

Summary
Da GPCRs attraktive arzneimittelfähige Ziele sind, ist das GPCR-Ligandenscreening daher für die Identifizierung von Bleiverbindungen und für Deorphanisierungsstudien unerlässlich. Im Rahmen dieser Bemühungen beschreiben wir PRESTO-Tango, eine Open-Source-Ressourcenplattform, die für die gleichzeitige Profilierung der vorübergehenden Rekrutierung von "Arrestin2" bei ca. 300 GPCRs mit einem TEV-basierten Reportertest verwendet wird.
Abstract
Als größte und vielseitigste Gen-Superfamilie und Vermittler einer Bandbreite zellulärer Signalwege stellen G-Protein-gekoppelte Rezeptoren (GPCRs) eines der vielversprechendsten Ziele für die pharmazeutische Industrie dar. Ergo, das Design, die Implementierung und die Optimierung von GPCR-Liganden-Screening-Assays, ist von entscheidender Bedeutung, da sie Fernsteuerungstools für die Arzneimittelentdeckung und die Manipulation der GPCR-Pharmakologie und -Ergebnisse darstellen. In der Vergangenheit haben G-Protein-abhängige Assays diesen Forschungsbereich typisiert, Liganden-induzierte Ereignisse erkannt und die Erzeugung von sekundären Botenstoffen quantifiziert. Seit dem Aufkommen der funktionellen Selektivität sowie einem erhöhten Bewusstsein für mehrere andere G-proteinunabhängige Pfade und die Einschränkungen im Zusammenhang mit G-proteinabhängigen Assays gibt es jedoch einen größeren Schub für die Schaffung alternativer GPCR Liganden-Screening-Assays. Zu diesem Unterfangen beschreiben wir die Anwendung einer solchen Ressource, der PRESTO-Tango-Plattform, einem luziferase-Reporter-basierten System, das die parallele und gleichzeitige Abfrage des menschlichen GPCR-Omeermöglicht, eine Leistung, die zuvor in Betracht gezogen wurde technisch und wirtschaftlich nicht machbar. Basierend auf einem G-Protein-unabhängigen Rekrutierungstest für die Rekrutierung von G-Protein-Arrestin2 macht die Universalität des von der GPCR vermittelten Handels und der Signalisierung von '-arrestin2- und Signalisierungs-Atier-Medien PRESTO-TANGO zu einem geeigneten Werkzeug für die Untersuchung von ca. 300 nicht-olfaktorischen menschlichen GPCRs, darunter etwa 100 Orphan-Rezeptoren. Die Empfindlichkeit und Robustheit von PRESTO-Tango machen es für primäre Hochdurchsatzbildschirme geeignet, die zusammengesetzte Bibliotheken verwenden, um neue GPCR-Ziele für bekannte Medikamente aufzudecken oder neue Liganden für Orphan-Rezeptoren zu entdecken.
Introduction
G-Protein-gekoppelte Rezeptoren (GPCRs) bilden die größte und vielfältigste Familie von Transmembranproteinen, die als Kommunikationsschnittstellen zwischen einer Zelle und ihrerUmgebung1 funktionieren. Die Vielseitigkeit von GPCRs wird durch ihre Fähigkeit, eine Vielzahl von Liganden zu erkennen – von Neurotransmittern bis zu Nukleotiden, Peptide bis hin zu Photonen und vielem mehr – sowie ihre Fähigkeit, zahlreiche nachgeschaltete Signalkaskaden zu regulieren, die an Zellwachstum, Migration, Differenzierung, Apoptose, Zellabschussusw.beteiligt sind, sowie ihre Fähigkeit, zahlreiche nachgeschaltete Signalkaskaden zu regulieren, die an Zellwachstum, Migration, Differenzierung, Apoptose, Zellabschuss usw. beteiligt sind,werden. Angesichts ihrer Allgegenwart und Beteiligung an einer Vielzahl von physiologischen Prozessen ist diese Rezeptorfamilie von größter therapeutischer Bedeutung, was durch die Tatsache belegt wird, dass mehr als ein Drittel der derzeit verfügbaren verordneten Medikamente auf GPCRs4abzielen. Diese bestehenden Therapeutika zielen jedoch nur auf eine kleine Teilmenge der Überfamilie ab (schätzungsweise 10 %), und die Pharmakologie vieler GPCRs bleibt ungeklärt. Darüber hinaus existieren mehr als 100 GPCRs als Orphan-Rezeptoren, da sie nicht mit einem endogenen Ligand5abgeglichen wurden. Daher ist das GPCR-Ligand-Screening entscheidend für die Entwaistisierung und Arzneimittelentwicklung, da es den Weg zur Lead-Entdeckung und -Optimierung und möglicherweise zur klinischen Testphase ebnet.
Methoden für das GPCR-Ligand-Screening sind traditionell in eine von zwei Kategorien gefallen, G-Protein-abhängige oder G-Protein-unabhängige funktionelle Assays6. Die GPCR-Signalisierung wird durch heterotrimere G-Proteine (G- und -Proteine) reguliert, die durch den Austausch von GTP gegen BIP aktiviert werden, die an die Untereinheit7gebunden sind. Signale des aktivierten Rezeptors werden von G-Proteinen über sekundäre Botenstoffe wie cAMP, Calcium, DAG und IP3 transduziert, um nachgeschaltete Signalisierung an downstreameffekten8zu vermitteln. Die Art der funktionellen Folgen der G-Protein-Signalisierung wurde genutzt, um zellbasierte Assays zu erstellen, die die Rezeptoraktivierung reflektieren. Diese Methoden, die proximale (direkte) oder distale (indirekte) Ereignisse in der G-Protein-Signalisierung messen, werden am häufigsten für das GPCR-Liganden-Screening verwendet und wurden hauptsächlich in Deorphanisierungsstudien eingesetzt6. Beispiele für Assays, die die GPCR-vermittelte G-Protein-Aktivierung direkt messen, sind der [35S]GTP-Bindungstest, der die Bindung eines radioaktiv markierten und nicht hydrolysierbaren GTP analog zur G-Untereinheit misst, und Förster/Biolumineszenz-Resonanzenergieübertragung (FRET/BRET) zur Überwachung von GPCR-G- und G-/G-Wechselwirkungen, die in den Jahren9,10, an Zugkraft gewonnen haben. Assays, die distale Ereignisse überwachen, sind die am häufigsten verwendeten Tools für die GPCR-Profilerstellung; Z. B. messen cAMP- und IP1/3-Assays die intrazelluläre Akkumulation von G-proteinabhängigen Sekundärbotenstoffen, während [Ca2+] Fluss- und Reporter-Assays mit spezifischen Reaktionselementen, die an der G-Protein-Aktivierung beteiligt sind (CRE, NFAT-RE, SRE, SRF-RE), Ereignisse weiter nach der Signalkaskade11untersuchen. Während die meisten der oben genannten Assays auf einem hohen Durchsatzniveau durchgeführt werden können, sind ziemlich empfindlich und rühmen sich bestimmter assayspezifischer Vorteile (z.B. Diskriminierung zwischen Voll-/Teilagonisten, neutralen Antagonisten und inversen Agonisten im Falle der GTP-Bindung oder Assay-Funktionalität auf lebenden Zellen wie [Ca2+] und IP1/3)6, es gibt leider keine vorhandenen G-Protein-abhängigen Methoden, die der Abfrage des gesamten medikamentösen GPCR-Ome sausen. Dies ist weitgehend auf die native Kopplung mehrerer G-Protein-Unterfamilien mit GPCRs zurückzuführen, was zu Signalisierungen an mehreren Kaskaden und der unbekannten G-Protein-Kopplung bei verwaisten GPCRs führt. Um dieses Problem zu mildern, wurden Assays entwickelt, um die promiscuous G-Protein-Kopplung durch eine einzige gemeinsame Signalisierungsauslesung wie cAMP und Ca2+zu erzwingen, obwohl die meisten von ihnen einen niedrigen Durchsatz12sind.
Ein wichtiger Aspekt des GPCR-Lebenszyklus ist die Beendigung der G-Protein-abhängigen Signalisierung, die zu einem großen Teil durch die Rekrutierung von S-Arrestinen erfolgt, die eine Dissoziation des G-Proteins induziert und letztlich den Rezeptor desensizipiert, das für die clathrinbeschichtete Internalisierung13bestimmt ist. Die allgegenwärtigsten Isoformen von '-arrestin sind die nicht-visuellen '-arrestin1 und '-arrestin2, auch als arrestin-2 und arrestin-3 bzw.14bezeichnet. Geben Sie G-Protein-unabhängige zellbasierte Assays ein, die dem GPCR-Liganden-Screening eine neue Dimension verleihen; Rezeptorenhandel, etikettenfreie ganze Zelle und Rekrutierungs-Assays sind bemerkenswerte Beispiele. GPCR-Handelstests verwenden fluorophormarkierte Liganden oder ko-internalisierte Antikörper, die auf den Rezeptorabzielen 15, während etikettenfreie Ganzzell-Assays Biosensoren verwenden, die zelluläre Veränderungen, die durch Ligandenbindung induziert werden, in quantifizierbare Ausgänge wie elektrische oder optische Signale16übersetzen. Bemerkenswert ist, dass die Quintessenz der GPCR---Arrestin-Interaktionen den Rekrutierungs-Assay von '-arrestin als attraktives Werkzeug im Repertoire der funktionalen Assays17. Das Tango-System, das erst vor einem Jahrzehnt von Barnea et al. entwickelt wurde, beinhaltet die Einführung von drei exogenen genetischen Elementen: eine Proteinfusion, bestehend aus dem "-arrestin2" mit einem Tabak-Ätzvirus-Protease (TEVp), einem Tetracyclin-Transaktivator (tTA), der über ein Tabak-Etch-Virus an einen GPCR gebunden ist Protease-Spaltungsstelle (TEVcs) und wird von einer Sequenz aus dem C-Terminus des V2-Vasopressinrezeptors (V2-Schwanz) zur Förderung der Verhaftung bei der Rekrutierung und einem Reporter-Luziferase-Gen, dessen Transkription durch die tTA-Transkriptionsfaktor-Translokation in den Kern, der nach der Rekrutierung von A-arrestin2 aus der Membranverankerung befreit wird (Abbildung 1)18. Quantitative Messwerte der GPCR-Aktivierung und der Rekrutierung von '-arrestin2 können anschließend durch Lesen auf Lumineszenz bestimmt werden. Ein bemerkenswerter Unterschied ist, dass der Tango zwar den Rezeptorhandel und die etikettenfreien Ganzzellmethoden relativ gering ist, aber mehrere Vorteile hat, einschließlich des selektiven Auslesens, das spezifisch für den Zielrezeptor ist, und der Empfindlichkeit aufgrund der Signalintegration, die ihn zu einem geeigneten Kandidaten für das Liganden-Screening in größerem Maßstabmachen 18.
Angesichts dieser strategischen Merkmale entwickelten Kroeze et al. PRESTO-Tango (Parallel Receptor-ome Expression and Screening via Transcriptional Output-Tango), eine Open-Source-Plattform mit hohem Durchsatz, die den Tango-Ansatz nutzt, um das medikamentöse GPCR-Ome parallel und gleichzeitig zu profilieren19. PRESTO-Tango nutzt die "promiscuous" Rekrutierung von '-arrestin2 für fast alle GPCRs und ist die erste seiner Art in Bezug auf zellbasierte funktionelle Assays, die ein schnelles "Erstrunden"-Screening von kleinen Molekülverbindungen an fast allen nicht-olfaktorischen GPCRs, einschließlich Waisen, unabhängig von der G-Protein-Unterfamilie ermöglicht.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Primäres Screening: Zellkultur und Plattenaussaat
- Zur Herstellung von Poly-L-Lysin(PLL)-beschichteten Platten 20 l/well einer 25-g/ml-Stammlösung pLL in weißen oder schwarzen 384-Well-optischen Bodenplatten mit einer elektronischen Mehrkanalpipette oder einem Reagenzienspender. Inkubieren Sie die Platten bei Raumtemperatur für 0,5-2 h.
HINWEIS: Wenn Sie die schwarzen 384-Well-Platten verwenden, erwarten Sie, dass das Hintergrundsignal niedriger ist als die weißen Platten. Schwarze Platten werden empfohlen, um die Durchblutung der Lumineszenz zwischen benachbarten Brunnen zu reduzieren. - Um die beschichteten Platten zu erhalten und die überschüssige PLL abzuwaschen, entfernen Sie die PLL, indem Sie sie über das Waschbecken streichen, tippen Sie trocken über ein Papiertuch und fügen Sie 40 L/Well verdünnter 1x-Lösung von Antibiotika-Antimykotik mit einer elektronischen Mehrkanalpipette oder einem Reagenzienspender hinzu. PLL-beschichtete Platten bei 4 °C lagern, bis sie zur Plattensaat bereit sind.
- Halten Sie HTLA-Zellen (freundlicherweise von Dr. Richard Axel zur Verfügung gestellt) – eine menschliche embryonale Nierenzelllinie (HEK293T) stabil auszudrückend -arrestin2-TEV und tTA-gesteuerte Luziferase – in vollständig Dulbeccos modifiziertem Eagle-Medium (DMEM), ergänzt mit 5% fetalem Rinderserum, 5 % des Rinderkalbserums, 2,5 g/ml Puromycin, 50 g/ml Hygromycin, 100 U/ml Penicillin und 100 g/ml Streptomycin bei 37 °C in einem befeuchteten Inkubator mit 5%CO2.
- Kultur HTLA-Zellen in 150 mm Geschirr und passieren Zellen zweimal pro Woche mit einem Verdünnungsfaktor von 1:10, mit optimaler Zelldurchgangszahl von 5–25. Stellen Sie sicher, dass eine ausreichende Anzahl von 150 mm Geschirr am Tag der 384-Well-Plattenaussaat, abhängig von der Skala des Primärsiebes, konfluent sind.
HINWEIS: Die Verwendung von HTLA-Zellen, die größer als Durchgang 25 sind, kann zu einer verminderten Lebensfähigkeit führen, was zu suboptimalen Ergebnissen führt. - Um HTLA-Zellen für den Primärsieb auszusäen, spülen Sie die konfluente 150 mm Schale(en) vorsichtig mit 1x phosphatgepufferter Saline (PBS), pH 7.4, ab. Lösen Sie Zellen mit ca. 6 ml 0,05% Trypsin/0,53 mM EDTA und übertragen Sie sie in ein Zentrifugenrohr, das mindestens die gleiche Menge des modifizierten Dulbecco-modifizierten Eagle-Mediums (DMEM) enthält, um das Trypsin zu neutralisieren.
- Drehen Sie HTLA-Zellen bei 500 x g für 3 min und setzen Sie das Zellpellet mit einer Dichte von 0,22 x 106 Zellen/ml in vollständiger DMEM wieder aus, wodurch die Zugabe von 2,5 g/ml Puromycin und 50 g/ml Hygromycin wegfällt, da sie die Transfektionswirksamkeit verringern können.
- Die notwendigen 384-Well-PLL-beschichteten Platten bei 37 °C inkubieren, um sie vor der Aussaat von Zellen zu erwärmen. Entfernen Sie die Aufbewahrungslösung von 1x Antimykotik antimykotika aus der 384-well PLL-beschichteten Platte(n), indem Sie die Platte über das Waschbecken flicken und sie zum Trocknen über ein Papiertuch kleben.
- Saatzellen in die 384-Well-PLL-beschichteten Platten mit einer Enddichte von 10.000 Zellen/Well, indem 45 l der 0,22 x 10 6-Zellen/ml-HTLA-Suspension mit einer elektronischen Mehrkanalpipette dosiert werden. Platten bei 37 °C über Nacht inkubieren. Wenn eine Transfektion am selben Tag bevorzugt wird, führen Samenzellen mit einer Dichte von 16.000 Zellen/Well die Transfektion 4 h später durch.
HINWEIS: Für eine hohe Transfektionseffizienz ist die Konfluenz von 50–70 % der Zellen optimal.
2. Primäres Screening: DNA-Plattenpräparation und Transfektionen
- Um die 384-Well-DNA-Quellenplatte für die Transfektion vorzubereiten, wie in Abbildung 2dargestellt, verteilen Sie die Plasmid-cDNAs, die die GPCR-Tango-Konstrukte kodieren, die für eine 96-Well-Platte mit einem anderen GPCR/Well von Interesse sind. Die Plasmid-DNA sollte in einem 0,1x Tris-EDTA (TE)-Puffer in einer Konzentration von 50 ng/l suspendiert werden.
HINWEIS: Die 96-Well-DNA-Platten können bei -20 °C versiegelt und gelagert und für mehrere Screening-Experimente wiederverwendet werden. Alle cDNA-kodierenden GPCR-Tango-Konstrukte sind kommerziell erhältlich (siehe Materialtabelle) und werden im neomycin-Plasmid pcDNA3.1 geklont. Das PRESTO-Tango GPCR Kit besteht aus vier 96-Well-Platten, die jeweils 80 GPCRs, ein paar Brunnen mit einem leeren Vektor als Negativkontrollen und Positive Control-Brunnen enthalten, die den Dopamin-Rezeptor D2 (DRD2) halten, und Brunnen, die ein Plasmid tragen, das für ein fluoreszierendes Protein (YFP) kodiert, um die Transfektionseffizienz zu verfolgen. - Übertragen Sie die DNA-Lösung mit hilfe einer Mehrkanalpipette manuell vom 96-Well auf die 384-Well-DNA-Quellenplatte, wodurch 10 l pro 384-Well hinzugefügt werden. Um sicherzustellen, dass jede Bedingung des Experiments in Quadruplikat untersucht wird, deckt die Hälfte der 96-Well-DNA-Platte (Zeilen A-D oder E-H) eine volle 384-Well-Platte ab, indem jeder GPCR in zwei Quadranten verteilt wird (erster Quadrant = - Verbindung, zweiter Quadrant = + Verbindung), so dass derselbe GPCR in 8 Brunnen der 384-Well-Platte transfiziert wird (siehe Abbildung 2 als Führungsteil).
- Zusammenbauen Sie die folgenden Transfektionsreagenzien, die für die Calciumphosphat-Fällungsmethode benötigt werden, wie von Jordan et al.20beschrieben: 0,1x TE-Puffer (1 mM Tris-HCl und 0,1 mM EDTA); 2.5 M CaCl2 Lösung; 2x Hepespuffer, pH 7,05 (50 mM HEPES, 280 mM NaCl, 1,5 mM Na2HPO4). Sterilisieren Sie alle Lösungen durch Filtration und lagern Sie bei 4 °C. Am Tag der Transfektion, lassen Sie die Reagenzien Raumtemperatur vor Gebrauch zu erreichen.
- Die 2,5 M CaCl2 Stofflösung in 0,1x TE (1:8 Verdünnung) auf eine Endkonzentration von 0,313 M CaCl2 und Wirbel verdünnen. Übertragen Sie 40 l von 0,313 M CaCl2 auf die 384-Well-DNA-Quellenplatte und mischen Sie sie durch Pipettieren mit einer handgeführten Mehrkanalpipette oder einem automatisierten 384-Kanal-Pipettor auf und ab.
- Der 384-Well-DNA-Quellenplatte 50 l 2x Hepespuffer hinzufügen, durch Pipettieren nach oben und unten wieder vermischen und 1 min stehen lassen; jeder 384-Well wird eine ausreichende Menge an DNA/Transfektionsgemisch für die Transfektion von neun 384-Well-Platten haben, abhängig von der Anzahl der Verbindungen, die getestet werden müssen. Übertragen Sie 10 l des DNA/Transfektionsgemisches von der 384-Well-DNA-Quellenplatte auf die gesäten HTLA-Zellen und inkubieren Sie die Platten über Nacht bei 37 °C.
3. Primäres Screening: Zellstimulation
- 24 Stunden später dekantieren Sie die transfizierten Zellmedien, indem Sie die 384-Well-Platte sanft über das Waschbecken streicheln und über ein Papiertuch oder mit einem Aspiratorkopf kleben. Fügen Sie langsam 40 L verhungernde Medien hinzu (DMEM ergänzt mit 1% dialysiertem fetalem Rinderserum (dFBS) und 1x Antibiotikum/Antimykotik), wobei darauf zu achten ist, dass die Zellen nicht direkt berührt werden.
- Pipetten Sie 20 l der Zinsverbindung in einer 3-fachen Konzentration (Endkonzentration des Arzneimittels in der Zellplatte beträgt 1x) in die abwechselnden Reihen mit (+) Stimulation und 20 l Fahrzeugpuffer für die Wechselreihen ohne (-) Verbindung. Geben Sie die Zellplatte bei 37 °C in 5%CO2 zurück und brüten Sie mindestens 16 h.
4. Primäres Screening: Lumineszenz-Lesung
- Bereiten Sie das Glo Reagenz, modifiziert von Baker und Boyce21: 108 mM Tris-HCl; 42 mM Tris-Base, 75 mM NaCl, 3 mM MgCl2, 5 mM Dithiothrei-tol (DTT), 0,2 mM Coenzym A, 0,14 mg/ml D-Luciferin, 1,1 mM ATP, 0,25% v/v Triton X-100, 2 mM Natriumhydrosulfit.
HINWEIS: Lagerlösungen der Reagenzien können im Voraus hergestellt werden, mit Ausnahme von D-Luciferin, das dem Glo-Reagenz in seiner pulverförmigen Form immer frisch zugesetzt wird. Wenn schwarze Platten verwendet wurden, kann die Menge an D-Luciferin bis zu 0,25 mg/ml erhöht werden. - Um 16–24 Uhr nach stimulationieren, dekantieren Sie die transfizierten Zellmedien, indem Sie die 384-Well-Platte sanft über das Waschbecken streichen und über ein Papiertuch kleben. Fügen Sie 20 L/Well von Glo-Reagenz hinzu und inkubieren Sie die Platte bei Raumtemperatur für 5–20 min. Lesen Sie die Platten mit einem Mikroplatten-Lumineszenzzähler, mit einer Integrationszeit von 1 s/well.
5. Primäres Screening: Datenanalyse
- Exportieren Sie die gespeicherten Dateien aus dem Lumineszenzzähler als Kalkulationstabelle; Ergebnisse werden in relativen Lumineszenzeinheiten (RLU) aufgezeichnet. Berechnen Sie anhand des Layouts der 384-Well-Platte die Aktivierung (Faltenänderung) jedes Rezeptors mit der folgenden Formel:
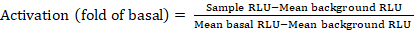
ANMERKUNG: Hier bezieht sich Sample RLU auf den Wert jeder der vier Replizierungsbohrungen des stimulierten (+ zusammengesetzten) Quadranten, der mittlere Hintergrund RLU ist der Mittelwert der Negativkontrollen auf der Platte, und die mittlere basale RLU bezieht sich auf den Mittelwert des unbehandelten Quadranten desselben Rezeptors (- Verbindung). Berechnen Sie außerdem die Standardabweichung der 4 Datenpunkte, um die Qualität der Ergebnisse zu überprüfen. Es wird empfohlen, eine log2-Transformation auf den Mittelwert der Faltenänderungen durchzuführen, um jegliche Heteroskedastizität zu korrigieren. Die log2-Basis ist eine praktische Wahl, um positive Treffer zu identifizieren. Empirisch die positiven Trefferschwellen festlegen; Es muss angemerkt werden, dass einige Rezeptoren so niedrig wie 2-fache Erhöhung und bis zu 40-fache Erhöhung für andere mit vollem Agonisten haben können. - Wählen Sie auf der Grundlage der Ergebnisse die GPCRs aus, die potenzielle positive Treffer für das sekundäre Screening sind.
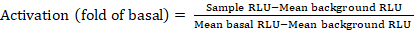
6. Sekundäres Screening: Zellaussaat und Transfektionen
- Subkultur-HTLA-Zellen in 100-mm-Schalen mit einer Gesamtzelldichte von 5 x 106 Zellen in 11 ml Gesamtmedien (4,55 x 105/ml) und bei 37 °C für 24 h inkubieren. Wenn eine Transfektion am selben Tag bevorzugt wird, führen Samenzellen mit einer Dichte von 7,5 x 106 Zellen die Transfektion 4 h später durch.
- Die für die Calciumphosphatfällung bei Raumtemperatur benötigten Reagenzien vorwärmen. Kombinieren Sie 450 l 0,1x TE-Puffer mit 50 l von 2,5 M CaCl2 und schnell Wirbel; diese Mengen sind spezifisch für eine 100 mm Schale, basierend auf dem Volumen des Wachstumsmediums, das es hält.
- In einer Röhre 500 l der TE/CaCl2-Lösung zu 10 g GPCR-cDNA und Wirbel hinzufügen. Fügen Sie 500 l 2x Hepes Pufferlösung in das Rohr, schütteln Sie kräftig (nicht Wirbel), und inkubieren für 1 min.
ANMERKUNG: 1 g eines beliebigen Plasmids, das für ein fluoreszierendes Protein kodiert (z. B. YFP, mCherry usw.), kann mit 9 g GPCR-cDNA für insgesamt 10 g kotranstranstranstranstranstranssektiert werden. Das fluoreszierende Protein wird verwendet, um die Transfektionseffizienz zu verfolgen, und diese minimale Menge wird den Test nicht stören. - Unmittelbar nach der kurzen Inkubation die 1 ml Lösung tropfenweise auf die Zellen abgeben. Die Platte vorsichtig hin und her schaukeln, um den Niederschlag gleichmäßig zu verteilen, wobei darauf zu achten ist, dass die Platte nicht gewirbelt wird, und bei 37 °C für 24 h inkubieren.
- Am nächsten Tag beobachten Sie die Transfektionseffizienz, indem Sie die Expression des fluoreszierenden Proteins unter einem fluoreszierenden Zellbilder betrachten; Transfektionen von mehr als 50 % Abdeckung sind ideal.
- Inkubieren Sie die notwendige 384-Well-PLL-beschichtete Platte(n) im Inkubator bei 37 °C, um sie vor der Aussaat von Zellen zu erwärmen. Entfernen Sie die Aufbewahrungslösung von 1x Antimykotik antimykotika aus der 384-well PLL-beschichteten Platte(n), indem Sie die Platte über das Waschbecken flicken und sie zum Trocknen über ein Papiertuch kleben.
- Spülen Sie die transfizierten Zellen vorsichtig mit Versene-Lösung (1X PBS, pH 7,4; 0,53 mM EDTA) und lösen Sie sich, indem Sie 3 ml 0,05% Trypsin/0,53 mM EDTA in die Schale geben. Übertragen Sie den Inhalt in ein Zentrifugenrohr, das mindestens eine gleiche Menge an vollständigem DMEM enthält, um das Trypsin zu neutralisieren.
- Drehen Sie die Zellen bei 500 x g für 3 min und suspendieren Sie die Zellen mit einer Dichte von 0,4 x 106 Zellen/ml in hungernden Medien. Saatzellen in die 384-Well-PLL-beschichtete Platte(n) mit einer Enddichte von 25.000 Zellen/Well, indem sie 45 l der Zellsuspension mit einer elektronischen Mehrkanalpipette dosieren. Bringen Sie die Platten für mindestens 4 h auf die 37 °C zurück, sodass sich die Zellen vor der Stimulation richtig an den Brunnen befestigen können.
7. Sekundäres Screening: Arzneimittelplattenpräparation für 16-Punkt (Halblog) Dosiskurve
- In einer 96-Well-Platte 270 L 1X HBSS-Medikamentenpuffer (1x Hank es Balanced Salt Solution [HBSS], 20 mM HEPES pH 7.4, 1x antimycotic) hinzufügen, mit Ausnahme der letzten Reihe (Zeile H) der Platte, wie in Abbildung 4dargestellt.
HINWEIS: Bei Peptiden, kolloidalen Molekülen und schlecht wasserlöslichen Verbindungen wird die Zugabe von 0,1–1% BSA empfohlen. Um die Oxidation von Medikamenten zu verhindern, können auch bis zu 0,01% Ascorbinsäure zugesetzt werden. - Aus dem Wirkstoffbestand eine Wirkstofflösung (als "hohe" Konzentration bezeichnet) durch Berechnung einer endgültigen 3x Konzentration (Endkonzentration des Arzneimittels in der Zellplatte wird 1x betragen). Zum Beispiel bereiten Sie für eine Dosis-Wirkungs-Kurve mit 10 M als höchste Konzentration die "hohe" Konzentration bei 30 m vor. Pipetten 300 l "Hohe" Konzentration in Brunnen in Reihe H.
- In einem anderen Rohr bereiten Sie die "Niedrige" Konzentration vor, die die "hohe" Konzentration dividiert durch 3,16 (Halblog) darstellt. Basierend auf dem vorherigen Beispiel würde die "Niedrige" Konzentration 9,49 M betragen. Pipetten 300 l "Niedrige" Konzentration in Brunnen in Reihe H, neben den "High" Brunnen.
HINWEIS: Die Gesamtzahl der 96-Bohrungen, die in Zeile H benötigt werden, hängt von der Anzahl der Zellen und stimulationsbedingungen ab. Vier Brunnen (zwei "High" und zwei "Low") haben reichlich Drogenlösung, um eine ganze 384 Well Platte zu stimulieren. - Führen Sie eine serielle Verdünnung durch Pipettierung von 30 l Medikamentenlösung von den "High" und "Low" Brunnen der Reihe H in die vorherige Reihe (Zeile G) und mischen Sie durch manuellepipetieren nach oben und unten, oder wie empfohlen, mit einer elektronischen Mehrkanalpipette mit der "Pipette und Mix" Funktion. Wiederholen Sie diesen Schritt bis zur ersten und am meisten verdünnten Zeile (Zeile A), während Sie Spitzen zwischen Verdünnungen ablegen.
HINWEIS: Auf Wunsch können die seriellen Verdünnungen vor Zeile A gestoppt werden, was eine interne Kontrolle ohne Medikament darstellt, mit anderen Worten, eine "wahre Null". - Anhand von Abbildung 4 als Referenz stimulieren Sie transfizierte Zellen, indem Sie 20 l der "Low"-Säulenverdünnungen von der 96-Well-Platte auf die Reihen A-O der zuvor gesäten 384-Well-Platte sowie 20 l der "Hohen" Säulenverdünnungen an die Brunnen B-P pipetieren. Inkubieren Sie die Platte bei 37 °C für ein Minimum von 16 h.
8. Sekundäres Screening: Lumineszenzlesen und Datenanalyse
- Um 16–24 Uhr nach stimulationieren, dekantieren Sie die transfizierten Zellmedien, indem Sie die 384-Well-Platte sanft über das Waschbecken streichen und über ein Papiertuch kleben. Fügen Sie 20 L/Well von Glo-Reagenz hinzu und inkubieren Sie die Platte bei Raumtemperatur für 5–20 min. Lesen Sie die Platten mit einem Mikroplatten-Lumineszenzzähler, mit einer Integrationszeit von 1 s/well.
- Exportieren Sie die gespeicherten Dateien aus dem Lumineszenzzähler als Kalkulationstabelle; Ergebnisse werden in relativen Lumineszenzeinheiten (RLU) aufgezeichnet. Übertragen Sie die Daten der 384-Well-Platte an eine Statistiksoftware, um die Ergebnisse mithilfe der integrierten XY-Analyse für die Passform der nichtlinearen Regressionskurve zu analysieren. Wählen Sie die integrierte 3-Parameter-Dosis-Wirkungs-Stimulationsfunktion "Log(agonist) vs. Response (drei Parameter)",

ANMERKUNG: Hier sind oben und unten Plateaus in den Einheiten der Y-Achse, bzw. die maximale Reaktion und Basalebene, EC50 ist die Konzentration des Agonisten, die 50% Antwort zwischen Oben und Unten erzeugt, und X bezieht sich auf die Log-Konzentration des Agonisten. Dieses Modell geht davon aus, dass die Dosis-Wirkungs-Kurve eine Standard-Hill-Neigung von 1 aufweist.

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Mit dem hier vorgestellten PRESTO-Tango-Protokoll wurde ein Chromaffin-Granulat (CG)-Extrakt mit 168 nicht-olfaktorischen GPCR-Zielen abgeschirmt, wobei die Mehrheit Waisenrezeptoren waren. Die Profilierung dieses Extraktes erfolgte durch die Untersuchung der Mobilisierung von '-arrestin2 an den gewählten Rezeptoren, basierend auf dem von Barnea et al.18 entworfenen Prinzip (Abbildung 1). Plasmid cDNA der GPCRs wurde aus dem PRESTO-Tango GPCR Kit entnommen und in zwei 96 Well-Platten im gewünschten Layout montiert. Insgesamt wurden vier 384-Well-Platten mit HTLA-Zellen transfiziert, da jede Hälfte der 96-Well-DNA-Platten in eine vollständige 384-Well-Platte umgewandelt wurde, was dazu führte, dass jeder Rezeptor in zwei Quadranten transfiziert wurde (insgesamt acht 384-Wells). Einer der beiden Quadranten wurde mit dem CG-Extrakt stimuliert; Anders ausgedrückt, stellten die abwechselnden Zeilen C–D, G–H, K-L und O-P die (+) Stimulation dar (Abbildung 2). Von den 168 GPCRs, die im primären Screening abgefragt wurden, waren nur zwei Rezeptoren Anwärter auf potenzielle aktive Ziele, insbesondere Dopaminrezeptor D3 (DRD3) und Opsin 5 (OPN5). DRD3 führte zu einer signifikanten Log2-Faltenänderung von 4,70, während OPN5 eine etwas geringere Antwort von 2,39 erzeugte, wobei beide die Schwelle für die Abgrenzung der Log2-Falzänderung erfüllten >2. Im Vergleich dazu wurde die positive Kontrolle für den Primärbildschirm DRD2 mit Quinpirole, einem selektiven Agonisten dieses Rezeptors, stimuliert und erzeugte eine Log2-Faltenänderung von 4,58 (Abbildung 3). Um diese Signalfenster zu reproduzieren und die Möglichkeit von falsch-positiven Treffern auszuschließen, wurde ein sekundärer Bildschirm mit den oben genannten Rezeptoren durchgeführt. Neben der Prüfung des CG-Extrakts, da DRD3 ein nicht-verwaister Rezeptor ist, wurde eine andere Bedingung als positive Kontrolle vorbereitet, insbesondere Stimulation mit Quinpirole, einem seiner selektiven Agonisten. Auf der anderen Seite, OPN5 ist ein Waisenrezeptor und als solche, kein Referenz-Agonist kann neben dem CG-Extrakt als positive Kontrolle getestet werden; nur Puffer wurde als Negativkontrolle getestet. Eine weitere pharmakologische Charakterisierung dieser beiden GPCRs erfolgte durch die Herstellung von 16-Punkt-Dosiskurven von 10-5 M bis 10-12,5 M. Insbesondere wurden die CG-Extrakt- und Quinpirole-Lagerlösungen bei 10 mM auf 30 m und 9,49 m verdünnt, die entsprechenden "Hohen" und "Niedrigen" Konzentrationen in der unteren Reihe (Reihe H) der 96-Well-Medikamentenplatte; Diese Formulierungen werden 10-5 M und 10-5,5 M, sobald 20 l in 40 l ausgehungertem Medium auf die transfizierten Zellen abgegeben werden, für insgesamt 60 l innerhalb jedes 384-Wells. Wie bereits beschrieben, wurden serielle Verdünnungen so durchgeführt, dass die verdünntesten Medikamentenformationen für 10-12 M und 10-12,5 M in der obersten Reihe (Reihe A) sind (Abbildung 4). Dosis-Wirkungs-Kurven aus dem sekundären Screening wurden mit GraphPad Prism erstellt, um Ligandenpotenz und Wirksamkeit zu bewerten. Im Vergleich zu Quinpirole erzeugte der CG-Extrakt ähnliche Signalfenster und EC50-Werte, was seine Gültigkeit als aktiver Treffer bei DRD3 bestätigte. Für OPN5 wurde jedoch eine flache Dosiskurve ähnlich der Negativkontrolle erzeugt, die sie als mögliches Ziel für den CG-Extrakt ausschließt (Abbildung 5).

Abbildung 1: Modularer Aufbau der TANGO-Konstrukte (A) und allgemeines Schema für den Rekrutierungstest für die Rekrutierung von S-Arrestin (Tango) (B). (A) Die GPCR Tango-Konstrukte bestehen aus verschiedenen Modulelementen in der folgenden Reihenfolge: einem HA-Signal/FLAG-Tag, dem GPCR-CDS, einem Vasopressin-Rezeptor 2 C-Terminal-Schwanz, einer TEV-Proteasespaltungsstelle und einem tTA-Transkriptionsfaktor. (B) Das Prinzip des Tango-Assays beinhaltet die vorübergehende Transfizierung der GPCR Tango-Plasmide in HTLA-Zellen, HEK293T-Zellen, die stabil ein Protease-Protease-Protease-Fusionsprotein von '-arrestin2-TEV exezieren, und ein Luziferase-Reportergen, dessen Expression durch tTA aktiviert wird. Die Aktivierung des GPCR wird schließlich zur Mobilisierung des '-arrestin2-TEV zum Rezeptor führen, wodurch die Protease in unmittelbarer Nähe zu seiner Spaltungsstelle wird. Als Ergebnis wird die tTA vom GPCR-Schwanz gespaltet, wodurch der Transkriptionsfaktor frei wird, um sich in den Kern zu transortieren und den Luziferase-Ausdruck zu aktivieren. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 2: Layouts der 96-Well-cDNA-Platte und der 384-Well-Zellplatte zur Transfektion und Stimulation im PRESTO-TANGO-Primärscreening. Die GPCR Tango-Konstrukte, die die Herstellung einer 384-Well-cDNA-Quellenplatte für die Transfektion darstellen, werden zunächst von der Hälfte einer 96-Well-Platte in eine vollständige 384-Well-Platte übertragen, wobei jeder Rezeptor in Oktuplicated transfiziert wird. In dieser Einstellung wird die Stimulation von Zellen mit (+) und ohne (-) das(n) Medikament(n) von Interesse in Quadruplicate für jeden einzelnen Rezeptor auftreten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 3: Grafische Darstellungen der Trefferidentifikation aus dem PRESTO-Tango-Primärscreening. Als Proof-of-Concept wurde die biologische Aktivität eines Chromaffingranulatextrakts (CG) auf dem GPCRome analysiert. HTLA-Zellen wurden in 384 Brunnenplatten mit 168 GPCR Tango-Konstrukten transfiziert und entweder mit dem CG-Extrakt (+ Verbindung) oder mit Fahrzeugpuffer (- Verbindung) stimuliert. pcDNA3.1 wurde als Negativkontrolle verwendet, und DRD2-Rezeptor stimuliert mit Quinpirole wurde als positive Kontrolle verwendet. Die Signalfenster (A) und die log2 Faltenänderung (B) bei der Rezeptoraktivierung wurden zwischen den Brunnen in Abwesenheit oder Anwesenheit von CG-Extrakt berechnet. Alle Fehlerbalken stellen SD dar (n = vier Messungen). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 4: Layout der 96-Well-Medikamentenplattenpräparation zur Stimulation im Sekundärscreening. Die Darstellung der Herstellung einer 96-Well-Medikamentenplatte zur Zellstimulation beginnen bei 10-5 M (Endkonzentration) in Reihe H, mit Halbprotokollintervallen zwischen 10-12,5 M in Reihe A. "High" und "Low" Wirkstoffsäulen werden verwendet, um abwechselnde Reihen der gesäten 384-Well-Platte zu stimulieren. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 5: Dosis-Kurven-Antworten für die Compound-Profilierung und Demonstration der Rekrutierung von '-arrestin2 bei GPCRs im sekundären Screening. HTLA-Zellen wurden vorübergehend mit rezeptoren DRD3 (A) und OPN5 (B) transfiziert. Beide transfizierten Bedingungen wurden mit einem CG-Extrakt in Halbprotokollschritten stimuliert, sowie die DRD3-spezifische agonistische Quinpirole als Positivkontrolle und Fahrzeugpuffer für OPN5 als Negativkontrolle. Alle Fehlerbalken stellen SD dar (n = drei Messungen). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Die konformdynamischen GPCRs sind Kraftpakete der Signaltransduktion. Die physiochemischen Eigenschaften der Bindungstaschen dieser heptahelischen Rezeptoren sowie ihre physiologische Relevanz unterstreichen die Notwendigkeit von GPCR-Ligand-Screening-Tools. Wie oben dargestellt, ist der PRESTO-Tango-Assay schnell, sensibel und benutzerfreundlich und verleiht sich der Arzneimittelentwicklung. Dieser Assay misst nicht nur die agonistisch-induzierte Aktivierung, sondern kann auch zur Quantifizierung der Aktivität von Antagonisten und allosterischen Modulatoren19verwendet werden. Angesichts der funktionellen Selektivität könnte ein Konzept, das nahelegt, dass verschiedene Wirkstoffstrukturen unterschiedliche Rezeptorsignalkaskaden an einem einzigen Rezeptor auslösen können, wobei der Vergleich der Aktivierung des G-Protein-Signalwegs mit G-Protein-abhängigen Assays mit der Rekrutierung von S-Arrestin mit PRESTO-Tango Hinweise auf die Entwicklung von Bleiverbindungen mit reduzierten negativen Nebenwirkungen liefern könnte. Insbesondere seine Unabhängigkeit vom Nachweis der G-Protein-Kopplung hilft dabei, Kopplungspartner für verwaiste GPCRs zu identifizieren, die zuvor nicht von G-Protein-abhängigen Assays nachgewiesen worden wären.
Um die Konsistenz und Robustheit der PRESTO-Tango-Bildschirme zu gewährleisten, muss in allen Schritten des Protokolls Vorsicht geboten sein, da die eingeführten Störungen aufgrund der Art dieser Plattform vergrößert werden. Natürlich gibt es allgemeine Maßnahmen, die allen HTS-Bildschirmen gemeinsam sind, die berücksichtigt werden sollten, wie die Verwendung von Reagenzien derselben Charge/Formulierung, um die gleiche Stabilität und biologische Aktivität während der gesamten Zeit zu gewährleisten, sowie die Gleichbleibenden auf dem HTS-System, wie z. B. die Zellsaatdichte und die Inkubationszeit von Arzneimitteln. Das miniaturisierte Format von PRESTO-Tango erfordert die Aufmerksamkeit auf einige kritische Punkte: Variation der Zellsädichte und deren homogene Verteilung (Klumpen versus Einzelzellsuspension) zwischen Brunnen, geringe Transfektionseffizienz und schlechte Zusammengesetzte Stimulation und -abgabe verhindern die tägliche Undplatten-Zu-Platte-Reproduzierbarkeit. Zu diesem Zweck trituieren Sie die HTLA-Zellsuspension, um die Lösung vor dem Seeding zu homogenisieren und eine 50–70%ige Zellkonfluenkation vor der Transfektion zu gewährleisten. Das Fahrzeug für die Lieferung der Verbindungen sollte überprüft werden, wobei Dimethylsulfoxid der häufigste Träger ist. Typischerweise ist die höchste Konzentration unserer Dosiskurven 10 M, aber dies kann sich je nach Art und Wirksamkeit der Verbindung ändern; Es ist wichtig, verschiedene Konzentrationen zu testen, um zelluläre Toleranz und Toxizität abzuleiten.
Angesichts der Tatsache, dass einige GPCRs eine hohe konstitutive Aktivität haben, kann ein Problem, das während des Screenings auftreten kann, ein reduzierter Dynamikbereich und ein Hintergrundsignal sein, das höher ist als erwartet. Dies kann etwas gemildert werden, indem sichergestellt wird, dass Serumhunger mit DMEM Medium durchgeführt wird, das mit 1% dFBS ergänzt wird. Es sollte berücksichtigt werden, dass bei einer hohen Lumineszenzleistung immer noch Durchblutungen in angrenzende Brunnen möglich sind, was zu irrtümlich berechneten Faltenänderungen führen kann. Nicht nachweisbare oder niedrige Signale (vorausgesetzt, eine Antwort wird erwartet) kann auf verschiedene Weise erklärt werden, nämlich schlechte Expression von GPCR(s) in HTLA-Zellen, die biologische Aktivität der Verbindung geht verloren, was sie ineffizient macht, oder die betreffenden Rezeptoren rekrutieren nicht an sich die Rekrutierung von -Arrestin2. Die Bewertung der Quantität und Qualität der transfizierten Plasmidrezeptor-DNA, die Prüfung anderer Präparate/Lots der infrageden unwirksamen Verbindung und die Durchführung orthologer Protein-Protein-Interaktionstechniken wie BRET/FRET oder Co-Immunoprecipitation sind einige Lösungsansätze für dieses Problem. Darüber hinaus könnte die Rezeptorexpression auch verbessert werden, indem Tango-Rezeptoren von Interesse in lentivirale Vektoren subkloniert und HTLA-Zellen transduziert werden, wodurch eine HTLA-GPCR-stabile Zelllinie erzeugt wird. Eine Verschiebung der erwarteten Potenz eines Agonisten während des Sekundärscreenings könnte implizieren, dass die Zur Stimulierung der Wirkstoffstimulation benötigte Wirkstoffstimulationszeit und/oder Konzentration der Verbindung nicht ausreicht oder dass die seriellen Verdünnungen der Arzneimittelplatte falsch vorbereitet wurden. Die Verwendung einer elektronischen Mehrkanalpipette oder eines automatisierten Pipettiersystems ohne Umdrehung entochern bei der Erstellung von seriellen Verdünnungen von Medikamenten könnte ein Problem bei der Arbeit mit klebrigen Verbindungen sein.
Bemerkenswerte Unterschiede zwischen dem ursprünglichen Tango-Assay, der von Barnea et al.18 entwickelt wurde, und der PRESTO-Tango-Plattform sind das Design des Rezeptors in einem modularen Format, bestehend aus codon-optimierten Sequenzen, die die Rezeptorexpression in Säugetierzellen verbessern, Epitop-Tags zur Validierung dieser Expression und Restriktionsstellen, die GPCRs, V2-Schwanz und TEVcs-tTA flankieren und die Exzise von Teilen ermöglichen. Am wichtigsten ist, dass PRESTO-Tango den Tango-Assay in Bezug auf Screening-Power und experimentelles Design übertrifft. Quadruplicate-Probentests von ca. 300 GPCRs werden nur in 8 384-Well-Platten durchgeführt, wobei negative Hintergrundkontrollen und positive Kontrollen zur Überwachung der Transfektionseffizienz berücksichtigt werden. Während PRESTO-Tango für das Screening des GPCR-Omes mit nur einer Verbindung von Interesse geeignet ist, kann auch eine Verhör mit mehreren Liganden durchgeführt werden, wenn auch zu erhöhten Kosten und Ressourcennutzung, wie z.B. mit gepoolten oder angeordneten kleinen Molekül-Compound-Bibliotheken oder biologischen Proben, die aus Mischungen verschiedener chemischer Einheiten bestehen. Zugegeben, dieses Problem kann durch die Verringerung der Anzahl der zu hinterfragenden Verbindungen durch die Durchführung chemischer Ähnlichkeits- und Diversitätsanalysen der betreffenden Zusammengesetztenbibliotheken gemildert werden. Während die PRESTO-Tango-Plattform eher für primäre Screening-Zwecke geeignet ist, kann die sekundäre Profilierung in kleinerem Maßstab, in mittleren oder niedrigen Durchsatzformaten, durchgeführt werden, um die funktionellen Folgen der Ligandenstimulation zu bestätigen. Wie bei allen anderen GPCR-Assays muss jedoch anerkannt werden, dass es keine geeigneten positiven Kontrollen für Orphan-Rezeptoren während des sekundären Screenings mit dem Tango-Assay gibt. Dennoch können potenzielle positive Treffer identifiziert werden, wenn die Ausgangsdaten mit einem berechneten Signalfenster und einem EC50-Wert an eine sigmoidale Dosis-Wirkungs-Kurve angepasst werden können. Es ist auch wichtig zu beachten, dass der Mechanismus der Ligandenaktivität, sei es für Waisen- oder Nicht-Waisenrezeptoren, nicht aufgeklärt werden kann, ohne parallele Assays auszuführen.
Da alle Komponenten von PRESTO-Tango bereits optimiert sind, einschließlich HTLA-Zelllinien- und GPCR-Tango-Konstrukten, ist neben der Wahl der zusammengesetzten Formulierung(en) nur wenig Spielraum für die Wirkstoffstimulation erforderlich. Auf Wunsch kann eine HTLA-Zelllinie, die einen Rezeptor stabil ausdrückt, leicht durch Klonen dieses GPCR-Tango-Rezeptors innerhalb des empfohlenen pIRESbleo3-Vektors (Clonetech) und durch Auswahl von Klonen mit Zeocin erzeugt werden. In Bezug auf den Swap von pcDNA3.1 zu pIRESbleo3 sollten Sie einfach das GPCR Tango-Konstrukt mit NotI und XbaI verdauen und an den Restriktionsstellen NotI und NheI in den Zielvektor einfügen. Ungeachtet dessen gibt es Möglichkeiten, diese Technologie anzupassen und zu optimieren. Eine der Säulen dieser Technologie sind HTLA-Zellen, eine HEK293T-Zelllinie, die ein Fusionsgen von '-arrestin2-TEV stabil ausdrückt, und ein tTA-abhängiger Luziferase-Reporter, der gnädig aus dem Labor von Richard Axel geliefert wird. Obwohl preSTO-Tango eine entscheidende Komponente ist, gibt es derzeit keine anderen Alternativen in Bezug auf den Ursprung der Zelllinie oder die Gene, die sie ausdrücken. Darüber hinaus können zukünftige konstruierte Zelllinien erzeugt werden, um andere TEV-Fusionsgene auszudrücken, um andere Proteine neben dem "-arrestin2" zu verfolgen, insbesondere solche, die zuvor nachweislich mit GPCRs interagieren oder in ihrem Wohnsitz gefunden wurden, wie z. B. 14-3-322, SAP9723und '-arrestin1, die vorherrschende Isoform von nicht-visuellen Arrestinen in Wirbeltieren24. Dies kann durch die Verwendung der elterlichen HTL-Zellen erreicht werden, die ausschließlich den luziferase-Reporter enthielten, der vom tetO7-Promotor gesteuert wurde. Eine Einschränkung für PRESTO-Tango ist die unspezifische Aktivierung des Reporter-Promoters. Basierend auf einem tetracyclinabhängigen Regulierungssystem (tet-System) steuert das Tetracyclin-responsive Element (TRE) die Expression des nachgeschalteten Luziferase-Reporters. Frühere Studien haben jedoch gezeigt, dass "leaky" Expression von Luziferase aufgrund der endogene Transkriptionsfaktoren25,26. Infolgedessen könnten einige Verbindungen den Reporter unabhängig von der Rekrutierung oder GPCR-Aktivierung aktivieren, wodurch die Anzahl der Falschmeldungen erhöht wird. Ein weiteres Problem, das auftaucht, auch gemeinsam mit anderen HTS-Methoden, sind "häufige Hitter", promiskuitive Verbindungen, die erhebliche Reaktionen in mehreren Zielen stimulieren27. Nichtsdestotrotz erleichtert das parallele Screening-Setup des PRESTO-Tango die Identifizierung dieser Artefakte, die weiter getestet werden können, um ihre Wirkung auf die Luziferaseaktivität zu bestätigen. Insgesamt hat PRESTO-Tango solide Grundlagen für die Untersuchung der Verhaftung bei der Rekrutierung von GPCRs und im größeren System der Arzneimittelentdeckung als utiles GPCR-Liganden-Screening- und Entwaistisierungsinstrument geschaffen.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die authours erklären keine konkurrierenden Interessen.
Acknowledgments
Diese Arbeit wurde von den Canadian Institutes of Health Research (CIHR-Stipendium #MOP142219) unterstützt.
Materials
| Name | Company | Catalog Number | Comments |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, BLACK, with lid, Sterile | NUNC | 12-566 | |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, White, with lid, Sterile | NUNC | 12-566-1 | |
| 384 Well Round Bottom, Polypropylene, Non-Treated, Blue, non-sterile, without lid | ThermoFisher | 12-565-390 | |
| Antibiotic-Antimycotic | Wisent | 450-115-EL | |
| D-Luciferin, sodium salt | GoldBio | LUCNA | |
| DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Corning | 10-013-CV | |
| Eppendorf Xplorer, 12-channel, variable, 15–300 µL | Eppendorf | 4861000155 | |
| Eppendorf Xplorer, 12-channel, variable, 5–100 µL | Eppendorf | 4861000139 | |
| Matrix Platemate 2x3 | ThermoFisher | 801-10001 | |
| MicroBeta 1450 Wallac | Perkin Elmer | ||
| Penicilin-Streptomycin | Wisent | 450-201-EL | |
| Poly-L-Lysine hydrobromide | Millipore-Sigma | P2636-500MG | |
| Roth Lab PRESTO-Tango GPCR Kit | Addgene | Kit #1000000068 |
References
- Liapakis, G., et al. The G-protein coupled receptor family: actors with many faces. Current pharmaceutical design. 18 (2), 175-185 (2012).
- Pierce, K. L., Premont, R. T., Lefkowitz, R. J. Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology. 3 (9), 639-650 (2002).
- Kroeze, W. K., Sheffler, D. J., Roth, B. L. G-protein-coupled receptors at a glance. Journal of cell science. 116, Pt 24 4867-4869 (2003).
- Rask-Andersen, M., Almén, M. S., Schiöth, H. B. Trends in the exploitation of novel drug targets. Nature Reviews Drug Discovery. 10 (8), 579-590 (2011).
- Ngo, T., et al. Identifying ligands at orphan GPCRs: current status using structure-based approaches. British journal of pharmacology. 173 (20), 2934-2951 (2016).
- Zhang, R., Xie, X. Tools for GPCR drug discovery. Acta pharmacologica Sinica. 33 (3), 372-384 (2012).
- Kimple, A. J., Bosch, D. E., Giguere, P. M., Siderovski, D. P. Regulators of G-Protein Signaling and Their G Substrates: Promises and Challenges in Their Use as Drug Discovery Targets. Pharmacological Reviews. 63 (3), 728-749 (2011).
- Wettschureck, N., Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiological Reviews. 85 (4), 1159-1204 (2005).
- Denis, C., Saulière, A., Galandrin, S., Sénard, J. -M., Galés, C. Probing heterotrimeric G protein activation: applications to biased ligands. Current Pharmaceutical Design. 18 (2), 128-144 (2012).
- Yin, H., et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. The Journal of Biological Chemistry. 284 (18), 12328-12338 (2009).
- Cheng, Z., et al. Luciferase Reporter Assay System for Deciphering GPCR Pathways. Current Chemical Genomics. 4, 84-91 (2010).
- Roth, B. L., Kroeze, W. K. Integrated Approaches for Genome-wide Interrogation of the Druggable Non-olfactory G Protein-coupled Receptor Superfamily. The Journal of Biological Chemistry. 290 (32), 19471-19477 (2015).
- Jean-Charles, P. -Y., Kaur, S., Shenoy, S. K. G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. Journal of Cardiovascular Pharmacology. 70 (3), 142-158 (2017).
- Smith, J. S., Rajagopal, S. The β-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. The Journal of Biological Chemistry. 291 (17), 8969-8977 (2016).
- Böhme, I., Beck-Sickinger, A. G. Illuminating the life of GPCRs. Cell Communication and Signaling. 7 (1), 16 (2009).
- Fang, Y. Label-Free Receptor Assays. Drug discovery today. Technologies. 7 (1), 5-11 (2011).
- Wang, T., et al. Measurement of β-Arrestin Recruitment for GPCR Targets. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences. , (2004).
- Barnea, G., et al. The genetic design of signaling cascades to record receptor activation. Proceedings of the National Academy of Sciences of the United States of America. 105 (1), 64-69 (2008).
- Kroeze, W. K., et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nature Structural & Molecular Biology. 22 (5), 362-369 (2015).
- Jordan, M., Schallhorn, A., Wurm, F. M. Transfecting Mammalian Cells: Optimization of Critical Parameters Affecting Calcium-Phosphate Precipitate Formation. Nucleic Acids Research. 24 (4), 596-601 (1996).
- Baker, J. M., Boyce, F. M. High-throughput functional screening using a homemade dual-glow luciferase assay. Journal of Visualized Experiments. (88), 50282 (2014).
- Li, H., Eishingdrelo, A., Kongsamut, S., Eishingdrelo, H. G-protein-coupled receptors mediate 14-3-3 signal transduction. Signal Transduction and Targeted Therapy. 1 (1), 16018 (2016).
- Walther, C., Ferguson, S. S. G. Minireview: Role of intracellular scaffolding proteins in the regulation of endocrine G protein-coupled receptor signaling. Molecular Endocrinology. 29 (6), Baltimore, Md. 814-830 (2015).
- Gurevich, E. V., Benovic, J. L., Gurevich, V. V. Arrestin2 expression selectively increases during neural differentiation. Journal of Neurochemistry. 91 (6), 1404-1416 (2004).
- Shaikh, S., Nicholson, L. F. B. Optimization of the Tet-On system for inducible expression of RAGE. Journal of Biomolecular Techniques JBT. 17 (4), 283-292 (2006).
- Blau, H. M., Rossi, F. M. Tet B or not tet B: advances in tetracycline-inducible gene expression. Proceedings of the National Academy of Sciences of the United States of America. 96 (3), 797-799 (1999).
- Roche, O., et al. Development of a Virtual Screening Method for Identification of "Frequent Hitters" in Compound Libraries. Journal of Medicinal Chemistry. 45 (1), 137-142 (2002).
