

Summary
GPCRは、魅力的な薬剤の標的であることを考えると、GPCRリガンドスクリーニングは、リード化合物の同定や脱孤児化研究に不可欠です。これらの取り組みに向けて、TEVベースのレポーターアッセイを用いて約300 GPCRで一過性β-arrestin2募集の同時プロファイリングに使用されるオープンソースのリソースプラットフォームPRESTO-Tangoについて説明します。
Abstract
細胞シグナル伝達経路の範囲の最大かつ最も汎用性の高い遺伝子スーパーファミリーおよびメディエーターとして、Gタンパク質共役受容体(GPCRs)は製薬業界にとって最も有望な標的の1つである。Ergoは、創薬やGPCR薬理学と成果を操作するための遠隔操作ツールを表すため、GPCRリガンドスクリーニングアッセイの設計、実施、最適化が重要です。これまで、Gタンパク質依存アッセイは、リガンド誘導事象を検出し、二次メッセンジャーの生成を定量化するこの分野を代表して行っていました。しかし、機能的選択性の出現以来、他のいくつかのGタンパク質非依存経路およびGタンパク質依存アッセイに関連する限界に対する意識の向上と同様に、代替の作成に向けてより大きな推進があるGPCRリガンドスクリーニングアッセイ。この取り組みに向けて、我々は、そのような資源の1つであるPRESTOタンゴプラットフォーム、ヒトGPCR-omeの並列かつ同時の尋問を可能にするルシファーゼレポーターベースのシステムの適用について述べている。技術的にも経済的にも実現不可能。Gタンパク質独立型β-arrestin2採用アッセイに基づいて、GPDRにおけるβ-arrestin2媒介性人身売買およびシグナリングの普遍性により、PRESTO-TANGOは約100人を含む約300の非嗅覚ヒトGPCRsを研究するための適当なツールとなる孤児受容体。PRESTOタンゴの感度と堅牢性は、既知の薬物の新しいGPCR標的を発見したり、孤児受容体の新しいリガンドを発見するために使用される複合ライブラリを使用して、一次ハイスループットスクリーンに適しています。
Introduction
Gタンパク質共役受容体(GPCRs)は、膜貫通タンパク質の最大かつ最も多様なファミリーを構成し、細胞とその環境間の通信インターフェースとして動作する1。GPCRsの多様性は、神経伝達物質からヌクレオチド、ペプチドから光子、および多くのリガンドの多様な配列を検出する能力と、細胞増殖、移動、分化、アポトーシス、細胞焼成などに関与する多数の下流シグナル伝達カスケードを調節する能力によって強調される。彼らの普遍性と多くの生理学的プロセスへの関与を考慮すると、この受容体ファミリーは、現在入手可能な処方薬の3分の1以上がGPCRs4を標的としているという事実によって示される、最も治療上の重要さである。しかし、これらの既存の治療法は、スーパーファミリーの小さなサブセット(推定10%)のみを標的とし、多くのGPCRsの薬理学は未解明のままである。さらに、100以上のGPCRsが、内因性リガンド5と一致していないとして、孤児受容体として存在する。したがって、GPCRリガンドスクリーニングは、リード発見と最適化への道を開き、おそらく臨床試験段階に道を開くため、脱ファン化と医薬品開発において重要です。
GPCRリガンドスクリーニング法は、従来、Gタンパク質依存性またはGタンパク質独立性機能アッセイ6の2つのカテゴリーのうちの1つに分類されている。GPCRシグナル伝達は、Gαサブユニット7に結合したGDPに対するGTPの交換によって活性化されるヘテロトリメリックGタンパク質(Gαβγ)によって調節される。活性化された受容体からのシグナルは、cAMP、カルシウム、DAG、およびIP3などの二次メッセンジャーを介してGタンパク質によって伝達され、下流エフェクター8で下流シグナル伝達を媒介する。Gタンパク質シグナル伝達の機能的な結果の性質は、受容体活性化を反映する細胞ベースのアッセイを作成するために利用されてきました。Gタンパク質シグナル伝達における近位(直接)または遠位(間接)事象を測定するこれらの方法は、GPCRリガンドスクリーニングに最も頻繁に使用され、主に脱孤児化研究6で採用されている。GPCR媒介Gタンパク質活性化を直接測定するアッセイの例としては、[35S]GTPγS結合アッセイが挙げられる。 Gαサブユニットに対する放射標識および非加水分解性GTPアナログと、Förster/生物発光共鳴エネルギー伝達(FRET/BRET、それぞれ)プローブとGPCR-GαおよびGα/Gγ相互作用を測定し、9年、10年にわたってより多くの牽引力を得ている。遠位イベントを監視するアッセイは、GPCRプロファイリングに最も一般的に使用されるツールです。例えば、cAMPおよびIP1/3アッセイは、Gタンパク質依存性二次メッセンジャーの細胞内蓄積を測定する一方で、[Ca2+]Gタンパク質活性化に関与する特異的応答要素を含むフラックスおよびレポーターアッセイ(CRE、NFAT-RE、SRE、SRF-RE)シグナルカスケード11をさらに下流の事象を調べる。前述のアッセイのほとんどはハイスループットレベルで実行できますが、 かなり敏感であり、特定のアッセイ固有の利点を誇る(例えば、GTPγS結合の場合の完全/部分的なアゴニスト、中立的アンタゴニストおよび逆アゴニスト間の差別、または[Ca2+]およびIP1/3)6などの生細胞上のアッセイ機能性6は、残念ながら既存のG依存性タンパク質方法は存在しない。これは主に、複数のGタンパク質サブファミリーがGPCBにネイティブに結合し、いくつかのカスケードでシグナル伝達を行い、オーファンGPDRで未知のGタンパク質結合を生じるためです。この問題を軽減するために、アッセイは、cAMPのような単一の共通シグナル伝達リードアウトを介して無差別Gタンパク質結合を強制するために開発された、およびCa2+、それらのほとんどは低スループット12であるが。
GPCRのライフサイクルの重要な側面は、Gタンパク質依存性シグナル伝達の終結であり、Gタンパク質の解離を誘導するβ-アスタミンの募集を通じて大部分が起こり、最終的にはクラトリンコーティングされた内在化を標的とする受容体を脱感作する。β-アレスティンの最もユビキタスに発現されるアイソフォームは、非視覚的β-アレスティン1およびβ-アレスティン2であり、また、それぞれ14のアレスティン-2及びアレスティン-3と示される。GPCRリガンドスクリーニングに新たな次元を追加するGタンパク質独立細胞ベースアッセイを入力してください。受容体の密売、標識のない全細胞、およびβ-アレプチンの募集アッセイは、すべて注目すべき例である。GPCR人身売買アッセイは、フルオロフォア標識リガンドまたは受容体15を標的とする共内在化抗体を採用する一方、ラベルフリーの全細胞アッセイは、リガンド結合によって誘導される細胞変化を電気信号または光学信号16などの定量可能な出力に変換するバイオセンサーを使用する。特に、典型的なGPCR-β-アレッシン相互作用は、機能アッセイ17のレパートリーにおける魅力的なツールとしてβ-アレッシン募集アッセイをファッションする。タンゴシステムは、わずか10年前にバルネアらによって最初に開発され、 3つの外因性遺伝要素の導入を伴う:β-arrestin2とタバコエッチウイルスプロテアーゼ(TEVp)からなるタンパク質融合、タバコエッチングを介してGPCRにつながれるテトラサイクリントランス活性化因子(tTA) ウイルスプロテアーゼ切断部位(TEVcs)と、逮捕を促進するためにV2バソプレシン受容体(V2尾)のC末語からの配列が先行し、その転写が誘発されるレポータールシメラーゼ遺伝子tTA転写因子は核への転座、β-アレプチン2募集後の膜アンカーから解放される(図1)18。GPCR活性化およびβ-arrestin2募集の定量的測定値は、その後、発光のための読み取りによって決定することができる。顕著な違いは、受容体の密売およびラベルフリーの全細胞法は比較的低いスループットであるが、タンゴは、標的受容体に特異的な選択的読み出しおよびシグナル統合による感受性を含むいくつかの利点を有し、これはより大きなスケール18でのリガンドスクリーニングに適した候補となる。
これらの戦略的特徴を考慮して、KroezeらはPRESTOタンゴ(パラレル受容体オーム発現および転写出力タンゴによるスクリーニング)を開発し、タンゴアプローチを使用して薬物可能GPCR-omeを並列かつ同時にプロファイルするハイスループットオープンソースプラットフォーム19を開発した。ほぼすべてのGPDRにβ-arrestin2の「無差別」募集を利用して、PRESTO-Tangoは細胞ベースの機能的アッセイの面で初めてのものである。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. 一次スクリーニング:細胞培養およびプレートシード
- ポリL-リジン(PLL)コーティングされたプレートを調製するために、電子マルチチャネルピペットまたは試薬ディスペンサーを使用して白または黒の384ウェル光学底板のPLLの20 μg/mLストック溶液の20 μL/ウェルを分配する。プレートを室温で0.5~2時間インキュベートします。
注: 黒い 384 ウェル プレートを使用する場合は、白いプレートと比較してバックグラウンド信号が低いことを期待してください。隣接するウェル間の発光のブリードスルーを減らすために、ブラックプレートをお勧めします。 - コーティングされたプレートを保存し、余分なPLLを洗い流し、それを流しの上にフリックしてPLLを取り除き、ペーパータオルの上で乾かして、電子マルチチャネルピペットまたは試薬ディスペンサーを使用して抗生物質抗ミキミティックの希釈された1x溶液の40 μL/ウェルを加えます。PLLコーティングされたプレートは、プレートの播種の準備ができるまで4°Cで保管してください。
- HTLA細胞(リチャード・アクセル博士が親切に提供)を維持するヒト胚性腎臓細胞株(HEK293T)は、β-アレスティン2-TEVおよびtTA駆動ルシファーゼ-完全なダルベッコの修飾イーグル培地(DMEM)で胎児の血清の5%を補い、 牛子牛血清5%、ピューロマイシン2.5μg/mL、ハイグロマイシン50μg/mL、100 U/mLペニシリン、および100μg/mLストレプトマイシンを5%CO2を含む加湿インキュベーターで37°Cで。
- HTLA細胞を150mmの皿で培養し、希釈係数1:10で週2回、最適な細胞通過数5~25で細胞を通過させます。プライマリ画面の規模に応じて、150 mmの皿が384ウェルプレートシードの日にコンフルエントであることを確認してください。
注: HTLA 細胞の使用法が 25 より大きいと生存率が低下し、最適ではない結果が得られる可能性があります。 - プライマリスクリーン用にHTLA細胞をシードするには、1xリン酸緩衝生理食塩水(PBS)pH 7.4でコンフルエント150mm皿をそっと洗い流します。約6 mLのトリプシン/0.53 mM EDTAを持つ細胞を取り外し、少なくとも同量の完全なダルベックコの改変イーグル培地(DMEM)を含む遠心管に移してトリプシンを中和する。
- HTLA細胞を500xgで3分間スピンダウンし、完全なDMEM中の0.22 x 106細胞/mLの密度で細胞ペレットを再懸濁し、2.5μg/mLのピューロマイシンおよび50μg/mLのハイグロマイシンの添加を省略して、トランスフェクション効果を低下させる可能性があります。
- 必要な384ウェルPLLコーティングプレートを37°Cでインキュベートし、細胞を播種する前に温めます。384ウェルPLLコーティングプレートから1x抗生物質抗ミキティックの貯蔵溶液を取り除き、プレートをシンクの上にフリックし、ペーパータオルの上にテーピングして乾燥させます。
- 電子マルチチャネルピペットを使用して0.22 x 106セル/mL HTLA懸濁液の45 μLを分配することにより、最終的な密度10,000セル/ウェルで384ウェルPLLコーティングプレートに細胞をシードします。一晩で37°Cでプレートをインキュベートする。当日トランスフェクションが好ましい場合は、16,000個の細胞/ウェルの密度で細胞をシードし、4時間後にトランスフェクションを行う。
注:高いトランスフェクション効率のために、50-70%の細胞の合流度が最適です。
2. 一次スクリーニング:DNAプレート調製およびトランスフェクション
- 図2に示すようにトランスフェクション用の384ウェルDNAソースプレートを調製するには、目的のGPCR-Tango構造をコードするプラスミドcDNAを96ウェルプレートに、異なるGPCR/ウェルで配布します。プラスミドDNAは、50 ng/μLの濃度で0.1xトリスEDTA(TE)バッファーに懸濁する必要があります。
注:96ウェルDNAプレートは-20°Cで密閉保存し、複数のスクリーニング実験に再利用することができます。GPCR-タンゴコンストラクトをコードするすべてのcDNAは市販されており(材料表を参照)、pcDNA3.1ネオマイシンプラスミドでクローン化されています。PRESTOタンゴGPCRキットは、それぞれ80個のGPCR、陰性対照として空のベクターを持つウェルのカップル、ドーパミン受容体D2(DRD2)を保持する陽性対照井戸、およびトランスフェクション効率を追跡するために蛍光タンパク質(YFP)をコードするプラスミドを運ぶウェルを含む4つの96ウェルプレートで構成されています。 - マルチチャネルピペットを使用して、96ウェルから384ウェルDNAソースプレートに手動でDNA溶液を移し、384ウェルあたり10 μLを追加します。実験の各条件が四重化でアッセイされることを確実にするために、96ウェルDNAプレート(行A-DまたはE-H)の半分は、各GPCRを2つの象限(第1象限=-化合物、第2象限=+化合物)に分配することによって、完全な384ウェルプレートを覆う。
- Jordan et al.20:0.1x TE バッファー (1 mM Tris-HCl および 0.1 mM EDTA) で説明されているように、リン酸カルシウム沈殿法に必要な次のトランスフェクション試薬を組み立てます。2.5 M CaCl2ソリューション;2xヘップスバッファー、pH 7.05(50 mM HEPES、280 mM NaCl、1.5 mM Na2HPO4)。ろ過によりすべての溶液を殺菌し、4 °Cで保管してください。トランスフェクションの日は、試薬が使用前に室温に達することを可能にする。
- 0.1x TE (1:8 希釈) で 2.5 M CaCl2ストック溶液を希釈し、0.313 M CaCl2と渦の最終濃度にします。0.313 M CaCl2の40 μLを384ウェルDNAソースプレートに移し、ハンドヘルドマルチチャンネルピペットまたは自動ベンチトップ384チャンネルピペットで上下にピペットして混合します。
- 384ウェルDNAソースプレートに2x Hepesバッファーの50 μLを追加し、上下にピペットして再び混ぜ、1分間放置します。各384ウェルは、試験する必要がある化合物の数に応じて、9つの384ウェルプレートのトランスフェクションのための十分な量のDNA/トランスフェクション混合物を有する。384ウェルDNAソースプレートから播種されたHTLA細胞にDNA/トランスフェクション混合物の10 μLを移し、37°Cでプレートを一晩インキュベートします。
3. 一次スクリーニング:細胞刺激
- 24時間後、384ウェルプレートを流しの上にそっとフリックしてペーパータオルの上、または吸引器の頭部でテーピングして、トランスフェクトされた細胞培地をデカントします。40 μLの飢餓培地(DMEMは1%透析したウシ血清(dFBS)と1x抗生物質/抗ミコティック剤を添加し、細胞に直接触れないように注意する。
- 目的の化合物のピペット 20 μL を 3 倍の濃度 (細胞板内の薬物の最終濃度は 1x)、交互の行に (+) 刺激を与え、(-)化合物を含まない交互の行に対する車両バッファーの 20 μL。5%CO2で37°Cのセルプレートを戻し、少なくとも16時間インキュベートする。
4. 一次審査:発光読み取り
- ベーカーとボイス21から変更されたGlo試薬を準備する: 108 mM Tris–HCl;42 mM Tris-Base, 75 mM NaCl, 3 mM MgCl2, 5 mM ジチオスレイトール (DTT), 0.2 mM 補酵素 A, 0.14 mg/ml D-ルシフェリン, 1.1 mM ATP, 0.25% v/v トリトン X-100, 2 mM ハイドロスルフィット.
注:試薬のストック溶液は、D-ルシフェリンを除いて、事前に作ることができます。ブラックプレートを使用した場合、D-ルシフェリンの量を0.25mg/mLまで増やすことができます。 - 刺激の後の16-24時間で、384ウェルプレートを流しの上にそっとフリックし、ペーパータオルの上にテーピングすることによって、トランスフェクトされた細胞培地をデカントします。Glo試薬を20 μL/wellに加え、室温でプレートを5〜20分間インキュベートします。
5. 一次審査:データ分析
- 保存したファイルをルミネセンス カウンタからスプレッドシートとしてエクスポートします。結果は相対発光単位(RLU)で記録されます。384ウェルプレートのレイアウトに基づいて、次の式を使用して各受容体の活性化(折り畳み変化)を計算します。
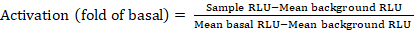
注:ここでサンプルRLUは、刺激された(+化合物)象限の4つの複製井戸のそれぞれの値を指し、平均背景RLUはプレート上の陰性対照の平均であり、平均基底RLUは、その同じ受容体の未処理の象限の平均を指します(- 化合物)。さらに、4つのデータポイントの標準偏差を計算して、結果の品質を検証します。変形性を是正するために、フォールドの平均に対して log2 変換を実行することをお勧めします。log2 ベースは、正のヒットを識別するのに役立つ実用的な選択肢です。経験的に正のヒットしきい値を設定します。いくつかの受容体は、2倍の増加と完全なアゴニストを持つ他の人のために最大40倍の増加まで低く持つことができることに留意する必要があります。 - 結果に基づいて、二次スクリーニングの潜在的な陽性ヒットであるGPCBを選択します。
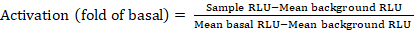
6. 二次スクリーニング:細胞の播種およびトランスフェクション
- 完全培地(4.55 x 105/mL)の11mLで5 x 106細胞の総細胞密度で100mm皿のHTLA細胞をサブ培養し、37°Cで24時間培養します。当日トランスフェクションが好ましい場合、細胞の濃度が7.5 x106の細胞で種子を入れ、4時間後にトランスフェクションを行う。
- 室温でリン酸カルシウム沈殿に必要な試薬を事前に温めます。0.1x TE バッファの 450 μL と 2.5 M CaCl2の 50 μL と素早く渦を組み合わせます。これらの量は、それが保持する成長培地の量に基づいて、100ミリメートル皿に固有です。
- チューブ内で、TE/CaCl2溶液の500 μLをGPCR cDNAおよび渦の10 μgに加えます。チューブに500 μLの2x Hepesバッファー溶液を加え、激しく振り(渦を起こさない)、1分間インキュベートします。
注:蛍光タンパク質をコードするプラスミドの1μg(YFP、mCherryなど)は、GPCR cDNAの9 μgを合計10 μgで共にトランスフェクトすることができます。蛍光タンパク質はトランスフェクション効率を追跡するために使用され、この最小量はアッセイを妨げない。 - 短いインキュベーションの直後に、1 mL溶液を細胞に滴下して分配する。プレートをゆっくりと前後に揺らし、沈殿物を均等に分配し、プレートを旋回しないように注意し、37°Cで24時間インキュベートします。
- 翌日、蛍光細胞のイメージャー下で蛍光タンパク質の発現を見てトランスフェクション効率を観察する。50%を超えるトランスフェクションが理想的です。
- 必要な384ウェルPLLコーティングプレートを37°Cのインキュベーターにインキュベーターでインキュベートし、細胞を播種する前に温めます。384ウェルPLLコーティングプレートから1x抗生物質抗ミキティックの貯蔵溶液を取り除き、プレートをシンクの上にフリックし、ペーパータオルの上にテーピングして乾燥させます。
- トランスフェクトされた細胞をVersene溶液(1X PBS、pH 7.4;0.53 mM EDTA)で穏やかに洗い、0.05%トリプシン/0.53 mM EDTAの3 mLを皿に加えて取り外します。少なくとも同量の完全なDMEMを含む遠心管に内容物を移してトリプシンを中和する。
- 500 x gで細胞を 3 分間回転させ、飢えた培地で 0.4 x 106細胞/mL の密度でセルを再懸濁します。電子マルチチャネルパイプを使用して細胞懸濁液の45 μLを分配することにより、25,000セル/ウェルの最終密度で384ウェルPLLコーティングプレートに細胞をシードします。プレートを37°Cに最低4時間戻し、細胞が適切に井戸に付着してから刺激に進みます。
7. 二次スクリーニング:16点(半log)線量曲線のための薬物プレート調製
- 96ウェルプレートに、図4に示すように、プレートの最後の行(行H)を除く1X HBSS薬物緩衝液(1xハンクのバランス塩溶液[HBSS]、20 mM HEPES pH 7.4、1x抗生物質抗抗抗抗毒素)の270μLを加える。
注:ペプチド、コロイド分子、水溶性化合物の場合、0.1~1%BSAの添加が推奨されます。薬物の酸化を防ぐために、アスコルビン酸を0.01%まで添加することもできる。 - 薬物ストックから、最終的な3倍濃度(細胞板中の薬物の最終濃度は1倍となる)を計算して、薬物溶液(「高」濃度と称する)を調製する。例として、10 μMの最高濃度の線量応答曲線の場合は、30 μMで「高」濃度を調製します。行Hの井戸に「高い」濃度のピペット300 μL。
- 別のチューブでは、「高」濃度を3.16(ハーフログ)で割った「低」濃度を調製します。前の例に基づいて、「低」濃度は9.49 μMになります。「ハイ」ウェルに隣接する行Hのウェルに「低」濃度のピペット300 μL。
注: 行 H に必要な 96 ウェルの合計数は、細胞の数と刺激条件によって異なります。4つの井戸(2つの「高」と2つの「低」)は、全体の384ウェルプレートを刺激するための十分な薬物溶液を持つことになります。 - 「高」と「低」のウェルから前の行(行G)に30 μLの薬物溶液をピペット化し、手動で上下にピペット処理するか、推奨通りに「ピペットとミックス」機能を備えた電子マルチチャンネルピペットを使用して混合してシリアル希釈を行います。希釈の間のヒントを捨てながら、最初の最も希釈された行(行A)までこのステップを繰り返します。
注: 必要に応じて、シリアル希釈は、行 A の前に停止することができ、薬物のない内部コントロール、つまり「真のゼロ」を表します。 - 図4を参考に、96ウェルプレートから前に播種された384ウェルプレートの行A-Oに「低」カラム希釈液の20μLをピペット化し、さらに「ハイ」カラム希釈液の20μLをウェルB-Pに20μLずつインキュベートして、最低16時間でプレートを37°Cにインキュベートして、トランスフェクトされた細胞を刺激します。
8. 二次スクリーニング:発光読み取りとデータ分析
- 刺激の後の16-24時間で、384ウェルプレートを流しの上にそっとフリックし、ペーパータオルの上にテーピングすることによって、トランスフェクトされた細胞培地をデカントします。Glo試薬を20 μL/wellに加え、室温でプレートを5〜20分間インキュベートします。
- 保存したファイルをルミネセンス カウンタからスプレッドシートとしてエクスポートします。結果は相対発光単位(RLU)で記録されます。384 ウェル プレートのデータを統計ソフトウェアに転送し、非線形回帰曲線適合の組み込み XY 分析を使用して結果を分析します。組み込みの3-パラメータ線量応答刺激機能「Log(アゴニスト)対応答(3つのパラメータ)」を選択し、

注:ここでは、上と下はY軸の単位でプラトー、それぞれ最大応答と基底レベル、EC50は、上と下の間に50%の応答を生成するアゴニストの濃度であり、Xはアゴニストの対数濃度を指します。このモデルは、線量応答曲線の標準ヒル勾配が 1 であると仮定します。

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
本明細書で提示されたPRESTOタンゴプロトコルを用いて、クロマフィン顆粒(CG)抽出物を168の非嗅覚GPCR標的に対してスクリーニングし、その大半は孤児受容体である。上記の抽出物のプロファイリングは、選択された受容体におけるβ-arrestin2動員を調べることによって、Barneaららによって設計された原理に基づいて行われた(図1)。目的のGPCRのプラスミドcDNAは、PRESTOタンゴGPCRキットから取り出し、所望のレイアウトで2つの96ウェルプレートに組み立てられました。合計で、HTLA細胞を播種した4つの384ウェルプレートがトランスフェクションされ、96ウェルDNAプレートの各半分が完全な384ウェルプレートに作られ、各受容体が2つの象限(合計8つの384ウェル)でトランスフェクトされた。2つの象限のうちの1つはCG抽出物で刺激された;異なる配置で、交互の行 C–D、G–H、K-L、および O–P は(+)刺激を表します (図 2)。一次スクリーニングで尋問された168のGPDRのうち、2つの受容体だけが潜在的な活性標的、特にドーパミン受容体D3(DRD3)およびオプシン5(OPN5)として候補となった。DRD3 は 4.70 のログ2倍の大きな変化を生じ、OPN5 はわずかに低い応答 2.39 を生成し、どちらも log2 のフォールド変更のしきい値カットオフを満たす >2。これに対し、一次スクリーンに対する陽性制御は、この受容体の選択的アゴニストであるクインピロールでDRD2刺激を受け、log2倍変化4.58を生成した(図3)。これらのシグナルウィンドウを再現し、偽陽性のヒットの可能性を排除するために、前述の受容体を用いて二次スクリーンを行った。CG抽出物の試験に加えて、DRD3が非孤児受容体であることを考えると、別の条件は、その選択的なアゴニストの一つであるキンピロールを用いた陽性対照として調製された。一方、 OPN5は孤児受容体であり、そのように、参照アゴニストは陽性対照としてCG抽出物と一緒にテストすることはできません。バッファのみが負のコントロールとしてテストされました。さらに、これら2つのGPCRの薬理学的特徴付けは、10-5 Mから10-12.5 Mまでの範囲の16ポイント線量曲線を調製することによって行われ、具体的には、10mMのCG抽出物およびクインピロールストック溶液を30μMおよび9.49μMに希釈した。これらの製剤は、20 μLが40 μLの飢餓培地でトランスフェクトされた細胞に分配されると、10-5 Mおよび10-5.5 Mになり、各384ウェル内で合計60 μLになります。前述のように、10-12Mおよび10-12.5Mに対する最も希薄な薬物形成が最上列(行A)になるように連続希釈を行った(図4)。二次スクリーニングからの線量応答曲線は、リガンドの効力と有効性を評価するためにGraphPadプリズムを使用して作成されました。クインピロールと比較して、CG抽出物は同様のシグナルウィンドウとEC50値を生成し、DRD3でのアクティブヒットとしての有効性を確認した。しかし、負のコントロールと同様の平坦な線量曲線をOPN5に対して作製し、それをCG抽出の可能な標的として除外した(図5)。

図1:タンゴ構造(A)のモジュール設計とβ-アレプチン(タンゴ)採用アッセイ(B)の一般スキーム。(A)GPCRタンゴは、HAシグナル/FLAGタグ、GPCR CDS、バソプレシン受容体2 C末端尾、TEVプロテアーゼ切断部位、およびtTA転写因子の様々なモジュール要素から構成される。(B) タンゴアッセイの原理は、HTLA細胞におけるGPCRタンゴプラスミドを一過性にトランスフェクトすることを含み、HEK293T細胞はβ-アレスティン2-TEVプロテアーゼ融合タンパク質およびtTAによって活性化されるルシメラーゼレポーター遺伝子を安定的に発現する。GPCRの活性化は最終的にβ-アレプチン2-TEVを受容体に動員し、プロテアーゼをその切断部位に近接させる。その結果、tTAはGPCRテールから切断され、転写因子が核内に転写され、ルシファーゼ発現を活性化する。この図の大きなバージョンを表示するには、ここをクリックしてください。

図2:PRESTO-TANGO一次スクリーニングにおけるトランスフェクションおよび刺激のための96ウェルcDNAプレートと384ウェルセルプレートのレイアウト。トランスフェクション用の384ウェルcDNAソースプレートの調製を描いたGPCR Tangoコンストラクトは、まず96ウェルプレートの半分から完全な384ウェルプレートに移され、各受容体は八重体でトランスフェクションされます。この設定では、(+)と(-)を含まない細胞の刺激は、目的の薬物(-)を個々の受容体の4倍にして起こる。この図の大きなバージョンを表示するには、ここをクリックしてください。

図3:PRESTO-タンゴ一次スクリーニングからのヒット識別のグラフィカル表現。概念実証として、GPCRome上のクロマフィン顆粒(CG)抽出物の生物学的活性を分析した。HTLA細胞は、168 GPCRタンゴコンストラクトを有する384ウェルプレートでトランスフェクトし、CG抽出物(+化合物)または車両緩衝液(-化合物)で刺激した。pcDNA3.1を陰性対照として用い、そして、クィンピロールで刺激されたDRD2受容体を陽性対照として用いた。シグナルウィンドウ(A)とlog2の倍変化(B)受容体活性化において、CG抽出物の不在または存在下のウェル間を計算した。すべての誤差範囲はSD(n = 4つの測定値)を表します。この図の大きなバージョンを表示するには、ここをクリックしてください。

図4:二次スクリーニングにおける刺激のための96ウェル薬物プレート調製物のレイアウト。細胞刺激のための96ウェル薬物プレートの調製を描いた、16ポイント線量曲線範囲の連続希釈は、行Hの10-5 M(最終濃度)から始まり、各ポイント間の半ログ間隔で10-12.5 M行A.「高」および「低」薬物列がシード384ウェルプレートの交互の列を刺激するために使用される。この図の大きなバージョンを表示するには、ここをクリックしてください。

図5:二次スクリーニングにおけるβ-arrestin2のGPDRへの実施の複合プロファイリングおよびデモンストレーションのための線量曲線応答。HTLA細胞は、一過性の受容体DRD3(A)およびOPN5(B)とトランスフェクトした。両方のトランスフェクション条件は、半ログ増分でCG抽出物、陽性対照としてDRD3特異的アゴニストキンピロール、および負の対照としてOPN5用の車両バッファで刺激された。すべての誤差範囲はSD(n = 3つの測定値)を表します。この図の大きなバージョンを表示するには、ここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
立体構造の動的GPCRsは、信号伝達の大国です。これらのヘプタケリカル受容体の結合ポケットの物理化学的性質と、それらの生理学的関連性は、GPCRリガンドスクリーニングツールの必要性を強調している。上記のように、PRESTOタンゴアッセイは迅速で敏感でユーザーフレンドリーであり、医薬品開発に適しています。このアッセイはアゴニスト誘導活性化を測定するだけでなく、アンタゴニストおよびアロステリックモジュレーター19の活性を定量化するためにも使用できる。機能的選択性に照らして、異なる薬物構造が単一の受容体で異なる受容体シグナル伝達カスケードを引き出すことを示唆する概念は、Gタンパク質依存アッセイを用いたGタンパク質経路の活性化とPRESTO-Tangoを用いたβ-アレプチン募集を比較することで、負の副作用を軽減した鉛化合物の設計に手掛かりを与えることができる。特に、Gタンパク質結合の検出からの独立性は、Gタンパク質依存アッセイでは以前は検出されていなかったであろう孤児GPDRの結合パートナーを同定するのに役立ちます。
PRESTO-Tangoスクリーンの一貫性と堅牢性を確保するためには、このプラットフォームの性質上、導入された摂動が拡大されるように、プロトコルのすべてのステップで注意を払う必要があります。もちろん、同じロット/製剤の試薬を使用して全体で同じ安定性と生物学的活性を確保し、HTSシステム上の条件を一貫して保つなど、考慮すべきすべてのHTSスクリーンに共通する一般的な措置があります。PRESTOタンゴの小型化された形式は、いくつかの重要な点に注意を払う必要があります:細胞播種密度の変動と井戸間の均質な分布(凝集対単一細胞懸濁液)、低トランスフェクション効率、および貧弱な化合物刺激および送達は、日々のプレート再現性を防ぎます。その効果のために、HTLA細胞懸濁液を三分体化して、播種前に溶液を均質化し、トランスフェクション前に50〜70%の細胞合流を確保します。化合物の送達のための車両は、ジメチルスルホキシドが最も一般的なキャリアである、検証する必要があります。通常、私たちの線量曲線の最高濃度は10 μMですが、これは化合物の性質と効力に応じて変化する可能性があります。細胞の耐性と毒性を推測するために様々な濃度をテストすることが重要です。
一部のGPCは構成活性が高いことを考えると、スクリーニング中に生じる可能性のある問題の1つは、ダイナミックレンジの低下と、予想以上に高いバックグラウンド信号です。これは、1%dFBSを添加したDMEM培地で血清飢餓が確実に行われることで、いくらか軽減することができます。発光出力が十分に高い場合、隣接するウェルにブリードスルーが存在し、誤って計算された折り目の変化をもたらすことを考慮する必要があります。検出不能または低いシグナル(応答が予想されると仮定して)は、いくつかの方法で説明することができます、すなわちHTLA細胞におけるGPCRの発現が悪い、化合物の生物学的活性が失われ、それを効果がない、または問題の受容体は本質的にβ-arrestin2をリクルートしない。それぞれ、トランスフェクトされたプラスミド受容体DNAの量と質を評価し、問題の他の調製物/多くの効果のない化合物をテストし、BRET/FRETまたは共免疫沈降などの直腸タンパク質相互作用技術を実行することは、この問題に対するいくつかの推奨される解決策である。さらに、目的のタンゴ受容体をレンチウイルスベクターにサブクローニングし、HTLA細胞をトランスデューシングし、HTLA-GPCR安定細胞株を生成することによって、受容体発現を改善することもできる。二次スクリーニング中にアゴニストの期待される効力の変化は、応答を刺激するために必要な薬物刺激時間および/または化合物の濃度が不十分であるか、または薬物プレート連続希釈が誤って調製されたことを意味する可能性がある。薬のシリアル希釈を作成するときの間にチップを変更することなく、電子マルチチャンネルピペットや自動ピペットシステムの使用は、粘着性化合物を使用する場合に問題になる可能性があります。
Barnea et18とPRESTO-Tangoプラットフォームによって開発された元のタンゴアッセイとPRESTO-Tangoプラットフォームの顕著な違いは、哺乳類細胞における受容体発現を改善するコドン最適化配列、前記発現を検証するエピトープタグ、およびGPCB、V2尾およびTEVcs-tTAに隣接する制限部位と、部分およびサブコンのエクスクチゾンを可能にする制限部位からなるモジュラー形式の受容体の設計を含む。最も重要なことは、PRESTOタンゴは、スクリーニング力と実験設計の面でタンゴアッセイを上回る。約300個のGPCBの4倍のサンプル試験は、わずか8つの384ウェルプレートで達成され、負のバックグラウンドコントロールと正の制御を考慮してトランスフェクション効率を監視します。PRESTOタンゴは、関心のある化合物を1つだけ含むGPCR-omeをスクリーニングするのに適していますが、プールまたはアレイ化された小分子化合物ライブラリや様々な化学実体の混合物からなる生物学的サンプルなど、コストの増加とリソースの使用にもかかわらず、複数のリガンドによる尋問も行うことができます。確かに、この問題は、問題の化合物ライブラリの化学的類似性と多様性分析を行うことによって、尋問する化合物の数を減らすことによって軽減することができる。PRESTO-Tangoプラットフォームは一次スクリーニング目的により適用可能であるが、二次プロファイリングは、リガンド刺激の機能的影響を確認するために、より小さなスケールで中程度または低スループットのフォーマットで行うことができる。しかし、他のすべてのGPCRアッセイと同様に、タンゴアッセイによる二次スクリーニング中に、孤児受容体に適切な陽性制御がないことを認めなければならない。それにもかかわらず、出力データを計算された信号ウィンドウとEC50値を持つシグモイド線量応答曲線に適合させることができる場合、潜在的な正のヒットを特定することができます。また、リガンド活性のメカニズムは、孤児または非孤児受容体の場合でも、並行アッセイを実行しなければ解明できないことに注意することも重要である。
PreSTO-Tangoのすべての成分は、HTLA細胞株およびGPCRタンゴ構築物を含め、すでに最適化されており、薬物刺激に使用される化合物製剤の選択とは別に、変更の余地はほとんどありません。所望であれば、受容体を安定的に発現するHTLA細胞株は、推奨pIRESbleo3ベクター内でGPCR-タンゴ受容体をクローニングし(Clonetech)、ゼオシンを用いてクローンを選択することにより容易に生成することができる。pcDNA3.1からpIRESbleo3へのスワップに関しては、単にNotIとXbaIでGPCRタンゴコンストラクトを消化し、制限部位NotIとNheIで目的地ベクトルに挿入します。それにもかかわらず、この技術を適応させ、最適化するための道があります。この技術の柱の一つはHTLA細胞、β-アレスティン2-TEV融合遺伝子を安定して発現するHEK293T細胞株と、リチャード・アクセルの研究室から優雅に供給されたtTA依存性ルシファーゼレポーターである。PRESTOタンゴの重要な成分である一方で、現在、細胞株起源やそれらが発現する遺伝子に関しては他の選択肢はない。さらに、将来の遺伝子操作細胞株は、他のTEV融合遺伝子を発現してβ-アレプチン2以外の他のタンパク質を追跡するために生成することができ、特に14-3-3 22、SAP9723、β-アレッシン1など、GPCRsに対して相互作用することが示されているか、または居住中に見つかったものは、脊椎動物24における非視覚的なアスタフィンのより一般的なアイソフォームである。これは、tetO7プロモーターによって制御されるルシファーゼレポーターのみを含む親HTL細胞を用いることによって達成することができる。PRESTOタンゴの1つの制限は、レポータープロモーターの非特異的活性化である。テトラサイクリン依存性調節系(tetシステム)に基づいて、テトラサイクリン応答性要素(TRE)は下流のルシファーゼレポーターの発現を制御する。しかし、これまでの研究では、内因性転写因子25,26によるルシファーゼの「漏れやすい」発現が実証されている。その結果、一部の化合物は、β-arrestin2の募集またはGPCR活性化とは無関係にレポーターを活性化させ、偽陽性の数を増加させる可能性がある。他のHTS法にも共通する別の問題は、「頻繁な打者」であり、いくつかの標的27における実質的な応答を刺激する無差別化合物である。それにもかかわらず、PRESTOタンゴの並列スクリーニングセットアップは、ルシファーゼ活性への影響を確認するためにさらにテストすることができるこれらのアーティファクトの同定を容易にする。全体として、PRESTOタンゴは、GPCRへの逮捕者募集の研究のための強固な基盤を提供し、創薬のより大きなスキームでは、利用GPCRリガンドスクリーニングおよび脱孤児化ツールとして提供してきました。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
オートアワーは競合する利益を宣言しません。
Acknowledgments
この研究は、カナダ保健研究所(CIHR助成金#MOP142219)によって支援されました。
Materials
| Name | Company | Catalog Number | Comments |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, BLACK, with lid, Sterile | NUNC | 12-566 | |
| 384 Well Optical Bottom Plates, Polystyrene Polymer Base, Cell Culture Treated, White, with lid, Sterile | NUNC | 12-566-1 | |
| 384 Well Round Bottom, Polypropylene, Non-Treated, Blue, non-sterile, without lid | ThermoFisher | 12-565-390 | |
| Antibiotic-Antimycotic | Wisent | 450-115-EL | |
| D-Luciferin, sodium salt | GoldBio | LUCNA | |
| DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Corning | 10-013-CV | |
| Eppendorf Xplorer, 12-channel, variable, 15–300 µL | Eppendorf | 4861000155 | |
| Eppendorf Xplorer, 12-channel, variable, 5–100 µL | Eppendorf | 4861000139 | |
| Matrix Platemate 2x3 | ThermoFisher | 801-10001 | |
| MicroBeta 1450 Wallac | Perkin Elmer | ||
| Penicilin-Streptomycin | Wisent | 450-201-EL | |
| Poly-L-Lysine hydrobromide | Millipore-Sigma | P2636-500MG | |
| Roth Lab PRESTO-Tango GPCR Kit | Addgene | Kit #1000000068 |
References
- Liapakis, G., et al. The G-protein coupled receptor family: actors with many faces. Current pharmaceutical design. 18 (2), 175-185 (2012).
- Pierce, K. L., Premont, R. T., Lefkowitz, R. J. Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology. 3 (9), 639-650 (2002).
- Kroeze, W. K., Sheffler, D. J., Roth, B. L. G-protein-coupled receptors at a glance. Journal of cell science. 116, Pt 24 4867-4869 (2003).
- Rask-Andersen, M., Almén, M. S., Schiöth, H. B. Trends in the exploitation of novel drug targets. Nature Reviews Drug Discovery. 10 (8), 579-590 (2011).
- Ngo, T., et al. Identifying ligands at orphan GPCRs: current status using structure-based approaches. British journal of pharmacology. 173 (20), 2934-2951 (2016).
- Zhang, R., Xie, X. Tools for GPCR drug discovery. Acta pharmacologica Sinica. 33 (3), 372-384 (2012).
- Kimple, A. J., Bosch, D. E., Giguere, P. M., Siderovski, D. P. Regulators of G-Protein Signaling and Their G Substrates: Promises and Challenges in Their Use as Drug Discovery Targets. Pharmacological Reviews. 63 (3), 728-749 (2011).
- Wettschureck, N., Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiological Reviews. 85 (4), 1159-1204 (2005).
- Denis, C., Saulière, A., Galandrin, S., Sénard, J. -M., Galés, C. Probing heterotrimeric G protein activation: applications to biased ligands. Current Pharmaceutical Design. 18 (2), 128-144 (2012).
- Yin, H., et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. The Journal of Biological Chemistry. 284 (18), 12328-12338 (2009).
- Cheng, Z., et al. Luciferase Reporter Assay System for Deciphering GPCR Pathways. Current Chemical Genomics. 4, 84-91 (2010).
- Roth, B. L., Kroeze, W. K. Integrated Approaches for Genome-wide Interrogation of the Druggable Non-olfactory G Protein-coupled Receptor Superfamily. The Journal of Biological Chemistry. 290 (32), 19471-19477 (2015).
- Jean-Charles, P. -Y., Kaur, S., Shenoy, S. K. G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. Journal of Cardiovascular Pharmacology. 70 (3), 142-158 (2017).
- Smith, J. S., Rajagopal, S. The β-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. The Journal of Biological Chemistry. 291 (17), 8969-8977 (2016).
- Böhme, I., Beck-Sickinger, A. G. Illuminating the life of GPCRs. Cell Communication and Signaling. 7 (1), 16 (2009).
- Fang, Y. Label-Free Receptor Assays. Drug discovery today. Technologies. 7 (1), 5-11 (2011).
- Wang, T., et al. Measurement of β-Arrestin Recruitment for GPCR Targets. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences. , (2004).
- Barnea, G., et al. The genetic design of signaling cascades to record receptor activation. Proceedings of the National Academy of Sciences of the United States of America. 105 (1), 64-69 (2008).
- Kroeze, W. K., et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nature Structural & Molecular Biology. 22 (5), 362-369 (2015).
- Jordan, M., Schallhorn, A., Wurm, F. M. Transfecting Mammalian Cells: Optimization of Critical Parameters Affecting Calcium-Phosphate Precipitate Formation. Nucleic Acids Research. 24 (4), 596-601 (1996).
- Baker, J. M., Boyce, F. M. High-throughput functional screening using a homemade dual-glow luciferase assay. Journal of Visualized Experiments. (88), 50282 (2014).
- Li, H., Eishingdrelo, A., Kongsamut, S., Eishingdrelo, H. G-protein-coupled receptors mediate 14-3-3 signal transduction. Signal Transduction and Targeted Therapy. 1 (1), 16018 (2016).
- Walther, C., Ferguson, S. S. G. Minireview: Role of intracellular scaffolding proteins in the regulation of endocrine G protein-coupled receptor signaling. Molecular Endocrinology. 29 (6), Baltimore, Md. 814-830 (2015).
- Gurevich, E. V., Benovic, J. L., Gurevich, V. V. Arrestin2 expression selectively increases during neural differentiation. Journal of Neurochemistry. 91 (6), 1404-1416 (2004).
- Shaikh, S., Nicholson, L. F. B. Optimization of the Tet-On system for inducible expression of RAGE. Journal of Biomolecular Techniques JBT. 17 (4), 283-292 (2006).
- Blau, H. M., Rossi, F. M. Tet B or not tet B: advances in tetracycline-inducible gene expression. Proceedings of the National Academy of Sciences of the United States of America. 96 (3), 797-799 (1999).
- Roche, O., et al. Development of a Virtual Screening Method for Identification of "Frequent Hitters" in Compound Libraries. Journal of Medicinal Chemistry. 45 (1), 137-142 (2002).
