parametere som type malignitet, tumorklasse og generell vurdering av immuninfiltrasjon (figur 2C). I sammenhengende avsnitt II ble det brukt to runder med mIF for merking av leverparenchymale og ikke-parenchymale celler (Figur 2A). I første runde ble normale og tumorfartøy visualisert ved hjelp av CD34-farging av endotelceller. I tillegg ble epitelceller (hepacytter og cholangiocytter) identifisert ved hjelp av cytokeratin 8/18, og fibrogene aktiverte hepatiske stellateceller ble identifisert som alfa glatte muskelactin positive (αSMA+) celler (figur 2C). Etter bildeoppkjøp ble vevsseksjoner strippet og reprobed med antistoffer mot makrofager (CD68), og myofibroblaster (desmin). For bedre å karakterisere tumorimmuninfiltraten, ble tilstøtende serieseksjon III farget ved hjelp av to runder med mIF for cellulære markører CD3, CD4, CD8, gaffelhodeboks P3 (FoxP3) og myeloperoxidase (MPO). I alle tilfeller ble DAPI brukt som en kjernefysisk motflekk. Til slutt ble avsnitt III farget med PSR-flekk og motfarget med hurtiggrønn for å visualisere flimmerkollagen og segmentere vevet i stroma og parenchyma (Figur 2C).
En hel lysbildeskanner utstyrt med et 20X objektivobjektiv ble brukt til å digitalisere beisede seksjoner og for å lage virtuelle lysbilder. Seks bilder ble hentet fra de tre seriedelene (Figur 2B) og de virtuelle lysbildene analyserte deretter ved hjelp av VIS-programvaren i henhold til den skjematiske representasjonen i figur 1.
Bildeanalyse
Bildeanalysen besto av fem trinn: 1) vevjustering; 2) vev deteksjon; 3) vev segmentering; 4) automatisert kvantifisering av COIer; og 5) vev varme kartlegging. Alle protokoller for bildeanalyse ble utviklet ved hjelp av Forfatter-modulen til bildeanalyseprogramvaren og refereres til i teksten som APP.
Justering av vev
Seks virtuelle lysbilder fra tre seriedeler, som spenner over 11 markører pluss H&E og PSR-flekker, ble lastet inn i Tissualign-modulen til bildeanalyseprogramvaren. Deretter ble bildene koblet sammen, justert og coregistered på en automatisert måte, og genererte et 11-plex pluss H&E og PSR virtuell komposittbilde, som inneholder alle lagene av de enkelte bildene (Figur 2A-C). Justering var nøyaktig i tilfelle av bilder som stammer fra tilstøtende seriedeler, som viser tilsvarende vevstrukturer plassert og arrangert på en homolog måte ved justering (Figur 2C og figur S1A). Videre var justeringen nøyaktig på det enkelte cellenivå for bilder som stammer fra samme avsnitt (Figur S1B). Tiden for automatisk justering avhenger av antall, størrelse, kompleksitet og likhet av bildene som skal justeres. Justeringen av de ovennevnte seks virtuelle lysbildene tok 15 min i vår VIS stasjon.

Figur 2: Farging av serievevsseksjoner og bildejustering. (A) Sammendrag av flekker gjort på tre seriedeler for visualisering av COI-er og TCer. Tall i parentes indikerer bildebetegnelse. For seksjon II og III ble vev strippet og reprobed med en annen cocktail av antistoffer. (B) Oversikt over seks individuelle hele vevsbilder før og etter vevsjustering (henholdsvis venstre og høyre). Skalalinje = 3500 μm. (C) Zoomet visning av justerte bilder. Skala bar = 80 μm. Vennligst klikk her for å vise en større versjon av denne figuren.
Vev deteksjon
Når bildene var koblet sammen og justert, forsøkte vi å identifisere TAPs (Figur 3A). For å designe en APP for automatisert påvisning av TAPer (APP 1, tabell 1), benyttet vi oss av to egenskaper som skiller TAPs fra piksler som ikke er forbundet med vev. For det første er DAPI-signalet (blått bånd) begrenset til kjernene, som ligger utelukkende i vevet, noe som betyr at alle DAPI+ piksler er et delsett av TAPs. For det andre har TAPs høyere autofluorescenssignal i de grønne og gule båndene sammenlignet med piksler som ikke er forbundet med vevet. Derfor utviklet vi APP 1 for vevsdeteksjon (Tabell 1), som oppdager TAPs basert på baseline signal i disse kanalene ved hjelp av enkle terstertteknikker. Terskler for de blå, grønne og gule båndene ble satt slik at TAPs hadde bakgrunnsintensitetsverdier over tersklene, mens piksler som ikke var forbundet med vevet hadde verdier nedenfor. APP 1 for vevsdeteksjon ble brukt på bilde IIA, som inneholder lag i blå, grønne og gule kanaler (Figur 3A). Som utganger av APP 1, ble en lys grønn maske lagt ned på toppen av TAPs, og en avkastning kalt "Vev" ble avgrenset (utgang, figur 3A). Videre ble området av vevet bestemt som en kvantitativ utgangsvariabel. Fordi APP 1 ikke inkorporerer pikslene som ikke er forbundet med vevet i avkastningen vev, ble de ekskludert fra etterfølgende analyse basert på denne avkastningen (figur 3A). Presisjonen til APP 1 ved identifisering av TAPer vises i figur 3A.
Vevssegmentering og avgrense av ROIer for TCer
Deretter fortsatte vi å definere forskjellige rom inne i roivevet ved å segmentere vevet i stroma versus parenchyma. Vi brukte PSR farget bilde (IIIC, Figur 2C),hvor stroma kan defineres som området forbundet med avsetning av flimmer kollagen (rødt bånd), parenchyma som området der flimmer kollagen er fraværende, og den raske grønne counterstaining dye råder (grønt bånd) (Figur 3B). Vi opprettet APP 2 (Tabell 1) for å digitalt avgrense TCer Stroma og Parenchyma. Denne APP fungerer på det forhåndsdefinerte AVKASTNINGEN Tissue (utgang, figur 3A)og bruker representative stroma og parenchyma områder for opplæring av klassifikatorverktøyet integrert i Bildeanalyse modulen. Den opplærte klassifikatoren tilordner pikslene til enten en stroma eller en parenchymaetikett (henholdsvis laks og grønt, figur 3B). Ved klassifisering av piksler utførte APP 2 morfologiske operasjoner med sikte på å definere ROIs Stroma og Parenchyma (Figur 3B og tabell 1). Ytelsen til APP 2 ved klassifisering av piksler og generering av de respektive ROIene vises i figur 3B. I tillegg kvantifiserer APP 2 området av stroma og parenchyma. Til slutt, selv om segmenteringen er gjort ved hjelp av PSR farget delen, kan de skisserte stroma og parenchyma regioner overføres til et bilde justert til PSR-bildet.

Figur 3: Automatisert vevsdeteksjon/segmentering og generering av respektive ROIer. (A) Bilde IIA ble brukt til å identifisere TAPs (venstre bilde, skala bar = 6000 μm). En lys grønn maske ble tildelt TAPer ved hjelp av APP 1 (Tabell 1) som genererer en avkastning kalt Tissue (utgang 1). Høyre, innfelt viser zoomet visning som viser presisjonen til APP 1 ved å oppdage TAPs. Skalastang = 350 μm. (B) ROI-vevet (utgang 1) er segmentert i stroma og parenchyma ved hjelp av APP 2. Bildet til venstre viser en visning av roivevesegmentert i roistroma (laks) og ROI parenchyma (grønn). Skalabar = 4500 μm. Til høyre, zoomet utsikt over innfelt for ROI Tissue, den opprinnelige PSR farging (bilde IIIC), og ROIs stroma og parenchyma. Skalabar = 250 μm. Vennligst klikk her for å se en større versjon av denne figuren.
Automatisert kvantifisering av COI-er
Deretter fortsatte vi å identifisere, lokalisere og kvantifisere COIer i ROIs Stroma og Parenchyma. APPs 3 til 8 (Tabell 1) ble opprettet for å finne og telle følgende COIer: CD4 + FoxP3 + , CD8 + CD68 + , MPO + , αSMA + og CD34 + celler, henholdsvis. APP 3 ble designet for å finne og telle CD4+ FoxP3 + celler (bilde IIIA, figur 2C) som surrogatmarkører for regulatoriske T-celler (Tregs). Denne protokollen oppdager samlokalisering av signalet fra kjernefysisk transkripsjonsfaktor FoxP3 (rødt bånd) og DNA-merking dye DAPI (blått band). Gitt at nylig aktivertE T-celler oppregulere FoxP3, for å berike for Tregs setter vi terskler for forhåndsvalg bare lyse FoxP3 + celler (FoxP3hi). Deretter, av alle forhåndsvalgte DAPI + FoxP3hi celler, bare de som var omgitt av lyse ringformede CD4 signaler (grønt bånd) ble merket og regnet som FoxP3hiCD4 + celler (rosa etikett, Figur 4A). Tettheten av FoxP3hiCD4 + celler i ROIs Stroma og Parenchyma ble bestemt som kvantitative utgangsvariabler av APP 3 (Figur 4A).
På samme måte ble APPs 4 til 6 designet for påvisning av CD8+, CD68+- og MPO+-celler. Disse APPer deler samme baseline design for å oppdage og kvantifisere COIer. Spesielt identifiseres COIer basert på signalintensitet fra den spesifikke cellepopulasjonens biomarkør, og deretter utføres flere etterbehandling morfologiske trinn for å avgrense individuelle celler (tabell 1). De enkelte cellene eller COIene er merket, talt, og deres vevkoordinater registrert. APPs 4 til 6 bestemmer også tettheten av COI-ene i ROIs Stroma og Parenchyma (figur 4B-D).
Kvaliteten på vår DAPI-farging var ikke god nok til å integrere kjernesegmentering i APPs 3 til 6, så vi kan ikke sikre at alle individuelt merkede objekter er individuelle celler. Av denne grunn uttrykte vi tettheten av celler i tellinger av merkede objekter/mm2 (Figur 4). Celleaggregater ble imidlertid delt inn i individuelle celler i etterbehandlingstrinnene som ble innebygd i APPs 3 til 6, og omfattende visuell inspeksjon viste at de fleste merkede objekter tilsvarte enkeltceller.
For å oppdage αSMA+ og CD34+ område utviklet vi henholdsvis APPs 7 og 8 (Tabell 1). Begge APPs oppdager det spesifikke signalet basert på terskler og bestemmer prosentandelen av positivt område i ROIs Stroma og Parenchyma (Figur 4E–F).
En av de mest interessante mulighetene for å generere virtuelle multiplekslysbilder er analysen av kolokaliseringsuttrykk. Vi genererte APP 10 for å oppdage samlokalisering mellom αSMA og desmin, to markører co-uttrykt av myofibroblasts i leveren. APP 10 bruker terskler for å finne piksler som er positive for αSMA, desmin og αSMA pluss desmin (Tabell 1). Som kvantitative utgangsvariabler bestemmer APP 10 αSMA+-området, desmin+-området og området kolokalisert uttrykk for disse to markørene (figur S3).

Figur 4: Identifikasjon og kvantifisering av COI-er i TC-stroma og parenchyma. -AJeg har ikke noe åsi. Automatisert deteksjon og kvantifisering av CD4+FoxP3+, CD8+, CD68+, MPO+, αSMA+, og CD34+ COIer i ROIs Stroma og Parenchyma ved hjelp av henholdsvis protokoller 3, 4, 5, 6, 7 og 8 (tabell 1). Vist til venstre er de opprinnelige bildene, i midten de behandlede bildene, og til høyre kvantifiseringene. For figur 4A–D, skalastang = 40 μm. For figur 4E og F, skala bar = 350 μm. Vennligst klikk her for å vise en større versjon av denne figuren.
Som et alternativ til å kvantifisere COI-ene i TCene Stroma og Parenchyma, bestemte vi tettheten av immunceller i de forskjellige ondartede knutene kalt 1 til 4 (Figur 5A, Hog jeg). Avkastningen for hver knute ble manuelt avgrenset som angitt i figur 5A. Karakteristiske vev immun signaturer preget hver knute, ytterligere avsløre iboende heterogenitet av TME.
Vev Varmekart
Som nevnt ovenfor lagrer APPs 3 til 8 vevskoordinatene til hvert individuelt merket objekt. Denne funksjonen gjør det mulig for den automatiserte genereringen av vevskart der regioner med høy tetthet av en gitt cellepopulasjon vises som hot spots (rød), og regioner med relativt lav tetthet som kalde flekker (mørk blå). Mellomliggende tetthetsverdier tilordnes farger i henhold til fargeskalaen som vises i figur 5. Vevsvarmekart ble generert av APPer som delte bildene i sirkler med 50 μm diameter og tildelt en farge i henhold til den relative tettheten av en gitt COI inne i sirkelen. Som vist i figur 5B–G,var posisjoneringsmønstrene og intensitetsfordelingen til de ulike KOIene i TME ganske variert. Videre, på nivået av individuelle knuter, var arrangementet av forskjellige populasjoner i vevsområdet unik (figur S2A-C). For å gi et eksempel på kraften i denne teknikken og visualisere den romlige organiseringen av hot spots fra forskjellige populasjoner i samme knute, ble hot spots fra individuelle celletyper manuelt hentet og kartlagt sammen på omrisset av knute 2 (Figur S2, Figur Dog Figur E).

Figur 5: Vevsvarmekart over COIer i TME. (A) Picrosirius Rød flekker viser plassering av knuter 1, 2, 3 og 4. -BJeg har ikke noe åsi. Vevsvarmekart for henholdsvis CD4+FoxP3+, CD8+, CD68+, MPO+, CD34+, og αSMA+COI. Mørk blå indikerer relativ lav tetthet, og rød indikerer relativ høy tetthet. Mellomliggende tetthetsverdier tilordnes farger i henhold til den viste fargeskalaen. (H og I) Kvantifisering av COI i henholdsvis 1, 2 og 3 + 4 organisert per celletype og per knute. Vennligst klikk her for å se en større versjon av denne figuren.

Supplerende figur S1: Validering av vevjustering. (A) CD34-farging (i rødt) gjort på avsnitt II (inngang 1) brukes til å generere en CD34-maske i grønt (utgang 1). Den grønne masken (utgang 1) legges over h&e-bildet fra den justerte seriedelen I (inngang 2). Fusjonsbildet viser perfekt korrespondanse med vaskulære strukturer. Skalabar = 50 μm. (B) Bilde IIIA som viser sammenslåingen av DAPI, CD4 og FoxP3 (inngang 1) ble brukt til å generere en etikett for CD4+ FoxP3 + celler (utgang 1 i magenta). Utgang 1 etiketten ble overført til justert bilde IIIB (inngang 2) og viser perfekt korrespondanse mellom parene FoxP3 / DAPI, og CD4 / CD3 i fusjonsbildet. Skalabar = 15 μm. Vennligst klikk her for å se en større versjon av denne figuren.

Supplerende figur S2: Zoomet visning av vev varmekart. (A-C) Vevsvarmekart for CD4+FoxP3+- CD8+- CD68+- og MPO+-celler i knuter 1–4. Skalastenger i knuter 1, 2 og 3 + 4 representerer henholdsvis 1500 μm, 700 μm og 500 μm. (D) Omrisset av knute 2 med svart fast linje. (E) Hot spots for CD4 + FoxP3 + , CD8 + CD68 + og MPO + celler i knute 2 ble hentet og kartlagt sammen på knuten 2 disposisjon definert i D. Vennligst klikk her for å se en større versjon av denne figuren.

Supplerende figur S3: Colocalization Analyse. (A) På venstre og midten er bilder av αSMA etiketten i grønn og desmin etikett i rødt henholdsvis. Til høyre er et αSMA/desmin dobbelt positivt område i gult. (B) Kvantifisering av αSMA+ område, desmin + område og αSMA/desmin dobbelt positivt område. Skalabar = 150 μm. Vennligst klikk her for å se en større versjon av denne figuren.
| App | Formål | Klassifisering | Klassifisering | Trinn for behandling etter behandling | Utgangsvariabler |
| Metoden | Funksjoner | ||||
| (pikselverdi) | |||||
| 1 | Vev deteksjon | Terskelen | Kanal DAPI (150) | o Merk objekter med kolokaliserte verdier over terskelen for de 3 kanalene | o ROI Vev |
| Kanal FITC/A488 (120) | o Lukk positivt objekt 5 piksler | o Vevsområde | |||
| Kanal TRITC/A568 (40) | o Lag ROI-vev | ||||
| 2 | Vev segmentering | Beslutning sandskog | RGB-R median | o Fyll hull | o ROI Stroma |
| RGB-G median | o Lag ROI Stroma | o Stroma-området | |||
| RGB-B median | o Lag ROI Parenchyma | o ROI Parenchyma | |||
| IHS-S median | o Parenchyma-området | ||||
| H&E Eosin median | |||||
| 3 | Finne og kvantifisere CD4+ FoxP3+-celler | Terskelen | Kanal-DAPI (>600) | o Merke objekter med kolokalisering av DAPI og Cy5/A647, omgitt av FITC/A488-signal | o Tellinger og tetthet av CD4+FoxP3+-celler i ROIs Stroma og Parenchyma |
| Kanal FITC/A488 poly utjevning (>850) | o Fjern objekter som er mindre enn 7 μm2 | o Koordinater for individuelle CD4+FoxP3+-celler | |||
| Kanal Cy5/A647(>800) | |||||
| 4 | Slik finner og kvantifiserer du CD8+-celler | Terskelen | Kanal-DAPI (<1200) | o Fjern positive gjenstander som er mindre enn 15 μm2 | o Tellinger og tetthet av CD8+-celler i ROIs Stroma og Parenchyma |
| Kanal Cy5/A647 median (>80) | o Lukk positive objekter 2 piksler | o Koordinater for individuelle celler | |||
| o Separate objekter | |||||
| 5 | Slik finner og kvantifiserer du CD68+-celler | Terskelen | Kanal FITC/A488 (>200) | o Fjern positive gjenstander som er mindre enn 20 μm2 | o Tellinger og tetthet av CD68+-celler i ROIs Stroma og Parenchyma |
| o Utvide positive objekter 3 piksler | o Koordinater for individuelle CD68+ celler | ||||
| o Separate objekter | |||||
| 6 | Slik finner og kvantifiserer du MPO+-celler | Terskelen | Kanal-DAPI (>400) | o Fjern objekter som er mindre enn 5 μm2 | o Tellinger og tetthet av MPO+-celler i ROIs Stroma og Parenchyma. |
| Kanal TRITC/A568 (900-4000) | o Utvide 3 piksler positive objekter | o Koordinater for individuelle MPO+-celler. | |||
| o Separate objekter | |||||
| 7 | Slik finner og kvantifiserer du αSMA+-området | Terskelen | Kanal TRITC/CF568 (>1050) | o Fjern positive gjenstander som er mindre enn 25 μm2 | o Tellinger og tetthet av αSMA+-området i ROIs Stroma og Parenchyma |
| o Utvide 3 piksler positive objekter | o Koordinater for αSMA+ piksler | ||||
| 8 | Slik finner og kvantifiserer du CD34+-området | Terskelen | Kanal-DAPI (<5000) | o Fjern positive gjenstander som er mindre enn 25 μm2 | o Tellinger og tetthet av CD34+ område i ROIs Stroma og Parenchyma |
| Kanal Cy5/A647 median (>120) | o Utvide 3 piksler positive objekter | o Koordinater for CD34+ piksler | |||
| 9 | Lag vevsvarmekart for en gitt cellepopulasjon | Objekt Varmekart | Objekt Varmekart | o Varmekart | |
| Tegning radius 50 μm | --- | ||||
| 10 | Kvantifiser ei kolokalisering mellom αSMA og Desmin | Terskelen | Kanal TRITC (CF568) (>1050) | o Merke objekter med terskelverdier ovenfor for TRITC (CF568) | o Kvantifisere kolokalisert uttrykk for αSMA og Desmin |
| Kanal Cy5 (A647) (>1000) | o Merke objekter med terskelverdier ovenfor for Cy5 (A647) | ||||
| o Merke objekter med kolokalisering av ovennevnte terskelverdier for TRITC (CF568) og Cy5 (A647) | |||||
| o Fjern positive gjenstander som er mindre enn 25 μm2 |
Tabell 1: Generelle parametere som brukes til utforming av APPer som brukes for bildeanalyse. Parameterne som er angitt i denne tabellen, justeres etter de unike egenskapene til bildene som brukes i denne analysen (f.eks. bakgrunn, artefakter osv.) og gjelder kanskje ikke for andre bilder. Fordi de nevnte etterbehandlingstrinnene ble definert for de spesifikke bildene som ble analysert i denne studien, er de med vilje ikke detaljerte. Brukeren bør tilpasse APPs til bildene som skal analyseres.
| Seksjon/farging | Primær antistoff | Sekundært antistoff |
| Avsnitt II/1st Befaring | Mus IgG2a anti-human αSMA Mus IgG1 anti-menneskelig CD34 Kanin anti-human Cytokeratin 8/18 |
Geit anti-mus IgG2a CF568 Rotte anti-mus IgG1 A647 Esel anti-kanin A488 |
| Avsnitt II/2nd Farging | Kanin anti-menneskelig Desmin Mus anti-menneskelig CD68 |
Esel anti-kanin A647 Esel anti-mus DyLight 755 |
| Avsnitt III/1st Beising | Mus anti-menneskelig CD4 Kanin anti-menneskelig FoxP3 Geit anti-menneskelig MPO |
Esel anti-mus A488 Esel anti-kanin A647 Esel anti-geit A568 |
| AvsnittIII/2. Beising | Kanin anti-menneskelig CD3 Mus anti-menneskelig CD8 |
Esel anti-mus DyLight 755 Esel anti-kanin A647 |
Tabell 2: Primærsekundære antistoffpar for mIF.
Discussion
Enkle, tilgjengelige og enkle å utføre multipleksingteknikker som tillater romlig oppløsning av immunceller i vevsseksjoner er nødvendig for å kartlegge immunlandskapet i kreft og andre immunologiske lidelser. Her beskriver vi en strategi som integrerer allment tilgjengelige merkings- og digitale analyseteknikker for å utvide multipleksjonsevnen og flerdimensjonal vurdering av bildebehandlingsanalyser12,13,17,19. Farging av tre seriedeler for forskjellige markører, og gjenbruk av seksjoner gjennom stripping og reprobing teknikker, gjorde oss i stand til å visualisere 11 parametere i tillegg til H & E og PSR flekker. Seks bilder fra disse seksjonene ble justert på en automatisert måte ved hjelp av vevjusteringsmodulen. Justeringen var nøyaktig på det enkelte cellenivå for bilder som stammer fra samme del og svært konkordans for bilder som stammer fra nærliggende seksjoner. Virtuell multipleksing gjorde det mulig for oss å bestemme hvordan markører visualisert i en del er romlig knyttet til markører visualisert i en annen sammenhengende del. Mens noen av flekker merket COIer, andre merket TCer, slik at vi kan kvantifisere COI er i de forskjellige TCene. Bruken av programvareverktøy for automatisert kvantifisering av COIer forenklet og akselerert behandling av bilder. Videre ble digital analyse brukt på hele vevsseksjoner i stedet for utvalgte synsfelt, noe som resulterte i en objektiv representasjon av TME. Videre, fordi vevkoordinatene til COI-er ble registrert, var det mulig å generere vevsvarmekart.
Det finnes flere områder i denne protokollen der feilsøking kan være nødvendig. For det første kan dårlig antigenhenting påvirke kvaliteten på mIF, derfor bør typen antigengjenfinningsbuffer og varighet optimaliseres for de spesifikke analyse-/biomarkørforholdene som brukes. For det andre bør typen blokkeringsløsning som brukes tilpasses vev/antigen/arter av primære og sekundære antistoffer. I våre hender, tillegg av 10% totalt serum fra arten der vevet kommer fra blokkerte Fc reseptorer, og dermed redusert uspesifikk antistoff binding. Tilsetning av 10% av serum fra arten de sekundære antistoffene ble reist i ville minimere direkte uspesifikk vedlegg av sekundære antistoffer mot vevsseksjonen. For det tredje er validering av spesifisiteten til de primære og sekundære antistoffene ved hjelp av de riktige positive og negative kontrollene avgjørende. Fjerde, økt autofluorescens i noen kanaler og diffusjon av DAPI ved primær antistoff stripping er også vanlig. For å løse den forbedrede autofluorescensen brukte vi primære/sekundære antistoffpar der det spesifikke signalet hadde intensitetsverdier minst 5x som i bakgrunnen. Til slutt kan noen høy affinitetsantistoffer ikke eluted med vanlige strippingprosedyrer. I dette tilfellet anbefaler vi å bruke slike antistoffer i den siste runden med merking. Brukeren må kanskje prøve forskjellige fargesekvenser for å finne den optimale konfigurasjonen for antistoffene av interesse. Effektiviteten av stripping bør bekreftes før du går videre til en andre eller tredje runde med merking.
Hovedbegrensningen og utfordringen med denne strategien er å finne de riktige kombinasjonene av primære og sekundære fluorescerende antistoffer for markører av interesse. Å finne primære antistoffer oppvokst i forskjellige arter eller med forskjellige isotyper som kan brukes samtidig, er begrenset av det som er kommersielt tilgjengelig. De fleste hele lysbildeskannere er utstyrt med lamper og filtre som tillater bildebehandling maksimalt fem kanaler, og sekundære antistoffer i de riktige artene og høyre fluoroforfor er ikke alltid tilgjengelig. Vi overvant delvis disse begrensningene ved hjelp av serielle flekker og sekvensiell merking. Flere antistoffkombinasjoner må kanskje testes for å komme frem til den beste kombinasjonen for markører av interesse. En annen begrensning er kvaliteten på DAPI farging, fordi stripping og reprobing kan ikke alltid tillate utføre kjernesegmentering.
Vevsjusteringsmodulen krever minimal opplæring og ingen programmeringsferdigheter fra brukerne. Programvaren teoretisk tillater justering av et ubegrenset antall bilder. Imidlertid avhenger presis justering av sammenhengen mellom seksjoner, hvor nærmere seksjoner som er mer histologisk konkordans er mer nøyaktig justert. Vi brukte Forfatter-modulen til VIS for å generere APPer. Grunnleggende kunnskap om bildeanalyse er nødvendig for å lage APPer, men dette er like tilfelle når du bruker annen bildeanalyseprogramvare. De unike fordelene med VIS sammenlignet med annen bildeanalyseprogramvare inkluderer automatisert justering av bilder fra seksjoner utarbeidet ved hjelp av forskjellige metoder (f.eks. IF, histochemistry, IHC). Dette gjør det mulig for kolokaliseringsstudier av flere markører av interesse ved hjelp av virtuell multipleksing. Videre tillater den fleksible og brukervennlige utformingen av APPer brukerspesifikk tilpasning. Automatisert kvantifisering og kartlegging, og muligheten for behandling av hele vevseksjoner, sparer tid og reduserer skjevhet sammenlignet med manuell telling ved visuell inspeksjon.
Denne strategien er et svært nyttig forskningsverktøy for vevimmunologi i sammenheng med kreft og autoimmunitet, men forblir ikke validert for klinisk bruk. Med ytterligere standardisering og validering kan den brukes i fremtiden for flere applikasjoner (f.eks. for å kartlegge immunlandskapet i kreft for å forutsi og overvåke responsen på immunterapeutiske midler). Det kan også tilpasses forskjellige inflammatoriske tilstander (f.eks. inflammatorisk tarmsykdom) for å kombinere patologisk evaluering med prognostiske biomarkører.
De viktigste kritiske trinnene i denne protokollen er effektiviteten/spesifisiteten til merkingen og robustheten til de utformede APPer-ene for tiltenkt bruk eller biomarkør. Derfor er regelmessig validering ved visuell inspeksjon, spesielt ved utforming av en ny APP, avgjørende. Effektiv bruk av flere runder med stripping og frastøting eller ulike typer flekker på samme seksjon er kritiske komponenter og kan være vev eller seksjonsspesifikke. Det er avgjørende å verifisere effektiviteten til slike prosesser før du fortsetter med stor batchanalyse.
Oppsummert tilbyr vi en strategi som maksimerer den kvantitative og romlige informasjonen som kan fås fra verdifulle kliniske vevsprøver. Ressursene, utstyret og kunnskapen som kreves for å implementere denne metodikken er allment tilgjengelig. Vi foreslår denne metodikken som en nyttig veiledning for planlegging av analyser som tar sikte på å identifisere, kvantifisere og kartlegge immuncellepopulasjoner i TME.
Disclosures
Forfatterne erklærer ingen interessekonflikter.
Acknowledgments
Vi takker studiedeltakeren. Vi takker Louise Rousseau, koordinator for HBP biobank for utvinning av vevsprøvene og all tilhørende klinisk informasjon. Vi anerkjenner molekylær patologi og cellebildekjerneanlegg ved CRCHUM og Michael Persch fra Visiopharm for utmerket teknisk assistanse. Finansiering: Denne studien ble støttet av tilskudd fra Canadian Liver Foundation, Fonds de recherche du Québec–Santé (FRQS) AIDS og Infectious Disease Network (Réseau SIDA-MI), og Canadian Network on Hepatitt C (CanHepC). CanHepC er finansiert av et felles initiativ fra Canadian Institutes of Health Research (CIHR) (NHC-142832) og Public Health Agency of Canada. M.F.M. fikk stipend fra Université de Montréal, Bourse Gabriel Marquis og FRQS. T.F. fikk doktorgradsstipend fra CIHR og CanHepC. S.T. har Roger-Des-Groseillers Chair i hepatobiliær og bukspyttkjertel onkologisk kirurgi, Université de Montréal.
Forfatterbidrag: M.F.M. designet, utførte eksperimenter og analyserte data. T.F. designet eksperimenter. A.C-B. teknisk veiledning. G.S. utførte all den patologiske vurderingen av studieemnet og ga innspill på alle de patologiske aspektene. L.M. utførte H&E-farging, optimalisert og utført bildeoppkjøp. M.N.A. utførte PSR-flekken og ga verdifull teknisk inngang. N.B. bidro til bildeanalysen. S.T. er hovedetterforsker for HBP biobank og er ansvarlig for å overvåke den generelle driften av biobanken. Han ga også uvurderlige innspill på alle aspekter av prosjektet og dets kliniske implikasjoner. M.F.M, T.F., og N.H.S. konseptualisert og designet studien. N.H.S. overvåket arbeidet og fikk finansiering. M.F.M., T.F., A.C-B og N.H.S. skrev manuskriptet. Alle forfattere anmeldte og godkjente manuskriptet.
Materials
| Name | Company | Catalog Number | Comments |
| Antigen Retrieval Solution: Sodium Citrate Buffer (10 mM Sodium Citrate, 0.05% v/v Tween 20, pH 6.0) | |||
| Blocking Solution: 1 % BSA, 10 % filtered human serum, 10 % filtered donkey serum, 0.1 % Tween 20, and 0.3% Triton in PBS | |||
| Bovine serum albumin (BSA) | Multicell | 800-095-EG | |
| Coplin jars (EASYDIP SLIDE STAINING SYSTEM) | Newcomersupply | 5300KIT | |
| Cover slides | Fisherbrand | 12-545E 22*50 | |
| Direct Red 80 | Sigma Aldrich | 365548 | |
| Donkey Serum | Sigma Aldrich | D9663 | |
| Ethanol 100% | |||
| Electric pressure cooker | Salton | ||
| Eosin | Leica Biosystems | 3801600 | CAUTION, eye irritation |
| Fast Green FCF | Sigma Aldrich | F7252 | CAUTION, harmful by inhalation, ingestion and skin absortion |
| FFPE section (4μm) slides | |||
| Glycine 0,1 M in PBS | |||
| Hematoxylin Stain Solution, Gil 1. Formulation, Regular Strength | Ricca Chemical Company | 3535-32 | |
| Holder (EasyDip Staining Jar Holder) | Newcomersupply | 5300RK | |
| Human Serum | Gemini | 22210 | |
| Humidity chamber | Millipore Sigma | Z670138-1EA | |
| Pap pen | abcam | ab2601 | |
| PBS | |||
| PBS-Tween 20 (0.1% v/v) | |||
| Permount Mounting Media | Fisher Chemical | SP15-500 | |
| Picric Acid 1.3 % | Sigma Aldrich | P6744 | CAUTION, skin and eye irritation |
| Picro-Sirius Red/Fast Green solution: Fast Green 0.1 % w/v + Sirius Red 0.2 % w/v in 1,3 % picric acid solution | |||
| Primary Antibody Anti-αSMA | Mouse IgG2a 1A4 | Sigma A2547 | Dilution 1/100 |
| Primary Antibody Anti-CD34 | Mouse IgG1 HPCA1/763 | Novus Biologicals NBP2-44568 | Dilution 1/250 |
| Primary Antibody Anti-Cytokeratin 8/18 | Rabbit EP17/EP30 | Agilent IR09461-2 | Ready to use |
| Primary Antibody Anti-CD68 | Mouse KP1 | Abcam ab955 | Dilution 1/200 |
| Primary Antibody Anti-Desmin | Rabbit Polyclonal | Invitrogen PA5-16705 | Dilution 1/200 |
| Primary Antibody Anti-CD4 | Mouse N1UG0 | Affymetrix 14-2444 | Dilution 1/250 |
| Primary Antibody Anti-FoxP3 | Rabbit 1054C | R & D MAB8214 | Dilution 1/100 |
| Primary Antibody Anti-MPO | Goat Polyclonal | R & D Systems AF3667 | Dilution 1/250 |
| Primary Antibody Anti-CD3 | Rabbit SP7 | Abcam ab16669 | Dilution 1/200 |
| Primary Antibody Anti-CD8 | Mouse C8/144B | Invitrogen 14-0085-80 | Dilution 1/200 |
| Secondary Antibody Donkey anti-mouse A488 | Polyclonal | Invitrogen A-21202 | Dilution 1/500 |
| Secondary Antibody Donkey anti-Rabbit A488 | Polyclonal | Invitrogen A-21206 | Dilution 1/500 |
| Secondary Antibody Donkey anti-goat A568 | Polyclonal | Invitrogen A-11057 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit A647 | Polyclonal | Invitrogen A-31573 | Dilution 1/500 |
| Secondary Antibody Rat anti-mouse IgG1 A647 | RMG1-1 | Biolegend 406618 | Dilution 1/500 |
| Secondary Antibody Goat anti-mouse IgG2a CF568 | Polyclonal | Sigma Aldrich SAB4600315 | Dilution 1/500 |
| Secondary Antibody Donkey anti-mouse DyLight 755 | Polyclonal | Invitrogen SA5-10171 | Dilution 1/500 |
| Secondary Antibody Donkey anti-rabbit DyLight 755 | Polyclonal | Invitrogen SA5-10043 | Dilution 1/500 |
| SDS | BioShop | SDS001,500 | CAUTION, oral skin and eye toxicity |
| Shandon multi-program robotic slide stainer | LabX | 11384903 | |
| Shandon Xylene Substitute, | Thermo Fisher Scientific | CA89413-336 | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Shaking water bath | |||
| SlowFade Gold antifade reagent with DAPI | Invitrogen | S36938 | |
| Sodium Citrate Dihydrate | Millipore Sigma | 1545801 | CAUTION, eye irritation |
| Stripping Buffer: mix 20 ml 10% w/v SDS with 12.5 ml 0.5 M Tris-HCl (pH 6.8), and 67.5 ml ultra-pure water. Under a fume hood, add 800 uL of 2-mercapto ethanol (114,4 mM final concentration) | |||
| Triton X-100 | Sigma Aldrich | T8787-50ML | |
| Tris-HCl | BioShop | 77-86-1 | |
| Tween 20 | Fisher Scientific | BP337-500 | |
| VIS Software | Visiopharm | ||
| Whole slide scanner Olympus BX61VS | Olympus | Microscope: Olympus Slide Scanner BX61VS, 5 slides scanner, motorized stage, autofocus. Camera: Lightsource: Xcite-120. Filters: BrightLine® Sedat filter set (# LED-DA/FI/TR/Cy5-4X4M-B-000, Semrock) | |
| Xylene | Sigma Aldrich | 214736-4L | CAUTION, Flammable, skin and eye irritation, Harmful when inhaled |
| Xylene : Ethanol solution (1:1 v/v) | |||
| 2-mercaptoethanol | Sigma | M6250 | CAUTION, harmful by ingestion, inhalation, fatal if sking absortion. Eye irritation. Use fume hood |
References
- Greten, F. R., Grivennikov, S. I. Inflammation and Cancer:Triggers, Mechanisms, and Consequences. Immunity. 51 (1), 27-41 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Binnewies, M., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine. 24 (5), 541-550 (2018).
- Taube, J. M., et al. Implications of the tumor immune microenvironment for staging and therapeutics. Modern Pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 31 (2), 214-234 (2018).
- Bindea, G., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 39 (4), 782-795 (2013).
- Galon, J., et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. The Journal of Pathology. 232 (2), 199-209 (2014).
- Finotello, F., Eduati, F. Multi-Omics Profiling of the Tumor Microenvironment: Paving the Way to Precision Immuno-Oncology. Frontiers in Oncology. 8, 430 (2018).
- Gerner, M. Y., Kastenmuller, W., Ifrim, I., Kabat, J., Germain, R. N. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 37 (2), 364-376 (2012).
- Giesen, C., et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nature Methods. 11 (4), 417-422 (2014).
- Porta Siegel, T., et al. Mass Spectrometry Imaging and Integration with Other Imaging Modalities for Greater Molecular Understanding of Biological Tissues. Molecular Imaging and Biology : MIB: the official publication of the Academy of Molecular Imaging. 20 (6), 888-901 (2018).
- Buchberger, A. R., DeLaney, K., Johnson, J., Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Analytical Chemistry. 90 (1), 240-265 (2018).
- Pirici, D., et al. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. Journal of Histochemistry and Cytochemistry. 57 (6), 567-575 (2009).
- Gendusa, R., Scalia, C. R., Buscone, S., Cattoretti, G. Elution of High-affinity (>10-9 KD) Antibodies from Tissue Sections: Clues to the Molecular Mechanism and Use in Sequential Immunostaining. Journal of Histochemistry and Cytochemistry. 62 (7), 519-531 (2014).
- van der Loos, C. M. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. Journal of Histochemistry and Cytochemistry. 56 (4), 313-328 (2008).
- Stack, E. C., Wang, C., Roman, K. A., Hoyt, C. C. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 70 (1), 46-58 (2014).
- Toth, Z. E., Mezey, E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. Journal of Histochemistry and Cytochemistry. 55 (6), 545-554 (2007).
- Robertson, D., Savage, K., Reis-Filho, J. S., Isacke, C. M. Multiple immunofluorescence labeling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell Biology. 9, 13 (2008).
- Segnani, C., et al. Histochemical Detection of Collagen Fibers by Sirius Red/Fast Green Is More Sensitive than van Gieson or Sirius Red Alone in Normal and Inflamed Rat Colon. PloS One. 10 (12), 0144630 (2015).
- Bolognesi, M. M., et al. Multiplex Staining by Sequential Immunostaining and Antibody Removal on Routine Tissue Sections. Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
