

Summary
Dies ist eine Methode, um "scarinant" rekombinante Vaknie-Viren mit Host-Range-Auswahl und visuelle Identifizierung von rekombinanten Viren zu generieren.
Abstract
Das Vaccinia-Virus (VACV) war maßgeblich an der Ausrottung des Variolavirus (VARV), des Erregers der Pocken, aus der Natur beteiligt. Seit seiner ersten Verwendung als Impfstoff wurde VACV als Vektor für therapeutische Impfstoffe und als onkolytisches Virus entwickelt. Diese Anwendungen nutzen das leicht zu manipulierende Genom und das breite Host-Sortiment von VACV als hervorragende Plattform, um rekombinante Viren mit einer Vielzahl von therapeutischen Anwendungen zu erzeugen. Mehrere Methoden wurden entwickelt, um rekombinante VACV zu erzeugen, einschließlich Markerauswahlmethoden und transient dominante Selektion. Hier stellen wir eine Verfeinerung einer Hostrange-Auswahlmethode in Verbindung mit der visuellen Identifizierung rekombinanter Viren vor. Unsere Methode nutzt den selektiven Druck, der von der antiviralen Proteinkinase R (PKR) des Wirts erzeugt wird, gekoppelt mit einem fluoreszierenden Fusionsgen, das mCherry-tagged E3L, einen von zwei VACV PKR-Antagonisten, exzessiiert. Die Kassette, einschließlich des Gens von Interesse und der mCherry-E3L Fusion, wird von Sequenzen flankiert, die aus dem VACV-Genom abgeleitet sind. Zwischen dem Gen von Interesse und mCherry-E3L ist eine kleinere Region, die identisch mit den ersten 150 Nukleotiden des 3' Arms ist, um homologe Rekombination und Verlust des mCherry-E3L-Gens nach der Selektion zu fördern. Wir zeigen, dass diese Methode eine effiziente, nahtlose Erzeugung von rVACV in einer Vielzahl von Zelltypen ermöglicht, ohne dass eine Arzneimittelauswahl oder ein umfangreiches Screening auf mutierte Viren erforderlich ist.
Introduction
Das Vaccinia-Virus (VACV) war entscheidend für die erste erfolgreiche Tilgung eines menschlichen Erregers, des Variolavirus (VARV), aus der Natur. Seit der Ausrottung des Variola-Virus sind Poxviren einschließlich VACV weiterhin nützliche therapeutische Viren für die Human- und Tiermedizin. Beispielsweise hat ein VACV-basierter Tollwutvirus-Impfstoff die Übertragung von sylvatischer Tollwut in Europa1 und den Vereinigten Staaten sehr wirksam verhindert2. In jüngerer Zeit haben rekombinante Poxviren, die eine Vielzahl von Anti-Tumor-Molekülen (z. B. einkettige Antikörper oder menschliches Erythropoietin) exzieren, ermutigende Erfolge als onkolytische Wirkstoffegesehen 3,4,5. VACV ist als Vektor besonders attraktiv, da es leicht für genetische Manipulationen geeignet ist, ein breites Wirtsspektrum besitzt und unter einer Vielzahl von Bedingungen stabil ist, was einen einfachen Transport und eine mögliche Lebensfähigkeit des Impfstoffs im Feld6,7ermöglicht. Während mehrere Techniken entwickelt wurden, um rekombinante VACV für Laborexperimente und Impfstoffgenerierung zu erzeugen, haben aktuelle Strategien zur Generierung dieser Viren bemerkenswerte Einschränkungen.
Aufgrund des Nutzens von VACV wurden mehrere Strategien zur Erzeugung rekombinanter Viren entwickelt. Die erste Strategie verwendet homologe Rekombination, um eine Kassette mit dem Transgen und einem wählbaren Markergen wie einem Antibiotikaresistenzgen einzuführen. Die Kassette wird flankiert von zwei Nukleotiden (nt) oder größeren Armen, die das Gen an einen bestimmten Ort im viralen Genom leiten, der dann stabil durch Doppel-Crossover-Ereignisse8,9,10integriert wird. Diese Strategie ist schnell und effizient; Es führt jedoch zu zusätzlichem genetischem Material in Form des Markergens, das unerwartete Effekte hervorrufen kann. Darüber hinaus gibt es eine praktische Obergrenze für die Anzahl der Transgene, die durch die Anzahl der verfügbaren eindeutigen wählbaren Marker begrenzt eingeführt werden können. Transient dominante Selektionsstrategien (TDS) haben dieses Problem angegangen, indem sie die Erzeugung von "scarless" rekombinanten Viren erleichtert haben11. Mit dieser Strategie werden ein Plasmid, das ein mutiertes VACV-Gen und ein wählbares Markergen enthält, in das virale Genom integriert, jedoch ohne zusätzliche flankierende VACV-DNA. Dieser Ansatz führt zu einer transienten Integration des gesamten Plasmids und zur Duplizierung des VACV-Gens als Folge der Integration durch ein einziges Crossover-Ereignis. Dieses Zwischenprodukt ist stabil, solange es unter Selektionsdruck aufrechterhalten wird, was eine Anreicherung dieses Konstrukts ermöglicht. Wenn die Auswahl entfernt wird, ermöglicht die VACV-Duplikation ein zweites Crossover-Ereignis, das zur Entfernung des Plasmids und der anschließenden Bildung des Wildtyps (Wt) oder des rekombinanten Virus im Verhältnis von ungefähr 50:50 führt. Während TDS rekombinante Viren erzeugt, ohne dass die stämmige Einführung fremder DNA erforderlich ist, müssen mehrere Virusklone durch Sequenzierungsanalyse auf die erwartete Mutation untersucht werden, ein potenziell zeitaufwändiger und kostspieliger Schritt.
Hier stellen wir einen Ansatz zur Erzeugung rekombinanter Poxviren vor, der die besten Aspekte jedes dieser Ansätze kombiniert, ähnlich einem Ansatz, der für die Replikation inkompetent modifizierte Impfung Ankara12,13,14beschrieben wurde. Diese Strategie kombiniert visuelle und Host-Bereichs-Auswahl, um schnell rekombinante Viren durch Doppel-Crossover-Ereignisse zu erzeugen, und anschließend das wählbare Markergen durch homologe Rekombination zu beseitigen. Dieser Ansatz ermöglicht die schnelle Generierung von Mutanten, die durch homologe Rekombination vermittelt werden, mit der "scarless" Natur von TDS-Ansätzen, ohne einen nachfolgenden Screening-Schritt zu erfordern, um Wildtyp- und mutierte Viren zu unterscheiden. Unsere Methode verwendet auch die Auswahl des Wirtsbereichs anstelle der Antibiotikaauswahl, wodurch das Risiko chemisch induzierter phänotypischer Veränderungen in der Zelllinie eliminiert wird. Für diesen Ansatz haben wir uns entschieden, den Wirt antivirale Proteinkinase R (PKR) als selektives Mittel zur Erzeugung rekombinanter VACV zu verwenden. PKR wird in den meisten Zelltypen als inaktives Monomer ausgedrückt15. Bei Bindung von doppelsträngiger RNA (dsRNA) an den N-terminalen dsRNA-bindenden Domänen dimerisiert PKR und wird16autophosphoryliert. Diese aktive Form von PKR phosphoryliert die Alpha-Untereinheit des eukaryotischen Initiationsfaktors 2 (eIF2), was letztlich die Abgabe von Initiator Methionyl-tRNA an das Ribosom hemmt, wodurch die intrazelluläre Translation verhindert und die Replikation vieler Virusfamilien weitgehend hemmt17,18.
Als Reaktion auf die breite und starke antivirale Aktivität von PKR haben viele Viren mindestens eine Strategie entwickelt, um die PKR-Aktivierung zu verhindern. Die meisten Poxviren exprimieren zwei PKR-Antagonisten, die von den E3L- und K3L-Genen in VACV kodiert werden und PKR durch zwei unterschiedliche Mechanismen antagonisieren19. E3 verhindert die PKR-Homodimerisierung durch Bindung doppelsträngiger RNA20,21, während K3 als Pseudosubstratinhibitor wirkt, indem es direkt an aktivierte PKR bindet und dadurch die Wechselwirkung mit seinem Substrat eIF2-22hemmt. Wichtig ist, dass diese beiden PKR-Antagonisten PKR nicht unbedingt von allen Arten hemmen. Zum Beispiel hemmte der K3-Homolog des Schafpockenvirus PKR stark von Schafen, während der Schafpocken-E3-Homolog keine nennenswerte PKR-Hemmung zeigte23,24. In dieser Studie stellen wir eine Methode zur Verwendung von PKR-vermitteltem selektiven Druck in Kombination mit Fluoreszenzauswahl vor, um ein VACV-Rekombinant zu erzeugen, das für E3L und K3L (VC-R4) gelöscht wurde und sich in PKR-fähigen Zellen, die von verschiedenen Arten stammen, nicht replizieren kann. Dieses rekombinante Virus bietet einen ausgezeichneten Hintergrund für die schnelle Generierung rekombinanter Viren, die Gene unter Kontrolle des nativen E3L-Promotors exezieren.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Generieren des Rekombinationsvektors
- Design-Primer, um die Auswahlkassette zu generieren. Entwerfen Sie jedes einzelne Amplikon mit überlappenden Sequenzen mit benachbarten Amplikonen und dem Vektor, um die isotherme enzymatische Montage von DNA-Molekülen, auch Gibson-Montage genannt, mit einem von mehreren Online-Primer-Design-Tools zu erleichtern.
HINWEIS: Dieses Protokoll kann auch mit herkömmlichen, auf Einschränkungen endonukleasebasierten Klonierungsmethoden abgeschlossen werden. In diesem Fall können Sie Primer mit den entsprechenden Einschränkungssites anstelle von überlappenden Sequenzen entwerfen. - Mit den in Schritt 1.1 entworfenen Primern verstärken die PCR die folgenden Elemente in der Reihenfolge von 5' bis 3'(Abbildung 1):500 Nukleotide der VACV-Genomregion 5' von E3L (5' Arm), EGFP oder dem Gen von Interesse, 150 Nukleotide aus der VACV-Genomregion sofort 3' e3L (kurzer 3'-Arm), ein synthetischer Früh-/Spät-Poxvirus-Promotor25, das mCherry-E3L-Fusionsgen und 500 Nukleotide aus der VACV-Genomregion 3' von E3L einschließlich des kurzen 3'-Arms (langer 3'-Arm).
- In einem PCR-Rohr die Reagenzien in folgender Reihenfolge für jedes Amplikon hinzufügen: 17 l DNase-freies Wasser, 1,2 l pro Primer (Anfangskonzentration = 10 Endkonzentration = 0,5 m), 5 l 5x PCR-Reaktionspuffer, Vorlagen-DNA (10 ng für Amplicons, verstärkt aus Plasmiden: EGFP- und E/L-Promoter-mCherry-E3L-Kassette; 100 ng für Amplicons, verstärkt aus viraler genomischer DNA: 5' und 3' Arme) und 0,5 l DNA-Polymerase. Passen Sie das Wasservolumen für ein Endreaktionsvolumen von 50 l an.
HINWEIS: Die Konzentration der Vorlagen-DNA sollte empirisch bestimmt werden, aber wir beginnen in der Regel mit 10 ng/Reaktion. - Legen Sie die Röhrchen in einen Thermocycler, und schmelzen Sie die DNA bei 98 °C für 30 s, und verwenden Sie dann 25 Runden eines dreistufigen PCR-Protokolls: 98 °C für 5 s, 55 °C für 10 s und 72 °C für 1 min.
HINWEIS: Bestimmen Sie die Schmelztemperatur basierend auf dem vom Hersteller vorgeschlagenen Tm für jeden Primersatz. Bestimmen Sie die geeignete Verlängerungszeit basierend auf der Länge jedes Amplicons (1 Minute/kb).
- In einem PCR-Rohr die Reagenzien in folgender Reihenfolge für jedes Amplikon hinzufügen: 17 l DNase-freies Wasser, 1,2 l pro Primer (Anfangskonzentration = 10 Endkonzentration = 0,5 m), 5 l 5x PCR-Reaktionspuffer, Vorlagen-DNA (10 ng für Amplicons, verstärkt aus Plasmiden: EGFP- und E/L-Promoter-mCherry-E3L-Kassette; 100 ng für Amplicons, verstärkt aus viraler genomischer DNA: 5' und 3' Arme) und 0,5 l DNA-Polymerase. Passen Sie das Wasservolumen für ein Endreaktionsvolumen von 50 l an.
- Visualisieren Sie die Amplifikationsprodukte auf einem 1% Agarose-Gel. Fügen Sie jedem DNA-Produkt 10 l und jedem Bohrspeicher 2 l Ladepuffer hinzu und laufen Sie bei 8 V/cm für 1 h.
- Gel reinigen jedes Amplicon mit einem DNA-Gel-Extraktionskit und dem Herstellerprotokoll. Elute die Amplicons aus der Säule durch Zugabe von 50 l DNase freies Wasser und sofort Zentrifugieren.
- Linearisieren Sie den pUC19 Klonvektor mit EcoRI Endonuklease-Verdauung. Zu einem Röhrchen 1 g pUC19, Wasser zu einem Volumen von 17 l, 2 l Reaktionspuffer und 1 l (20 Einheiten) EcoRIhinzufügen. Bei 37 °C für 1 h inkubieren.
- Visualisieren Sie die Amplifikationsprodukte auf einem 1% Agarose-Gel lauf bei 8 V/cm für 1 h. Verbrauchen Sie das Band aus dem Gel, und reinigen Sie das Produkt mit dem DNA-Gel-Extraktionskit, wie in Schritt 1.4 beschrieben.
- Ligate alle einzelnen, gelgereinigten Amplicons und den linearisierten Vektor mit einem Master-Mix-Kit.
- Zu einem PCR-Rohr, fügen Sie 0,2 pmol linearisierte pUC19 und jeder Amplicon (5' Arm, EGFP, kurze 3' Arm, E / L Promoter-mCherry-E3L Kassette, 3'arm). Fügen Sie DNase-freies Wasser zu einem Endvolumen von 10 l hinzu, und fügen Sie dann 10 L DNA-Baugruppen-Master-Mix hinzu. Proben bei 50 °C für 1 h inkubieren.
- Chemisch kompetente E. coli mit 2 l des montierten Produkts aus Schritt 1.6 wie zuvor beschrieben26,27transformieren. Platte 100 l der transformierten Zellen auf LB-Agaroseplatten, die 100 g/ml Ampicillin enthalten. Die Platten über Nacht bei 37 °C inkubieren.
- Wählen Sie gut isolierte Kolonien und übertragen Sie einzelne Kolonien in Röhren, die Luria-Brühe mit 100 g/ml Ampicillin enthalten. Die Röhren über Nacht bei 37 °C bebrüten, während sie bei 225 U/min schütteln.
- Isolieren Sie die Plasmide aus der Nachtkultur mit einem Plasmid-Miniprep-Kit. Überprüfen Sie die Konzentration und Reinheit der DNA mit einem Spektralphotometer. Ein A260/A280-Verhältnis zwischen 1,8 und 2,0 ist akzeptabel.
- Reichen Sie die Plasmide für die Sanger-Sequenzierung ein, um festzustellen, ob das gewünschte Klonprodukt korrekt ist. Bewahren Sie die DNA bei -20 °C auf.
2. Generierung des rekombinanten Virus
- Infizieren Sie eine konfluente Monoschicht geeigneter Zellen mit dem Virus, das bei einer Vielzahl von Infektionen von 1,0 (MOI = 1,0) in einer 6-Well-Platte rekombiniert werden soll. Inkubieren Sie die infizierten Zellen bei 37 °C und 5%CO2 für 1 h. Dann das Medium ansaugen und durch frisches DMEM ersetzen. Inkubieren Sie die infizierten Zellen bei 37 °C und 5%CO2.
HINWEIS: Für die Replikation eignen sich kompetente Viren wie ein Impfvirus ohne K3L22, eine Zelllinie wie die europäische Kaninchennierenzelllinie RK13 (ATCC #CCL-37) oder BSC-40 ist geeignet. Für die Replikation mangelhafter Viren, wie das in diesem Papier beschriebene Virus, das sowohl die PKR-Antagonisten E3L als auch K3L enthält, ist jedoch eine ergänzende Zelllinie erforderlich, die diese beiden Gene in Trans- oder PKR-Knock-down- oder Knock-out-Zellen exprozisiert. - Transfetiere die infizierten Zellen mit 500 ng des Vektors, der in Schritt 1.10 erzeugt und validiert wurde, mit einem kommerziell erhältlichen Transfektionsreagenz nach dem Herstellerprotokoll. Inkubieren Sie die Zellen bei 37 °C und 5%CO2 für 48 h.
HINWEIS: Wenn ein Impfvirus verwendet wird, das sowohl E3L als auch K3L enthält, wird der von PKR vermittelte selektive Druck die Auswahl rekombinierter Viren antreiben und die Expression des mCherry-E3L-Fusionsproteins in diesen Zellen aufrechterhalten. Auf Wunsch sollte es auch möglich sein, PCR nur den Einsatz zu verstärken, der für die Transfektion anstelle des gesamten Plasmids verwendet werden soll. - 48 Stunden nach der Infektion, Ernten Sie die infizierte Monolayer. In einigen Fällen können die Zellen durch Pipetten geerntet werden, aber wenn sie noch fest anhaften, ernten Sie sie mit einem Zellschaber. Die Zellen dreimal einfrieren und die Lysate dann für 15 s bei 50% Amplitude beschallen. Bewahren Sie dieses Lysat bei -80 °C auf, bis es einsatzbereit ist.
- Das in Schritt 2.3 geerntete Lysat in Schritt 2.3 von 10-1 bis 10-6 wird seriell 10-fach verdünnt, indem 120 l des Lysats zu 1080 l DMEM (10-1)addiert und dann 120 l dieser Verdünnung zu 1080 l DMEM (10-2) addiert und dieser Prozess vier weitere Male wiederholt wird. Fügen Sie 1 ml jeder Verdünnung zu einem individuellen, konfluenten Brunnen einer PKR-kompetenten Zelllinie, in diesem Fall RK13-Zellen, hinzu.
- Inkubieren Sie die infizierten Zellen bei 37 °C und 5%CO2 für 1 h. Dann das Medium ansaugen und durch frische DMEM-Inkubation der infizierten Zellen bei 37 °C und 5%CO2ersetzen.
- 24 bis 48 Stunden nach der Infektion, identifizieren rekombinante Viren durch Fluoreszenzmikroskopie. Plaques von rekombinanten Viren drücken rote Fluoreszenz durch Integration des mCherry-E3L-Fusionsgens aus (Abbildung 2). Wenn zunächst ein Virus ohne PKR-Inhibitoren verwendet wurde, enthalten alle Plaques rekombinantes Virus.
- Plaque reinigen rekombinante Viren dreimal auf RK13-Zellen. Nach der letzten Runde der Plaquereinigung sollten alle Plaques rote Fluoreszenz ausdrücken.
- Infizieren Sie eine konfluente 6-Well-Platte von RK13-Zellen, die die VACV-PKR-Inhibitoren E3L und K3L (RK13+E3L+K3L-Zellen28) mit dem plaquegereinigten roten Fluoreszenzvirus ab Schritt 2.6 exzieren. Ziel für ca. 50-100 Plaques pro Brunnen.
HINWEIS: Diese Zellen liefern die VACV PKR-Antagonisten trans und lindern den PKR-vermittelten selektiven Druck, das mCherry-E3L-Fusionsgen aufrechtzuerhalten, wodurch die "scarless" Generation des rekombinanten Virus gefördert wird. - Identifizieren Sie kollabierte Viren mittels Fluoreszenzmikroskopie mit einem EVOS2-Mikroskop und einem GFP-Filterwürfel (Erregung: 470/22, Emission: 525/50) und einem RFP-Filterwürfel (Erregung: 531/40, Emission: 593/40).
ANMERKUNG: Die Häufigkeit, mit der das mCherry-E3L-Fusionsgen verloren geht, beträgt etwa 2,5 % (Tabelle 2). Wenn EGFP nicht als Markergen enthalten ist, sind Plaques von mutierten Viren, die das mCherry-E3L-Fusionsgen verloren haben, farblos. - Plaque reinigen nur grün (VC-R4) oder farblose Plaques (E3L) dreimal auf RK13 +E3L+K3L Zellen. Stellen Sie sicher, dass keine Plaques rot fluoreszieren.
- Bestätigen Sie den Verlust von mCherry-E3L und das Vorhandensein der erwarteten Mutation durch PCR- und Sanger-Sequenzierung.
HINWEIS: Wenn das Gen oder die Mutation von Interesse keine PKR-hemmende Aktivität hat, müssen rekombinante Viren auf RK13+E3L+K3L-Zellen oder einer gleichwertigen PKR-hemmenden oder PKR-mangelhaften Zelllinie angebaut werden (Abbildung 3).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Wir verwendeten das in Abbildung 1 dargestellte Verfahren, um einen VACV zu erzeugen, der sowohl die PKR-Antagonisten E3L als auch K3L fehlte, indem wir E3L durch EGFP in einem Virus ersetzten, das bereits für K3L (vP872) gelöscht wurde. Abbildung 2 zeigt rote fluoreszierende Plaques in PKR-kompetenten RK13-Zellen, die auf eine virale Expression von mCherry-E3L hinweisen, sowie EGFP, ausgedrückt in RK13+E3L+K3L-Zellen, die den Verlust von E3L und den Zusammenbruch des mCherry-E3L-Auswahlmarkers bestätigen. Abbildung 3 bestätigt, dass sich dieses rekombinante Virus, VC-R4, ohne beide PKR-Antagonisten nicht in PKR-fähigen RK13-Zellen replizieren kann, während das übergeordnete Virus, vP872, das E3L ausdrückt, replikationsfähig ist. Um zu bestätigen, dass diese Unfähigkeit, sich in RK13-Zellen zu replizieren, nur auf den Verlust von E3L zurückzuführen war, haben wir EGFP in VC-R4 durch E3L ersetzt, um ein revertantes Virus mit demselben Auswahlprotokoll zu erzeugen. Abbildung 3 bestätigt auch, dass sich dieser hallende Virus so effizient repliziert wie vP872 in RK13-Zellen. Interessanterweise wurden farblose Plaques, die mit dem Kollaps des mCherry-E3L-Auswahlmarkers übereinstimmen, vor der Auswahl in RK13+E3+K3-Zellen identifiziert, die in der Regel zur Auswahl "scarless" Rekombinanten erforderlich sind, wahrscheinlich aufgrund der erweiterten Sequenzidentität zwischen der mCherry-E3L-Rekombinationskassette und dem E3L-Gen, das in VC-R4 eingesetzt wird. Daher, um die Effizienz der Rekombination und die Rate des Zusammenbruchs zu bestimmen, haben wir gewählt, um Viren zu produzieren, die den Poxvirus PKR-Antagonisten K3L exezieren, um das Problem des frühen Zusammenbruchs zu vermeiden23. Abbildung 4 zeigt das Auftreten von farblosen Plaques (Pfeilspitzen) nach einer Infektion von RK13+E3L+K3L-Zellen. Tabelle 1 zeigt die Ergebnisse von drei unabhängigen Experimenten, bei denen durchschnittlich 12,6 % der Nachkommenvirionen mit dem transfizierten Plasmid rekombiniert wurden, ähnlich wie zuvor gemeldete Frequenzen29,30,31. Tabelle 2 zeigt die Häufigkeit farbloser Plaques relativ zu gesamtplaques in RK13+E3L+K3L-Zellen, was die Kollapsrate und den Verlust des mCherry-E3L-Auswahlmarkers bei einer Häufigkeit von etwa 1,8 % zeigt.
Abbildung 1: Diagramm von p837-GOI-mCherry-E3L sowie die Host-Range- und visuelle Rekombinationsstrategie. (A) 5' Arm (schwarz) und 3' Arm (grau) flankieren den E3L-Lokus (braun) in VACV. (B) In p837-GOI-mCherry-E3L flankieren diese Arme eine Kassette mit dem Gen von Interesse (GOI), in diesem Fall EGFP, (grün) getrennt von einem mCherry-E3L (rot) Fusionsgen unter Kontrolle des synthetischen frühen/späten Poxvirus-Promotors25 blau) durch einen kurzen 3'-Arm (grau). Diese äußeren Arme treiben eine homologe Rekombination zwischen VACV und dem p837-GOI-mCherry-E3L an. Schwarze Pfeilspitzen zeigen die Standorte der überlappenden Primer an, die verwendet werden, um dieses Plasmid durch Gibson-Klonen zu erzeugen. (C) Wenn der selektive PKR-Druck entfernt wird, können Viren ausgewählt werden, die eine intramolekulare Rekombination zwischen den kurzen und langen 3'-Armen durchlaufen haben. (D) Das Virus (VC-R4) enthält nur das Gen, das im E3L-Lokus von Interesse ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 2: Fluoreszierende Mikrographien einer (oben) rekombinanten Virus-Plaque 24 Stunden nach Rekombination mit p837-GOI-mCherry-E3L, die sowohl mCherry (links) als auch EGFP (rechts) in RK13-Zellen exemitt. (Unten) Mikrograph einer rekombinanten Virus-Plaque 48 Stunden nach PKR-vermitteltem selektiven Druck wurde in RK13++-Zellen entfernt, die EGFP (rechts) exemitzen, aber nicht mCherry (links). Die Skalenstange zeigt für alle Panels 650 m an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
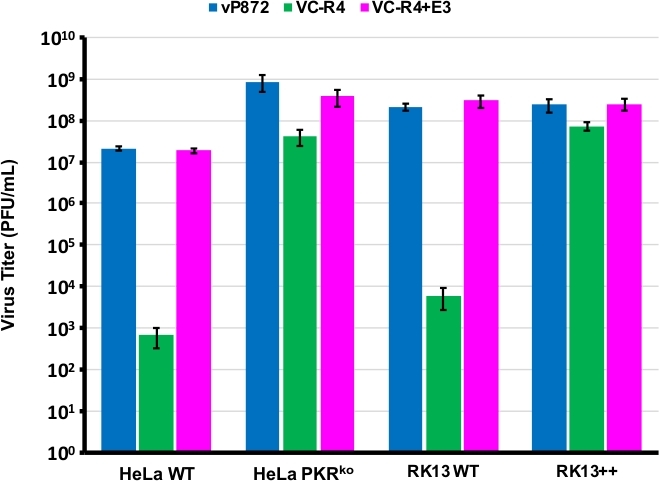
Abbildung 3: VC-R4 kann nicht in PKR-fähigen Zellen repliziert werden. Die angegebenen Zelllinien wurden mit vP872 (blau), VC-R4 (grün) oder VC-R4+E3L (Magenta) bei MOI = 0,1 infiziert. 48 Stunden nach der Infektion wurden die infizierten Zellen geerntet und durch serielle Verdünnung auf RK13+E3L+K3L-Zellen gezerrert. Titers werden in PFU/mL gemeldet, Fehlerbalken stellen die Standardabweichung von drei Replikationsexperimenten dar. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 4: Verlust der mCherry-E3L-Expression in RK13+E3L+K3L-Zellen. Overlay von fluoreszierenden und Phasenkontrast-Mikrographen von VC-R4+K3L-mCherry-E3L-infizierten RK13+E3L+K3L-Zellen. Drei Plaques drücken mCherry (Kreise) aufgrund des Zusammenbruchs der Auswahlkassette, die VC-R4+K3L ergibt, nicht mehr aus. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
| Experiment 1 | Experiment 2 | Experiment 3 | |
| Rote Plaketten (RK13) | 30 | 11 | 18 |
| Plaketten insgesamt (RK13+E3L+K3L) | 225 | 64 | 249 |
| Rekombinationsrate | 13.30% | 17.20% | 7.20% |
Tabelle 1: Rekombinationshäufigkeit von VACV mit dem Plasmid p837-K3L-mCherry-E3L.
| Experiment 1 | Experiment 2 | Experiment 3 | |
| Plaketten insgesamt (RK13+E3L+K3L) | 115 | 44 | 210 |
| Farblose Plaketten (RK13+E3L+K3L) | 3 | 1 | 1 |
| Rekombinationsrate | 2.60% | 2.30% | 0.50% |
Tabelle 2: Häufigkeit des mCherry-E3L-Verlustes von VC-R4+K3L-mCherry-E3L in RK13+E3+K3-Zellen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Hier stellen wir eine Variation einer transienten Markerauswahlstrategie 32 vor, um rekombinante Impfstoffviren zu erzeugen, ohne fremde DNA im endgültigen rekombinanten Virus zu speichern. Unsere Strategie verwendet selektiven Druck, der vom antiviralen Protein PKR des Wirts vermittelt wird, anstatt andere Formen des selektiven Drucks wie Antibiotika. Die Verwendung von antiviralen Wirtsgenen eliminiert die Möglichkeit chemisch induzierter phäkotypischer Veränderungen in den Zellen oder ein erhöhtes Mutationsrisiko aufgrund von Selektionsmedikamenten. Darüber hinaus gibt es, anders als bei der Arzneimittelauswahl, keine Verzögerungsphase für unseren Ansatz, da PKR konstitutiv in allen Zellen ausgedrückt wird. Die sekundäre visuelle Selektion auf Basis der mCherry-Expression verbessert auch die Spezifität dieser Methode, indem sichergestellt wird, dass in der ersten Phase nur Plaques, die das Transgen exezieren, entnommen werden, und ist ebenso effizient wie ein negativer selektiver Marker bei der Auswahl reifer rekombinanter Viren, die das mCherry-E3L-Gen verloren haben.
Die wichtigsten Schritte für diese Rekombinationsstrategie sind die Erzeugung des geeigneten Rekombinationsvektors und die entsprechende Plaque-Reinigung, um sicherzustellen, dass das ausgewählte Virus klonal ist. In diesem Artikel schlagen wir "Gibson Assembly" vor, um den Rekombinationsvektor zu generieren. Diese Strategie ist äußerst effizient und ermöglicht die Montage aller Fragmente, die den Rekombinationsvektor an einem Tag umfassen. Da jedoch der kurze 3'-Arm und der lange 3'-Arm identische Sequenzen haben, haben diese Fragmente das Potenzial, während der Klonreaktion zusammengefügt zu werden, und einige Vektoren enthalten möglicherweise nicht die mCherry-E3L-Kassette. Nach unserer Erfahrung ist dies selten, aber die Bestätigung der Struktur des Vektors nach dem Klonen ist notwendig. Wir haben auch Rekombinationsvektoren für diese Strategie mit traditionellen Endonuklease- und Ligase-Methoden generiert. Diese Strategie vermeidet das oben beschriebene Problem, kann aber arbeitsintensiver sein. Die Plaque-Reinigung ist im Allgemeinen einfach und hängt in erster Linie von der Verwendung geeigneter freizügiger Zellen für die anfängliche Rekombination, PKR-kompetenten Zellen für die anfängliche Plaque-Reinigung ab, um sicherzustellen, dass sich nur rekombinante Viren replizieren können, und dann wieder permissive Zellen, um die intramolekulare Rekombination und den Verlust des wählbaren Markers zu erleichtern. Eine besondere Aufmerksamkeit für Zelllinien ist daher entscheidend für die erfolgreiche und effiziente Anwendung dieser Strategie.
In dieser Studie zeigen wir die Verwendung dieser Methode zur Erzeugung eines VACV-Rekombinantens, das sowohl für die PKR-Antagonisten E3L als auch für K3L gelöscht wurde und EGFP unter Kontrolle des E3L-Promotors ausdrückt. In Zukunft wird dieses Virus als effizienter Hintergrund für zukünftige rekombinante Viren dienen, da es nicht in der Lage ist, sich in PKR-fähigen Zellen zu replizieren. Daher wird es einen starken PKR-vermittelten selektiven Druck geben, um die mCherry-E3L-Rekombinationskassette in Nachkommen-Virionen zu treiben und gleichzeitig im Wesentlichen die Replikation von nicht rekombinanten Viren zu verhindern. Darüber hinaus ist der Verlust von EGFP durch Aufnahme der Rekombinationskassette ein nützlicher sekundärer Auswahlmarker, um sicherzustellen, dass gepflückte Plaques nicht mit einem nicht rekombinanten Virus koinfiziert werden. Wir beobachteten Rekombinationsraten, die mit den zuvor gemeldeten Raten für VACV übereinstimmen, aber die visuellen Fluoreszenzmarker erhöhen die Effizienz der Erzeugung rekombinanter Viren, indem sie sicherstellen, dass die Wahrscheinlichkeit erhöht wird, dass die geeigneten rekombinanten Viren ausgewählt werden. Unsere Beobachtung von farblosen Plaques nach zwei Selektionsrunden an PKR-kompetenten Zellen, vermutlich aufgrund der erhöhten Länge identischer Sequenz zwischen E3L und dem mCherry-E3L-Markergen, legt nahe, dass die Rate des mCherry-E3L-Verlustes durch Erhöhung oder Verkleinerung der Länge des kurzen 3' Arms "abgestimmt" werden kann. Die primäre Einschränkung dieser Technik ist die Verwendung von PKR als selektiver Druck für Rekombinanten. Die effizienteste Nutzung dieser Rekombinationsstrategie ist die Generierung dieser Viren in einem Hintergrund ohne PKR-Antagonisten. Der farbmetrische Selektionsmarker ermöglicht es jedoch, diese Rekombinationsstrategie auch ohne die von PKR vermittelte Auswahl zu verwenden, einfach durch Plaque-Reinigung von mCherry-ausdrucksfreien Plaques. Während der Mangel an PKR-vermitteltem selektiven Druck die Effizienz des ersten Screening-Schritts verringern wird, ist der Prozentsatz der mCherry-Ausdrucksplaques immer noch hoch genug, dass eine farbbasierte Auswahl realisierbar ist. Somit kann diese Methode verwendet werden, um fast jedes Gen in das Poxvirus-Genom einzufügen.
Wie die Einfügung von EGFP zeigt, kann mit diesem Ansatz jedes Gen unter Kontrolle des nativen Promotors schnell in den E3L-Lokus eingeführt werden, vorausgesetzt, dass PKR-Nullzellen oder ergänzende Zelllinien für nachgeschaltete Experimente verwendet werden, wenn das Transgen kein PKR-Antagonist ist. Diese Strategie, kombiniert mit dem VC-R4-Virus, über das wir hier berichten, fügt eine neue und potente Methode hinzu, um schnell und zuverlässig rekombinante Impfungsviren zu erzeugen, die den von uns vermittelten selektiven Druck und die visuelle Identifizierung von Rekombinanten zu beginn des Prozesses verwenden.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren erklären keine konkurrierenden finanziellen Interessen.
Acknowledgments
Dieses Projekt wurde von den National Institutes of Health (AI114851) an SR finanziert.
Materials
| Name | Company | Catalog Number | Comments |
| 2X-Q5 Master Mix | NEB | M0492L | High-fidelity polymerase used in PCR |
| Ampicillin | ThermoFisher Scientific | 11593027 | Bacterial selective agent |
| Disposable Cell Scrapers | ThermoFisher Scientific | 08-100-242 | Cell scraper to harvest infected cells |
| EVOS FL Auto 2 Cell imaging system | ThermoFisher Scientific | AMAFD2000 | Fluorescent microscope |
| EVOS Light Cube, GFP | ThermoFisher | AMEP4651 | GFP Cube |
| EVOS Light Cube, RFP | ThermoFisher | AMEP4652 | RFP Cube |
| GenJet | SignaGen Laboratories | SL100489 | Transfection reagent |
| Luria Bertani (LB) Broth | Gibco | 10855021 | Bacterial growth medium |
| Monarch DNA gel extraction kit | NEB | T1020L | Gel purification kit used to purify amplicons and linearized vectors |
| Monarch Plasmid Miniprep kit | NEB | T1010L | Miniprep kit ussed to purify plasmids |
| NanoDrop One | ThermoFisher Scientific | ND-ONE-W | Spectrophotometer used to measure RNA and DNA concentration |
| NEBuilder Master Mix | NEB | E2621L | Isothermal enzymatic assembly kit used to generate the recombination vector |
| Q500 Sonicator | Qsonica | Q500-110 | Sonicator for virus lysates |
| RK13 cells | ATCC | CCL-37 | Rabbit kidney cells |
| VWR Multiwell Cell Culture plates | VWR | 10062-892 | Cell culture plates |
References
- Brochier, B., et al. Large-scale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature. 354 (6354), 520-522 (1991).
- Pastoret, P. P., Brochier, B. The development and use of a vaccinia-rabies recombinant oral vaccine for the control of wildlife rabies; a link between Jenner and Pasteur. Epidemiology and Infection. 116 (3), 235-240 (1996).
- Chan, W. M., McFadden, G. Oncolytic Poxviruses. Annual review of virology. 1 (1), 119-141 (2014).
- Nguyen, D. H., et al. Vaccinia virus-mediated expression of human erythropoietin in tumors enhances virotherapy and alleviates cancer-related anemia in mice. Molecular Therapy. 21 (11), 2054-2062 (2013).
- Frentzen, A., et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proceedings of the National Academy of Sciences of the United States of America. 106 (31), 12915-12920 (2009).
- Pastoret, P. P., Vanderplasschen, A. Poxviruses as vaccine vectors. Comparative Immunology, Microbiology and Infectious Diseases. 26 (5-6), 343-355 (2003).
- COLLIER, L. H. The development of a stable smallpox vaccine. The Journal of Hygiene. 53 (1), 76-101 (1955).
- Weir, J. P., Bajszár, G., Moss, B. Mapping of the vaccinia virus thymidine kinase gene by marker rescue and by cell-free translation of selected mRNA. Proceedings of the National Academy of Sciences of the United States of America. 79 (4), 1210-1214 (1982).
- Mackett, M., Smith, G. L., Moss, B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proceedings of the National Academy of Sciences of the United States of America. 79 (23), 7415-7419 (1982).
- Nakano, E., Panicali, D., Paoletti, E. Molecular genetics of vaccinia virus: demonstration of marker rescue. Proceedings of the National Academy of Sciences of the United States of America. 79 (5), 1593-1596 (1982).
- Falkner, F. G., Moss, B. Transient dominant selection of recombinant vaccinia viruses. Journal of Virology. 64 (6), 3108-3111 (1990).
- Staib, C., Drexler, I., Ohlmann, M., Wintersperger, S., Erfle, V., Sutter, G. Transient Host Range Selection for Genetic Engineering of Modified Vaccinia Virus Ankara. BioTechniques. 28 (6), 1137-1148 (2000).
- Staib, C., Drexler, I., Sutter, G. Construction and Isolation of Recombinant MVA. Vaccinia Virus and Poxvirology. , 77-99 (2004).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Pfaller, C. K., Li, Z., George, C. X., Samuel, C. E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Current Opinion in Immunology. 23 (5), 573-582 (2011).
- Bevilacqua, P. C., George, C. X., Samuel, C. E., Cech, T. R. Binding of the protein kinase PKR to RNAs with secondary structure defects: Role of the tandem A - G mismatch and noncontigous helixes. Biochemistry. 37 (18), 6303-6316 (1998).
- Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E., Hinnebusch, A. G. Tight Binding of the Phosphorylated Subunit of Initiation Factor 2 (eIF2) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor eIF2B Is Required for Inhibition of Translation Initiation. Molecular and Cellular Biology. 21 (15), 5018-5030 (2001).
- Rothenburg, S., Georgiadis, M. M., Wek, R. C. Evolution of eIF2α kinases: Adapting translational control to diverse stresses. Evolution of the Protein Synthesis Machinery and Its Regulation. , 235-260 (2016).
- Bratke, K. A., McLysaght, A., Rothenburg, S. A survey of host range genes in poxvirus genomes. Infection, Genetics and Evolution. 14, 406-425 (2013).
- Chang, H. W., Watson, J. C., Jacobs, B. L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proceedings of the National Academy of Sciences. 89 (11), 4825-4829 (1992).
- Romano, P. R., et al. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Molecular and Cellular Biology. 18 (12), 7304-7316 (1998).
- Beattie, E., Tartaglia, J., Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology. 183 (1), 419-422 (1991).
- Park, C., Peng, C., Brennan, G., Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Annals of the New York Academy of Sciences. 1438 (1), 18-29 (2019).
- Rothenburg, S., Brennan, G. Species-Specific Host-Virus Interactions: Implications for Viral Host Range and Virulence. Trends in Microbiology. , (2019).
- Chakrabarti, S., Sisler, J. R., Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques. 23 (6), 1094-1097 (1997).
- Chung, C. T., Niemela, S. L., Miller, R. H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution (recombinant DNA). Biochemistry. 86, 2172-2175 (1989).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Methods in Enzymology. 218, 621-627 (1993).
- Rahman, M. M., Liu, J., Chan, W. M., Rothenburg, S., McFadden, G. Myxoma Virus Protein M029 Is a Dual Function Immunomodulator that Inhibits PKR and Also Conscripts RHA/DHX9 to Promote Expanded Host Tropism and Viral Replication. PLOS Pathogens. 9 (7), 1003465 (2013).
- Evans, D. H., Stuart, D., McFadden, G. High levels of genetic recombination among cotransfected plasmid DNAs in poxvirus-infected mammalian cells. Journal of Virology. 62 (2), 367-375 (1988).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. Journal of Virology. 61 (6), 1788-1795 (1987).
- Spyropoulos, D. D., Roberts, B. E., Panicali, D. L., Cohen, L. K. Delineation of the viral products of recombination in vaccinia virus-infected cells. Journal of Virology. 62 (3), 1046-1054 (1988).
- Liu, L., et al. Transient dominant host-range selection using Chinese hamster ovary cells to generate marker-free recombinant viral vectors from vaccinia virus. BioTechniques. 62 (4), 183-187 (2017).
