

Summary
Étant donné que les RPGC sont des cibles médicamenteuses attrayantes, le dépistage du ligand GPCR est donc indispensable pour l’identification des composés de plomb et pour les études de désorphanisation. À l’égard de ces efforts, nous décrivons PRESTO-Tango, une plate-forme de ressources open-source utilisée pour le profilage simultané du recrutement transitoire d’un PGS2 à environ 300 GPCR à l’aide d’un essai de journaliste basé sur TEV.
Abstract
En tant que superfamille génique la plus grande et la plus polyvalente et les médiateurs d’une gamme de voies de signalisation cellulaires, les récepteurs couplés à la protéine G (GPCR) représentent l’une des cibles les plus prometteuses pour l’industrie pharmaceutique. Ergo, la conception, la mise en œuvre et l’optimisation des essais de dépistage de ligand GPCR est cruciale, car ils représentent des outils de contrôle à distance pour la découverte de médicaments et pour manipuler la pharmacologie et les résultats du GPCR. Dans le passé, les essais dépendants de la protéine G caractérisaient ce domaine de recherche, détectant les événements induits par le ligand et quantifiant la génération de messagers secondaires. Cependant, depuis l’avènement de la sélectivité fonctionnelle, ainsi qu’une prise de conscience accrue de plusieurs autres voies indépendantes des protéines G et des limites associées aux essais dépendants de la protéine G, il y a une plus grande poussée vers la création d’alternatives Essais de dépistage de ligand GPCR. Vers cette entreprise, nous décrivons l’application d’une telle ressource, la plate-forme PRESTO-Tango, un système luciferase basé sur un journaliste qui permet l’interrogatoire parallèle et simultané du GPCR-ome humain, un exploit qui était auparavant considéré techniquement et économiquement irréalisable. Sur la base d’un essai de recrutement indépendant de G-protein ' 'arrestin2, l’universalité de la traite et de la signalisation à médiation de la protéine G2 fait de PRESTO-TANGO un outil approprié pour étudier environ 300 GPCR humains non olfactifs, dont environ 100 récepteurs orphelins. La sensibilité et la robustesse de PRESTO-Tango le rendent approprié pour les écrans primaires à haut débit utilisant des bibliothèques composées, employés pour découvrir de nouvelles cibles de GPCR pour les médicaments connus ou pour découvrir de nouveaux ligands pour les récepteurs orphelins.
Introduction
Les récepteurs couplés à la protéine G (GPCR) constituent la famille la plus importante et la plus diversifiée de protéines transmémbranées, fonctionnant comme interfaces de communication entre une cellule et son environnement1. La polyvalence des GPCRs est mise en évidence par leur capacité à détecter un large éventail de ligands, des neurotransmetteurs aux nucléotides, peptides aux photons, et beaucoup plus, ainsi que leur capacité à réguler de nombreuses cascades de signalisation en aval impliquées dans la croissance cellulaire, la migration, la différenciation, l’apoptose, la mise à feu cellulaire,etc. 2,3. Compte tenu de leur ubiquité et de leur implication dans une multitude de processus physiologiques, cette famille de récepteurs est de la plus haute importance thérapeutique, mise en évidence par le fait que plus d’un tiers des médicaments prescrits actuellement disponibles ciblent les GPCR4. Cependant, ces thérapies existantes ne ciblent qu’un petit sous-ensemble de la superfamille (environ 10%), et la pharmacologie de nombreux GPCRs reste non polluée. En outre, plus de 100 GPCRs existent comme récepteurs orphelins, car ils n’ont pas été jumelés avec une ligand endogène5. Ainsi, le dépistage de ligand GPCR est essentiel dans la déorphanasisation et le développement de médicaments, car il ouvre la voie à la découverte et à l’optimisation des plombs, et peut-être à la phase d’essai clinique.
Les méthodes de dépistage du ligand GPCR sont traditionnellement tombées dans l’une des deux catégories, les essais fonctionnels indépendants de G-protein ou de G-protein6. La signalisation GPCR est réglementée par des protéines G hétérotrimeriques (G), qui sont activées par l’échange de GTP contre le PIB lié sur le sous-unitéG 7. Les signaux du récepteur activé sont transduits par des protéines G via des messagers secondaires, tels que cAMP, Calcium, DAG et IP3, pour servir de médiateur en aval aux effecteurs en aval8. La nature des conséquences fonctionnelles de la signalisation de protéine G a été exploitée pour créer des essais cellulaires qui reflètent l’activation des récepteurs. Ces méthodes, qui mesurent les événements proximal (directs) ou distal (indirects) dans la signalisation des protéines G, sont le plus fréquemment utilisées pour le dépistage du ligand GPCR et ont été principalement utilisées dans des études de désorphanasisation6. Parmi les exemples d’essais qui mesurent directement l’activation de la protéine G négociée par le GPCR, mentionnons l’essai de liaison [35S]GTP-S, qui mesure la liaison d’un TPL GTP par radiolacs et non hydrolysable analogique au sous-unité de G, et les sondes de transfert d’énergie par résonance de la société de développement et de bioluminescence (FRET/BRET, respectivement) pour surveiller les interactions GPCR-G ET Gô/G, qui ont gagné plus de traction au cours des années9,10. Les essais qui surveillent les événements distal sont les outils les plus couramment utilisés pour le profilage du GPCR; par exemple, les essais de cAMP et d’IP1/3 mesurent l’accumulation intracellulaire des messagers secondaires dépendants de la protéine G, tandis que [Ca2 ]flux et essais de reporter impliquant des éléments de réponse spécifiques impliqués dans l’activation de la protéine G (CRE, NFAT-RE, SRE, SRF-RE) examinent les événements plus en aval de la cascade de signalisation11. Alors que la plupart des essais susmentionnés peuvent être effectués à un niveau de haut débit, sont assez sensibles, et se vantent de certains avantages spécifiques à l’analyse (par exemple, la discrimination entre les agonistes complets/partiels, les antagonistes neutres et les agonistes inverses dans le cas de la liaison GTP-S, ou la fonctionnalité d’analyse sur les cellules vivantes telles que [Ca2] et IP1/3)6, il n’y a malheureusement pas de méthodes existantes dépendantes de la protéine G qui s’agencent à l’interrogatoire de l’ensemble du GPCR-ome. Cela est en grande partie dû à l’accouplement indigène de plusieurs sous-familles de protéines G aux GPCRs, ce qui entraîne la signalisation à plusieurs cascades et le couplage inconnu de G-protéine au GPCRs orphelins. Pour atténuer ce problème, des essais ont été développés pour forcer le couplage promiscuité de la protéine G par le biais d’une seule lecture commune de signalisation, comme le cAMP, et le Ca2MD,bien que la plupart d’entre eux soient à faible débit12.
Un aspect important du cycle de vie du GPCR est l’arrêt de la signalisation dépendante de la protéine G, qui se produit en grande partie par le recrutement de 'arrestins qui induit la dissociation de la protéine G, et finalement désensibilisant le récepteur, qui est ciblé pour l’internalisation enduite de clathrine13. Les isoformes les plus omniprésents exprimés de l’arrêt sont les non-visuels 'arrestin1 et 'arrestin2, également désignés comme arrestin-2 et arrestin-3, respectivement14. Entrez les essais à base de cellules indépendantes de G-protein, qui ajoutent une nouvelle dimension au dépistage de ligand DE GPCR ; le trafic de récepteurs, la cellule entière sans étiquette et les essais de recrutement d’arrestation sont tous des exemples notables. Les essais de trafic GPCR emploient des ligands étiquetés fluorophore ou des anticorps co-intériorisés ciblant le récepteur15,tandis que les essais de cellules entières sans étiquette utilisent des biocapteurs qui traduisent les changements cellulaires induits par la liaison ligand en sorties quantifiables, telles que les signaux électriques ou optiques16. Notamment, la quintessence GPCR- -arrestin interactions façon de l’essai de recrutement '-arrestin comme un outil attrayant dans le répertoire des essais fonctionnels17. Le système Tango, développé pour la première fois par Barnea et coll. il y a seulement dix ans, implique l’introduction de trois éléments génétiques exogènes : une fusion protéique consistant en un 2e jour avec une protéase du virus de l’échage du tabac (TEVp), un transactivateur de tétracycline (TTA) qui est attaché à un GPCR par l’intermédiaire d’un virus de l’échage du tabac le site de clivage de protease (TEVcs) et est précédé d’une séquence du C-terminus du récepteur de vasopressine V2 (queue V2) pour favoriser le recrutement d’arrestin, et d’un gène de luciferase reporter dont la transcription est déclenchée par le translocation du facteur de transcription tTA au noyau, qui est libéré de l’ancrage de la membrane à la suite du recrutement de 'arrestin2 ' (figure 1)18. Les lectures quantitatives de l’activation du GPCR et du recrutement de l’arrêt2 peuvent ensuite être déterminées par la lecture pour la luminescence. Une distinction notable est que si le trafic de récepteurs et les méthodes cellulaires entières sans étiquette sont relativement faible débit, le Tango a plusieurs avantages, y compris la lecture sélective qui est spécifique au récepteur cible et la sensibilité due à l’intégration du signal, qui en font un candidat approprié pour le dépistage de ligand sur une plus grande échelle18.
Compte tenu de ces caractéristiques stratégiques, Kroeze et coll. ont développé PRESTO-Tango (Parallel Receptor-ome Expression and Screening via Transcriptional Output-Tango), une plate-forme open-source à haut débit qui utilise l’approche Tango pour profiler le GPCR-ome médicamentable d’une manière parallèle et simultanée19. Exploitant le recrutement « promiscuité » de l’arrêt2 à presque tous les GPCR, PRESTO-Tango est le premier de son genre en termes d’essais fonctionnels à base de cellules, permettant un dépistage rapide « de premier tour » des composés de petites molécules dans presque tous les GPCRs non olfactifs, y compris les orphelins, indépendamment du couplage sous-familial de la G-protéine.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Dépistage primaire : culture cellulaire et ensemencement des plaques
- Pour préparer des plaques recouvertes de poly-L-lysine (PLL), distribuez 20 L/puits d’une solution de stock de 25 g/mL de PLL dans des plaques de fond optique blanches ou noires de 384 puits à l’aide d’une pipette multicanal électronique ou d’un distributeur de réactifs. Incuber les plaques à température ambiante pendant 0,5 à 2 h.
REMARQUE : Si vous utilisez les plaques noires de 384 puits, attendez-vous à ce que le signal de fond soit plus bas que les plaques blanches. Il est recommandé de réduire le saignement de luminescence entre les puits adjacents. - Pour préserver les plaques enduites et laver l’excès de PLL, retirez le PLL en le faisant glisser sur l’évier, tapez-vous sur un essuie-tout et ajoutez 40 L/puits de solution diluée de 1x d’antimycotique antibiotique à l’aide d’une pipette multicanal électronique ou d’un distributeur réactif. Conserver les assiettes enduites de PLL à 4 oC jusqu’à ce qu’elles soient prêtes pour l’ensemencement des plaques.
- Maintenir les cellules HTLA (aimablement fournies par le Dr Richard Axel) une lignée humaine de cellules rénales embryonnaires (HEK293T) exprimant de façon stable le moyen de l’aigle modifié (DMEM) de Dulbecco, complété par 5 % du sérum bovin fœtal, 5 % du sérum de veau bovin, 2,5 g/mL de puromycine, 50 g/mL d’hygromycine, 100 P/ML pénicilline et 100 g/mL de streptomycine à 37 oC dans un incubateur humidifié contenant 5 % de CO2.
- Cellules de culture HTLA dans des plats de 150 mm et passer les cellules deux fois par semaine à un facteur de dilution de 1:10, avec le nombre optimal de passage cellulaire de 5-25. Assurez-vous qu’un nombre suffisant de plats de 150 mm sont confluents le jour de l’ensemencement des plaques de 384 puits, selon l’échelle de l’écran primaire.
REMARQUE : L’utilisation des cellules HTLA supérieure au passage 25 peut entraîner une viabilité réduite, ce qui donne des résultats sous-optimaux. - Pour déposer les cellules HTLA pour l’écran primaire, rincez doucement le plat confluent de 150 mm(es) avec 1x phosphate-tamponné saline (PBS), pH 7.4. Détachez les cellules avec environ 6 ml de Trypsin/0,53 mM EDTA, et transférez-les à un tube de centrifugeuse contenant au moins une quantité égale du milieu Eagle modifié (DMEM) modifié de Dulbecco pour neutraliser la trypsine.
- Tourner vers le bas les cellules HTLA à 500 x g pendant 3 min et résimettrez le granule cellulaire à une densité de 0,22 x 106 cellules/mL en DMEM complet, en omettant l’ajout de 2,5 g/mL de puromycine et de 50 g/mL d’hygromycine car ils peuvent diminuer l’efficacité de la transfection.
- Incuber les plaques nécessaires de 384 puits recouvertes de PLL à 37 oC pour les réchauffer avant les cellules d’ensemencement. Retirez la solution de stockage de 1x antibiotique-antimycotique de la plaque de 384 puits recouverte de PLL en faisant glisser la plaque sur l’évier et en l’encolant sur un essuie-tout pour sécher.
- Cellules de graines dans les plaques de 384 puits enrobées de PLL à une densité finale de 10 000 cellules/puits en distribuant 45 L de la suspension 0,22 x 106 cellules/mL HTLA à l’aide d’un tuyau multicanal électronique. Plaques incubées à 37 oC pendant la nuit. Si une transfection le jour même est préférée, les cellules de semence à une densité de 16.000 cellules/puits et effectuer la transfection 4 h plus tard.
REMARQUE : Pour une efficacité transfection élevée, la confluence cellulaire de 50 à 70 % est optimale.
2. Dépistage primaire : préparation des plaques d’ADN et transfections
- Pour préparer la plaque source d’ADN de 384 puits pour la transfection comme le montre la figure 2, distribuez les ADRC plasmides codant les constructions d’intérêt GPCR-Tango dans une plaque de 96 puits, avec un GPCR/well différent. L’ADN plasmide doit être suspendu dans un tampon Tris-EDTA (TE) de 0,1x à une concentration de 50 ng/L.
REMARQUE : Les plaques d’ADN de 96 puits peuvent être scellées et stockées à -20 oC, et réutilise pour de multiples expériences de dépistage. Toutes les constructions de GPCR-Tango codantes de l’ADC sont disponibles dans le commerce (voir la Table des Matériaux)et sont clonées dans le plasmide néomycine pcDNA3.1. Le kit PRESTO-Tango GPCR se compose de quatre plaques de 96 puits, qui comprennent 80 GPCR chacune, un couple de puits avec un vecteur vide comme contrôles négatifs, et des puits de contrôle positifs qui détiennent le récepteur de dopamine D2 (DRD2), et des puits qui portent un plasmide codant une protéine fluorescente (YFP) pour suivre l’efficacité de la transfection. - À l’aide d’une pipette multicanal, transférer manuellement la solution d’ADN du puits 96 à une plaque d’ADN de 384 puits, ajoutant 10 ll par puits de 384. Pour s’assurer que chaque condition de l’expérience est analysée en quadruplicate, la moitié de la plaque d’ADN de 96 puits (lignes A-D ou E-H) couvrira une plaque complète de 384 puits en distribuant chaque GPCR dans deux quadrants (premier quadrant - composé, deuxième quadrant et composé), de sorte que le même GPCR sera transfected dans 8 puits de la plaque de 384 puits (voir la figure 2 comme guide).
- Assembler les réactifs transfection suivants nécessaires à la méthode des précipitations de phosphate de calcium, tel que décrit par Jordan et coll.20: tampon TE de 0,1x (1 mM Tris-HCl et 0,1 mM EDTA); 2,5 M CaCl2 solution; 2x Hepes buffer, pH 7.05 (50 mM HEPES, 280 mM NaCl, 1.5 m Na2HPO4). Stériliser toutes les solutions en filtration et conserver à 4 oC. Le jour de la transfection, permettre aux réactifs d’atteindre la température ambiante avant l’utilisation.
- Diluer la solution de stock CaCl2 de 2,5 M en 0,1x TE (1:8 dilution) à une concentration finale de 0,313 M CaCl2 et vortex. Transférer 40 L de 0,313 M CaCl2 à la plaque source d’ADN de 384 puits et mélanger en tuyautant de haut en bas avec une pipette multicanal portée ou un tuyauttor automatisé de 384 canaux.
- Ajouter 50 L de tampon d’hépes 2x à la plaque source d’ADN de 384 puits, mélanger à nouveau en tuyautant de haut en bas et laisser reposer pendant 1 min; chaque puits de 384 aura une quantité suffisante de mélange d’ADN/transfection pour la transfection de neuf plaques de 384 puits, selon le nombre de composés qui doivent être testés. Transférer 10 L du mélange ADN/transfection de la plaque source d’ADN de 384 puits aux cellules HTLA ensemencées et incuber les plaques pendant la nuit à 37 oC.
3. Dépistage primaire : Stimulation cellulaire
- Vingt-quatre heures plus tard, décanter le support cellulaire transfecté en clignotant doucement la plaque de 384 puits sur l’évier et en l’encochant sur un essuie-tout, ou avec une tête d’aspirateur. Ajoutez lentement 40 L de supports affamés (DMEM complétés par 1% de sérum bovin fœtal dialysé (dFBS) et 1x antibiotique/antimycotique), en prenant soin d’éviter de toucher directement les cellules.
- Pipet 20 L du composé d’intérêt à une concentration de 3x (concentration finale du médicament dans la plaque cellulaire sera de 1x) dans les lignes alternées avec la stimulation (A) et 20 l de tampon de véhicule pour les lignes alternées sans composé (-). Remettre la plaque cellulaire à 37 oC en CO2 à 5 % et incuber pendant au moins 16 h.
4. Dépistage primaire : lecture de Luminescence
- Préparer le réactif Glo, modifié à partir de Baker et Boyce21: 108 mM Tris-HCl; 42 mM Tris-Base, 75 mM NaCl, 3 mM MgCl2, 5 mM Dithiothrei-tol (DTT), 0,2 mM Coenzyme A, 0,14 mg/ml D-Luciferin, 1,1 mM ATP, 0,25% v/v Triton X-100, 2 mM sodium hydrosulfite.
REMARQUE : Les solutions stock des réactifs peuvent être faites à l’avance, à l’exception de D-Luciferin, qui est toujours fraîchement ajouté au réactif Glo sous sa forme en poudre. Si des plaques noires ont été utilisées, la quantité de D-Luciferin peut être augmentée jusqu’à 0,25 mg/mL. - À 16-24 h après la stimulation, décanter le support cellulaire transfected en clignotant doucement la plaque de 384 puits au-dessus de l’évier et en l’encaissant sur une serviette en papier. Ajouter 20 l/puits de réactif Glo et incuber la plaque à température ambiante pendant 5 à 20 min. Lisez les plaques à l’aide d’un compteur de luminescence microplate, avec un temps d’intégration de 1 s/puits.
5. Dépistage primaire : Analyse des données
- Exporter les fichiers enregistrés du compteur de luminescence comme feuille de calcul; résultats seront enregistrés dans des unités de luminescence relative (RLU). Sur la base de la disposition de la plaque de 384 puits, calculer l’activation (changement de pli) de chaque récepteur à l’aide de la formule suivante :
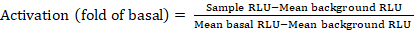
REMARQUE : Ici, l’échantillon RLU se réfère à la valeur de chacun des quatre puits de reproduction du quadrant stimulé (composé), le fond moyen RLU est le moyen des contrôles négatifs sur la plaque, et le RLU basal moyen se réfère à la moyenne du quadrant non traité de ce même récepteur (- composé). En outre, calculer l’écart standard des 4 points de données pour vérifier la qualité des résultats. Il est recommandé d’effectuer une transformation de journal2 sur la moyenne des changements de pli pour corriger toute hétéroskedasticité ; la base log2 est un choix pratique pour aider à identifier les coups positifs. Empiriquement, les seuils de frappe positifs sont fixés; il faut noter que certains récepteurs peuvent avoir une augmentation aussi faible que 2 fois et jusqu’à 40 fois augmenter pour d’autres avec l’agoniste complet. - Sur la base des résultats, sélectionnez les RPGC qui sont des succès positifs potentiels pour le dépistage secondaire.
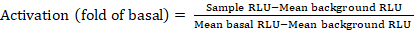
6. Dépistage secondaire : ensemencement cellulaire et transfections
- Sous-culture cellules HTLA dans des plats de 100 mm à une densité cellulaire totale de 5 x 106 cellules dans 11 ml de support complet (4,55 x 105/mL) et incuber à 37 oC pour 24 h. Si une transfection le jour même est préférée, les cellules de semence à une densité de 7,5 x 106 cellules et effectuent la transfection 4 h plus tard.
- Préchauffer les réactifs nécessaires aux précipitations de phosphate de calcium à température ambiante. Combinez 450 L de tampon TE de 0,1x avec 50 ll de 2,5 M CaCl2 et vortex rapidement; ces quantités sont spécifiques pour un plat de 100 mm, en fonction du volume de croissance moyen qu’il détient.
- Dans un tube, ajouter 500 L de la solution TE/CaCl2 à 10 g de GPCR cDNA et vortex. Ajouter 500 L de solution tampon Hepes 2x dans le tube, secouer vigoureusement (ne pas vortex), et incuber pendant 1 min.
REMARQUE : 1 g de tout plasmide codant une protéine fluorescente (p. ex. YFP, mCherry, etc.) peut être co-transfect avec 9 g de GPCR cDNA pour un total de 10 g. La protéine fluorescente est utilisée pour suivre l’efficacité de la transfection, et cette quantité minimale n’interfèrera pas avec l’essai. - Immédiatement après l’incubation courte, distribuez la solution de 1 ml dans le sens des cellules. Rochez doucement la plaque d’avant en arrière pour répartir uniformément le précipité, en prenant soin de ne pas faire tourbillonner la plaque, et incuber à 37 oC pendant 24 h.
- Le lendemain, observez l’efficacité de la transfection en examinant l’expression de la protéine fluorescente sous un imageur fluorescent; les transfections supérieures à 50% de couverture sont idéales.
- Incuber la plaque(s) pLÉA-enduite de 384 puits dans l’incubateur à 37 oC pour la réchauffer avant l’ensemencement des cellules. Retirez la solution de stockage de 1x antibiotique-antimycotique de la plaque de 384 puits recouverte de PLL en faisant glisser la plaque sur l’évier et en l’encolant sur un essuie-tout pour sécher.
- Rincez doucement les cellules transfectées avec la solution Versene (1X PBS, pH 7,4; 0,53 mM EDTA), et détachez en ajoutant 3 mL de 0,05% trypsin/0,53 mM EDTA au plat. Transférer le contenu dans un tube de centrifugeuse contenant au moins une quantité égale de DMEM complet pour neutraliser la trypsine.
- Tourner les cellules à 500 x g pendant 3 min et réanimer les cellules à une densité de 0,4 x 106 cellules/mL dans les médias affamés. Cellules de graines dans la plaque de 384 puits enrobée de PLL à une densité finale de 25 000 cellules/puits en distribuant 45 l de la suspension cellulaire à l’aide d’une pipette électronique multicanal. Remettre les plaques à la température de 37 oC pour un minimum de 4 h, ce qui permet aux cellules de se fixer correctement aux puits avant de passer à la stimulation.
7. Dépistage secondaire : Préparation de plaque de drogue pour une courbe-dose de 16 points (demi-journal)
- Dans une plaque de 96 puits, ajoutez 270 L de tampon de médicaments HBSS 1X (1x Hank’s Balanced Salt Solution [HBSS], 20 mM HEPES pH 7.4, 1x antibiotique-antimycotique), à l’exclusion de la dernière rangée (ligne H) de la plaque, comme le montre la figure 4.
REMARQUE : Pour les peptides, les molécules colloïdales et les composés mal solubles dans l’eau, l’ajout de 0,1 à 1 % de BSA est suggéré. Pour prévenir l’oxydation des médicaments, jusqu’à 0,01% d’acide ascorbique peut également être ajouté. - À partir du stock de drogue, préparer une solution médicamenteuse (appelée « concentration élevée ») en calculant une concentration finale de 3x (la concentration finale du médicament dans la plaque cellulaire sera de 1x). Par exemple, pour une courbe dose-réponse avec 10 M comme sa plus forte concentration, préparer la concentration « élevée » à 30 m. Pipet 300 L de concentration "Haute" dans les puits dans la rangée H.
- Dans un autre tube, préparer la concentration « faible », qui représente la concentration « élevée » divisée par 3,16 (demi-journal). D’après l’exemple précédent, la concentration « faible » serait de 9,49 M. Pipet 300 L de concentration "faible" dans les puits de la rangée H, à côté des puits "High".
REMARQUE : Le nombre total de 96 puits nécessaires dans la rangée H dépendra du nombre de cellules et des conditions de stimulation. Quatre puits (deux "High" et deux "Low") auront une solution de drogue suffisante pour stimuler une plaque de puits 384 entière. - Effectuez une dilution en série en passant par une solution de drogue de 30 l de « Haut » et « Faible » de la rangée H à la rangée précédente (rangée G) et mélangez en faisant de la canalisation manuelle de haut en bas, ou comme recommandé, à l’aide d’une pipette électronique multicanal avec la fonction « Pipette and Mix ». Répétez cette étape jusqu’à la première et la plus diluée rangée (ligne A), tout en jetant des extrémités entre les dilutions.
REMARQUE : Si désiré, les dilutions en série peuvent être arrêtées avant la rangée A, représentant un contrôle interne sans drogue, c’est-à-dire un « vrai zéro ». - En utilisant la figure 4 comme référence, stimuler les cellules transfected en canalisation 20 L des dilutions de colonne « faible » de la plaque de 96 puits aux rangées A-O de la plaque précédemment ensemencée de 384 puits, ainsi que 20 l de la « haute » plaque de dilution aux puits B-P. Incubate la plaque à 37 oC pour un minimum de 16 h.
8. Dépistage secondaire : lecture de Luminescence et analyse des données
- À 16-24 h après la stimulation, décanter le support cellulaire transfected en clignotant doucement la plaque de 384 puits au-dessus de l’évier et en l’encaissant sur une serviette en papier. Ajouter 20 l/puits de réactif Glo et incuber la plaque à température ambiante pendant 5 à 20 min. Lisez les plaques à l’aide d’un compteur de luminescence microplate, avec un temps d’intégration de 1 s/puits.
- Exporter les fichiers enregistrés du compteur de luminescence comme feuille de calcul; résultats seront enregistrés dans des unités de luminescence relative (RLU). Transférez les données de la plaque de 384 puits à un logiciel de statistiques pour analyser les résultats à l’aide de son analyse XY intégrée pour l’ajustement de courbe de régression non linéaire. Sélectionnez la fonction intégrée de stimulation dose-réponse à 3 paramètres « Log (agoniste) vs réponse (trois paramètres) »,

REMARQUE: Ici, top et fond sont des plateaux dans les unités de l’axe Y, respectivement la réponse maximale et le niveau basal, EC50 est la concentration de l’agoniste qui génère 50% de réponse entre Top et Bottom, et X se réfère à la concentration de journal de l’agoniste. Ce modèle suppose que la courbe dose-réponse a une pente Hill standard de 1.

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
À l’aide du protocole PRESTO-Tango présenté ici, un extrait de granule de chromaffin (CG) a été examiné contre 168 cibles GPCR non olfactives, la majorité étant des récepteurs orphelins. Le profilage de l’extrait dit a été effectué en examinant la mobilisation d’arrestation dans les récepteurs choisis, sur la base du principe conçu par Barnea et coll.18 (figure 1). Plasmid cDNA des GPCRs d’intérêt a été tiré de la trousse PRESTO-Tango GPCR et assemblé en deux 96 plaques de puits dans la disposition souhaitée. Au total, quatre plaques de 384 puits ensemencées de cellules HTLA ont été transfectedes, car chaque moitié des plaques d’ADN de 96 puits a été transformée en une plaque complète de 384 puits, ce qui a entraîné la transfectedité de chaque récepteur dans deux quadrants (huit 384 puits au total). L’un des deux quadrants a été stimulé avec l’extrait de CG ; en d’autres termes, l’alternance des lignes C-D, G-H, K-L et O-P représentait la stimulation (md)(figure 2). Sur les 168 GPCRs qui ont été interrogés lors du dépistage primaire, seuls deux récepteurs étaient des candidats comme cibles actives potentielles, en particulier le récepteur de la dopamine D3 (DRD3) et l’opsine 5 (OPN5). DRD3 a produit un changement significatif de journal2fold de 4,70, tandis que l’OPN5 a produit une réponse légèrement inférieure de 2,39, tous deux répondant à la coupure de seuil de la modification de pli de journal2 -gt;2. En comparaison, le contrôle positif de l’écran primaire a été stimulé par DRD2 avec du quinpirole, un agoniste sélectif de ce récepteur, et a produit un changement de pli journal2 de 4,58 (figure 3). Pour reproduire ces fenêtres de signal et éliminer la possibilité de coups faussement positifs, un écran secondaire a été conduit avec les récepteurs susmentionnés. En plus de tester l’extrait de CG, étant donné que DRD3 est un récepteur non orphelin, une autre condition a été préparée comme un contrôle positif, spécifiquement la stimulation avec quinpirole, l’un de ses agonistes sélectifs. D’autre part, OPN5 est un récepteur orphelin et en tant que tel, aucun agoniste de référence ne peut être testé aux côtés de l’extrait de CG comme un contrôle positif; seul le tampon a été testé comme un contrôle négatif. D’autres caractérisation pharmacologiques de ces deux GPCR ont été effectuées en préparant des courbes de dose de 16 points allant de 10-5 M à 10-12,5 M. Plus précisément, les solutions de stock d’extrait et de quinpirole CG à 10 mM ont été diluées à 30 m et 9,49 m, les concentrations correspondantes de « haute » et de « basse » dans la rangée inférieure (rangée H) de la plaque de drogue de 96 puits; ces formulations deviendront de 10à 5 M et de10 à 5,5 M une fois que 20 l seront distribuées aux cellules transfectées dans 40 l de milieu affamé, pour un total de 60 l dans chaque puits de 384. Comme nous l’avons déjà décrit, les dilutions en série ont été effectuées de telle sorte que les formations de médicaments les plus diluées pour 10-12 M et 10-12,5 M sont dans la première rangée (rangée A) (figure 4). Des courbes dose-réponse du criblage secondaire ont été créées à l’aide de GraphPad Prism pour évaluer la puissance et l’efficacité du ligand. Par rapport au quinpirole, l’extrait de CG a produit des fenêtres de signal similaires et des valeurs EC50, confirmant sa validité en tant que coup actif à DRD3. Cependant, une courbe à dose plate semblable au contrôle négatif a été produite pour OPN5, l’excluant comme une cible possible pour l’extrait de CG(figure 5).

Figure 1 : Conception modulaire des constructions TANGO (A) et du schéma général pour l’essai de recrutement de l’arrêt (Tango) (B). (A) Les constructions GPCR Tango se composent de divers éléments de module dans l’ordre suivant : un signal HA/FLAG tag, le CDS GPCR, un récepteur de vasopressine 2 C-terminal queue, site de clivage de protéase TEV, et un facteur de transcription tTA. (B) Le principe de l’essai Tango implique de transfecter transitoirement les plasmides de Tango GPCR dans les cellules HTLA, les cellules HEK293T exprimant de façon stable une protéine de fusion de protéase de protease de 2-TEV et un gène de journaliste de luciferase dont l’expression est activée par tTA. L’activation du GPCR finira par se traduire par la mobilisation du 2-TEV à l’arrêt, ce qui amènera la protéase à proximité de son site de clivage. En conséquence, le tTA est clidé de la queue gpCR, libérant le facteur de transcription pour translocaliser dans le noyau et activer l’expression de luciferase. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 2 : Dispositions de la plaque cDNA de 96 puits et de la plaque cellulaire de 384 puits pour la transfection et la stimulation dans le dépistage primaire PRESTO-TANGO. Représentant la préparation d’une plaque de source cDNA de 384 puits pour la transfection, les constructions de TANGO DE GPCR sont d’abord transférées de la moitié d’une plaque de 96 puits dans une plaque complète de 384 puits, chaque récepteur étant transfected dans l’octuplicate. Dans ce contexte, la stimulation des cellules avec (-) et sans (-) le médicament (s) d’intérêt se produira dans le quadruplicate pour chaque récepteur individuel. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 3 : Représentations graphiques de l’identification à succès de la projection primaire PRESTO-Tango. Comme preuve de concept, l’activité biologique d’un extrait de granule de chromaffin (CG) sur le GPCRome a été analysée. Les cellules HTLA ont été transfectedes dans 384 plaques de puits avec 168 constructions de Tango GPCR, et stimulées avec l’extrait de CG (composé) ou avec la mémoire tampon du véhicule (- composé). pcDNA3.1 a été utilisé comme un contrôle négatif, et le récepteur DRD2 stimulé avec du quinpirole a été utilisé comme un contrôle positif. Les fenêtres de signal (A) et le changement de pli de journal2 (B) dans l’activation des récepteurs ont été calculées entre les puits en l’absence ou la présence de l’extrait de CG. Toutes les barres d’erreur représentent SD (n - quatre mesures). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 4 : Disposition de la préparation de plaque de drogue de 96 puits pour la stimulation dans le criblage secondaire. Représentant la préparation d’une plaque de drogue de 96 puits pour la stimulation cellulaire, les dilutions en série pour une plage de courbe de dose de 16 points commencent à10-5 M (concentration finale) dans la rangée H, avec des intervalles de demi-journal entre chaque point jusqu’à10-12,5 M dans la rangée A. "High" et "Low" colonnes de médicaments sont utilisés pour stimuler les lignes alternées de la plaque ensemencée 384-puits. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.

Figure 5 : Réponses dose-courbe pour le profilage composé et la démonstration du recrutement de PG2 à GPCRs dans le dépistage secondaire. Les cellules HTLA ont été transitoirement transmétiquement infectées par les récepteurs DRD3 (A) et OPN5 (B). Les deux conditions transfected ont été stimulées avec un extrait de CG dans des incréments de demi-log, aussi bien que le quinpirole agoniste spécifique de DRD3 comme contrôle positif, et le tampon de véhicule pour OPN5 comme contrôle négatif. Toutes les barres d’erreur représentent SD (n - trois mesures). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Les GPCR dynamiques sont des centrales de la transduction du signal. Les propriétés physiochimiques des poches liantes de ces récepteurs héptéliriques, ainsi que leur pertinence physiologique soulignent la nécessité d’outils de dépistage de ligand GPCR. Tel que présenté ci-dessus, l’essai PRESTO-Tango est rapide, sensible et convivial, se prêtant au développement de médicaments. Non seulement cet essai mesure-t-il l’activation induite par l’agoniste, mais il peut également être utilisé pour quantifier l’activité des antagonistes et des modulateurs allosteriques19. À la lumière de la sélectivité fonctionnelle, un concept qui suggère que différentes structures médicamenteuses peuvent provoquer différentes cascades de signalisation des récepteurs à un seul récepteur, en comparant l’activation de la voie de la protéine G à l’aide d’essais dépendants de la protéine G avec le recrutement de 'arrestin utilisant PRESTO-Tango pourrait fournir des indices pour la conception des composés de plomb avec des effets secondaires négatifs réduits. Notamment, son indépendance de la détection de l’accouplement de la protéine G aide à identifier les partenaires de couplage pour les GPCRs orphelins qui n’auraient pas été précédemment détectés par les essais dépendants de la protéine G.
Pour assurer la cohérence et la robustesse des écrans PRESTO-Tango, il faut faire attention à toutes les étapes du protocole, car les perturbations introduites seront amplifiées en raison de la nature de cette plate-forme. Bien sûr, il existe des mesures générales communes à tous les écrans HTS qui devraient être prises en considération, comme l’utilisation de réactifs du même lot / formulation pour assurer une stabilité identique et l’activité biologique tout au long, ainsi que de maintenir les conditions sur le système HTS cohérente comme la densité d’ensemencement cellulaire et le temps d’incubation des médicaments. Le format miniaturisé de PRESTO-Tango exige une attention à quelques points critiques : la variation de la densité d’ensemencement cellulaire et leur distribution homogène (suspension par opposition à cellule unique) entre les puits, faible efficacité transfection, et une faible stimulation et livraison composées empêcheront la reproductibilité quotidienne et de la plaque à la plaque. À cet effet, triturez la suspension de cellules HTLA pour homogénéiser la solution avant l’ensemencement et assurer une confluence cellulaire de 50 à 70 % avant la transfection. Le véhicule pour la livraison des composés doit être vérifié, avec le sulfoxide de diméthyle étant le transporteur le plus commun. Typiquement, la concentration la plus élevée de nos courbes de dose est de 10 m, mais cela peut changer en fonction de la nature et de la puissance du composé; il est important de tester diverses concentrations pour déduire la tolérance cellulaire et la toxicité.
Étant donné que certains GPCR ont une activité constitutive élevée, un problème qui peut se poser pendant le dépistage est une plage dynamique réduite et un signal de fond qui est plus élevé que prévu. Ceci peut être quelque peu atténué en veillant à ce que la famine de sérum soit exécutée avec le milieu de DMEM complété par 1% dFBS. Il faut tenir compte du fait que si la production de luminescence est suffisamment élevée, il peut encore y avoir un saignement dans les puits adjacents, ce qui peut entraîner des changements de plis calculés par erreur. Les signaux indétectables ou faibles (en supposant qu’une réponse est attendue) peuvent être expliqués de plusieurs façons, à savoir une mauvaise expression de GPCR(s) dans les cellules HTLA, l’activité biologique du composé est perdue le rendant inefficacité, ou le récepteur (s) en question ne recrutent pas intrinsèquement '-arrestin2. Respectivement, l’évaluation de la quantité et de la qualité de l’ADN transfected de récepteur plasmide, testant d’autres préparations/lots du composé inefficacieux en question, et exécutant des techniques orthologues d’interaction protéine-protéine telles que BRET/FRET ou co-immunoprecipitation sont quelques solutions suggérées à ce problème. En outre, l’expression des récepteurs pourrait également être améliorée en sous-fermant les récepteurs Tango (s) d’intérêt dans les vecteurs lentiviral et en transduisant des cellules HTLA, générant une lignée cellulaire stable HTLA-GPCR. Un changement dans la puissance prévue d’un agoniste pendant le dépistage secondaire pourrait impliquer que le temps de stimulation de drogue et/ou la concentration du composé nécessaire pour stimuler une réponse est insuffisant, ou que les dilutions série de la plaque de drogue ont été mal préparées. L’utilisation d’une pipette multicanal électronique ou d’un système automatisé de tuyauterie sans changer de pointe entre les deux lors de la création de dilutions en série de médicaments pourrait être un problème lorsque vous travaillez avec des composés collants.
Les différences notables entre l’analyse originale de Tango développée par Barnea et al.18 et la plate-forme PRESTO-Tango incluent la conception du récepteur dans un format modulaire, composé de séquences codon-optimisées, qui améliore l’expression des récepteurs dans les cellules de mammifères, les étiquettes d’épitope pour valider ladité, et les sites de restriction qui flanquent GPCRs, queue V2 et TEVcs-tTA, permettant l’excision des parties et la subcloning. Plus important encore, PRESTO-Tango surpasse l’essai Tango en termes de puissance de dépistage et de conception expérimentale. Les tests d’échantillons quadruplicate d’environ 300 GPCR sont effectués dans seulement 8 plaques de 384 puits, tout en tenant compte de contrôles de fond négatifs et de contrôles positifs pour surveiller l’efficacité de la transfection. Bien que PRESTO-Tango soit adapté au dépistage du GPCR-ome avec un seul composé d’intérêt, l’interrogatoire avec plusieurs ligands peut également être effectué, bien qu’à un coût et une utilisation accrus des ressources, comme avec des bibliothèques de composés de petites molécules mises en commun ou à la portée ou des échantillons biologiques qui se composent de mélanges de diverses entités chimiques. Certes, cette question peut être atténuée en réduisant le nombre de composés à interroger en effectuant des analyses de similitude chimique et de diversité des bibliothèques composées en question. Bien que la plate-forme PRESTO-Tango soit plus applicable aux fins du dépistage primaire, le profilage secondaire peut être effectué à plus petite échelle, dans des formats moyens ou à faible débit, pour confirmer les conséquences fonctionnelles de la stimulation ligand. Cependant, comme avec tous les autres essais GPCR, il faut reconnaître qu’il n’y a pas de contrôles positifs appropriés pour les récepteurs orphelins lors du dépistage secondaire avec l’essai Tango. Néanmoins, des coups positifs potentiels peuvent être identifiés si les données de sortie peuvent être fixées à une courbe de dose-réponse sigmoïdale, avec une fenêtre de signal calculée et une valeur EC50. Il est également important de noter que le mécanisme de l’activité ligand, que ce soit pour les récepteurs orphelins ou non orphelins, ne peut pas être élucidé sans exécuter des essais parallèles.
Avec tous les composants de PRESTO-Tango déjà optimisés, y compris la lignée cellulaire HTLA et GPCR Tango construit, peu de place pour la modification est nécessaire en dehors du choix de la formulation composée (s) à utiliser pour la stimulation des médicaments. Si désiré, une lignée cellulaire HTLA exprimant de façon stable un récepteur peut être facilement générée par le clonage dit récepteur GPCR-Tango dans le vecteur recommandé pIRESbleo3 (Clonetech), et la sélection des clones à l’aide de zeocine. En ce qui concerne l’échange de pcDNA3.1 à pIRESbleo3, il suffit de digérer la construction GPCR Tango avec NotI et XbaI et d’insérer dans le vecteur de destination sur les sites de restriction NotI et NheI. Malgré tout, il existe des moyens d’adapter et d’optimiser cette technologie. L’un des piliers de cette technologie sont les cellules HTLA, une lignée cellulaire HEK293T exprimant de façon stable un gène de fusion de l’ANT-arrestin2-TEV et un journaliste de luciferase dépendant de tTA, gracieusement fourni par le laboratoire de Richard Axel. Bien qu’il s’inscrivent dans une composante cruciale de PRESTO-Tango, il n’existe actuellement aucune autre solution de rechange en termes d’origine de la lignée cellulaire, ni les gènes qu’ils expriment. En outre, les futures lignées cellulaires d’ingénierie peuvent être générées pour exprimer d’autres gènes de fusion TEV pour suivre d’autres protéines en plus de 23 , en particulier celles qui ont été précédemment montrés pour interagir ou trouvés en résidence à GPCRs, tels que 14-3-322, SAP9723, et '-arrestin1, qui est l’isoforme la plus répandue des arrestations non visuelles dans les vertébrés24. Ceci peut être réalisé en utilisant les cellules parentales HTL qui contenaient uniquement le journaliste de luciferase contrôlé par le promoteur tetO7. Une limitation à PRESTO-Tango est l’activation non spécifique du promoteur journaliste. Basé sur un système de régulation dépendant de la tétracycline (système de tétracycline), l’élément tétracycline-sensible (TRE) contrôle l’expression du journaliste en aval de luciferase. Cependant, des études antérieures ont démontré l’expression « fuite » de luciferase due aux facteurs endogènes de transcription25,26. En conséquence, certains composés pourraient activer le journaliste indépendamment de l’activation de recrutement ou de GPCR, ce qui augmenterait le nombre de faux positifs. Une autre question qui émerge, également commun à d’autres méthodes HTS, sont "frappeurs fréquents", composés promiscuité qui stimulent des réponses substantielles dans plusieurs cibles27. Néanmoins, la mise en place parallèle du dépistage PRESTO-Tango facilite l’identification de ces artefacts, qui peuvent être testés plus en détail pour confirmer leur effet sur l’activité de luciferase. Au total, PRESTO-Tango a fourni des bases solides pour l’étude du recrutement d’arrestations aux GPCRs, et dans le plus grand schéma de découverte de médicaments, comme un outil utile de dépistage et de désorphanasisation du GPCR.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les authours ne déclarent aucun intérêt concurrent.
Acknowledgments
Ces travaux ont été appuyés par les Instituts de recherche en santé du Canada (subvention des IRSC #MOP142219).
Materials
| Name | Company | Catalog Number | Comments |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, BLACK, with lid, Sterile | NUNC | 12-566 | |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, White, with lid, Sterile | NUNC | 12-566-1 | |
| 384 Well Round Bottom, Polypropylene, Non-Treated, Blue, non-sterile, without lid | ThermoFisher | 12-565-390 | |
| Antibiotic-Antimycotic | Wisent | 450-115-EL | |
| D-Luciferin, sodium salt | GoldBio | LUCNA | |
| DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Corning | 10-013-CV | |
| Eppendorf Xplorer, 12-channel, variable, 15–300 µL | Eppendorf | 4861000155 | |
| Eppendorf Xplorer, 12-channel, variable, 5–100 µL | Eppendorf | 4861000139 | |
| Matrix Platemate 2x3 | ThermoFisher | 801-10001 | |
| MicroBeta 1450 Wallac | Perkin Elmer | ||
| Penicilin-Streptomycin | Wisent | 450-201-EL | |
| Poly-L-Lysine hydrobromide | Millipore-Sigma | P2636-500MG | |
| Roth Lab PRESTO-Tango GPCR Kit | Addgene | Kit #1000000068 |
References
- Liapakis, G., et al. The G-protein coupled receptor family: actors with many faces. Current pharmaceutical design. 18 (2), 175-185 (2012).
- Pierce, K. L., Premont, R. T., Lefkowitz, R. J. Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology. 3 (9), 639-650 (2002).
- Kroeze, W. K., Sheffler, D. J., Roth, B. L. G-protein-coupled receptors at a glance. Journal of cell science. 116, Pt 24 4867-4869 (2003).
- Rask-Andersen, M., Almén, M. S., Schiöth, H. B. Trends in the exploitation of novel drug targets. Nature Reviews Drug Discovery. 10 (8), 579-590 (2011).
- Ngo, T., et al. Identifying ligands at orphan GPCRs: current status using structure-based approaches. British journal of pharmacology. 173 (20), 2934-2951 (2016).
- Zhang, R., Xie, X. Tools for GPCR drug discovery. Acta pharmacologica Sinica. 33 (3), 372-384 (2012).
- Kimple, A. J., Bosch, D. E., Giguere, P. M., Siderovski, D. P. Regulators of G-Protein Signaling and Their G Substrates: Promises and Challenges in Their Use as Drug Discovery Targets. Pharmacological Reviews. 63 (3), 728-749 (2011).
- Wettschureck, N., Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiological Reviews. 85 (4), 1159-1204 (2005).
- Denis, C., Saulière, A., Galandrin, S., Sénard, J. -M., Galés, C. Probing heterotrimeric G protein activation: applications to biased ligands. Current Pharmaceutical Design. 18 (2), 128-144 (2012).
- Yin, H., et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. The Journal of Biological Chemistry. 284 (18), 12328-12338 (2009).
- Cheng, Z., et al. Luciferase Reporter Assay System for Deciphering GPCR Pathways. Current Chemical Genomics. 4, 84-91 (2010).
- Roth, B. L., Kroeze, W. K. Integrated Approaches for Genome-wide Interrogation of the Druggable Non-olfactory G Protein-coupled Receptor Superfamily. The Journal of Biological Chemistry. 290 (32), 19471-19477 (2015).
- Jean-Charles, P. -Y., Kaur, S., Shenoy, S. K. G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. Journal of Cardiovascular Pharmacology. 70 (3), 142-158 (2017).
- Smith, J. S., Rajagopal, S. The β-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. The Journal of Biological Chemistry. 291 (17), 8969-8977 (2016).
- Böhme, I., Beck-Sickinger, A. G. Illuminating the life of GPCRs. Cell Communication and Signaling. 7 (1), 16 (2009).
- Fang, Y. Label-Free Receptor Assays. Drug discovery today. Technologies. 7 (1), 5-11 (2011).
- Wang, T., et al. Measurement of β-Arrestin Recruitment for GPCR Targets. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences. , (2004).
- Barnea, G., et al. The genetic design of signaling cascades to record receptor activation. Proceedings of the National Academy of Sciences of the United States of America. 105 (1), 64-69 (2008).
- Kroeze, W. K., et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nature Structural & Molecular Biology. 22 (5), 362-369 (2015).
- Jordan, M., Schallhorn, A., Wurm, F. M. Transfecting Mammalian Cells: Optimization of Critical Parameters Affecting Calcium-Phosphate Precipitate Formation. Nucleic Acids Research. 24 (4), 596-601 (1996).
- Baker, J. M., Boyce, F. M. High-throughput functional screening using a homemade dual-glow luciferase assay. Journal of Visualized Experiments. (88), 50282 (2014).
- Li, H., Eishingdrelo, A., Kongsamut, S., Eishingdrelo, H. G-protein-coupled receptors mediate 14-3-3 signal transduction. Signal Transduction and Targeted Therapy. 1 (1), 16018 (2016).
- Walther, C., Ferguson, S. S. G. Minireview: Role of intracellular scaffolding proteins in the regulation of endocrine G protein-coupled receptor signaling. Molecular Endocrinology. 29 (6), Baltimore, Md. 814-830 (2015).
- Gurevich, E. V., Benovic, J. L., Gurevich, V. V. Arrestin2 expression selectively increases during neural differentiation. Journal of Neurochemistry. 91 (6), 1404-1416 (2004).
- Shaikh, S., Nicholson, L. F. B. Optimization of the Tet-On system for inducible expression of RAGE. Journal of Biomolecular Techniques JBT. 17 (4), 283-292 (2006).
- Blau, H. M., Rossi, F. M. Tet B or not tet B: advances in tetracycline-inducible gene expression. Proceedings of the National Academy of Sciences of the United States of America. 96 (3), 797-799 (1999).
- Roche, O., et al. Development of a Virtual Screening Method for Identification of "Frequent Hitters" in Compound Libraries. Journal of Medicinal Chemistry. 45 (1), 137-142 (2002).
