

Summary
Este é um método para gerar vírus de vaccinia recombinante "sem scarless" usando seleção de alcance de host e identificação visual de vírus recombinantes.
Abstract
O vírus da vaccinia (VACV) foi fundamental na erradicação do vírus variola (VARV), o agente causador da varíola, da natureza. Desde sua primeira utilização como vacina, o VACV tem sido desenvolvido como vetor para vacinas terapêuticas e como um vírus oncolítico. Essas aplicações aproveitam o genoma facilmente manipulado do VACV e a ampla gama de hospedeiros como uma excelente plataforma para gerar vírus recombinantes com uma variedade de aplicações terapêuticas. Vários métodos foram desenvolvidos para gerar VACV recombinante, incluindo métodos de seleção de marcadores e seleção dominante transitória. Aqui, apresentamos um refinamento de um método de seleção de alcance de host, juntamente com a identificação visual de vírus recombinantes. Nosso método aproveita a pressão seletiva gerada pelo hospedeiro da proteína antiviral kinase R (PKR) juntamente com um gene de fusão fluorescente expressando mCherry-tagged E3L, um dos dois antagonistas VACV PKR. O, incluindo o gene de interesse e a fusão mCherry-E3L é ladeado por seqüências derivadas do genoma VACV. Entre o gene de interesse e mCherry-E3L é uma região menor que é idêntica aos primeiros ~150 nucleotídeos do braço de 3', para promover a recombinação homóloga e a perda do gene mCherry-E3L após a seleção. Demonstramos que este método permite uma geração eficiente e perfeita de rVACV em uma variedade de tipos de células sem exigir seleção de medicamentos ou triagem extensiva para vírus mutantes.
Introduction
O vírus da vaccinia (VACV) foi fundamental para a primeira erradicação bem sucedida de um patógeno humano, o vírus variola (VARV), da natureza. Desde o extermínio do vírus variola, os vírus da varíola, incluindo o VACV, continuam a ser vírus terapêuticos úteis para a medicina humana e animal. Por exemplo, uma vacina contra o vírus da raiva baseada em VACV tem sido muito eficaz na prevenção da transmissão da raiva silvestre na Europa1 e nos Estados Unidos2. Mais recentemente, os vírus da varíola recombinantes que expressam uma variedade de moléculas antitumorais (por exemplo, anticorpos de cadeia única ou eritropoietina humana) têm visto sucesso encorajador como agentes oncolíticos3,4,5. O VACV é particularmente atraente como vetor porque é facilmente passível de manipulação genética, possui uma ampla gama de hospedeiros, e é estável sob uma variedade de condições, permitindo fácil transporte e viabilidade vacinal no campo6,7. Embora várias técnicas tenham sido desenvolvidas para gerar VACV recombinante para experimentos laboratoriais e geração de vacinas, as estratégias atuais para gerar esses vírus têm limitações notáveis.
Devido à utilidade do VACV, várias estratégias para gerar vírus recombinantes foram desenvolvidas. A primeira estratégia emprega recombinação homóloga para introduzir um incluindo o transgene e um gene marcador selecionável, como um gene de resistência a antibióticos. O é ladeado por dois ~500 nucleotídeos (nt) ou braços maiores direcionando o gene para um local específico no genoma viral, que é então integrado por eventos de crossover duplo8,9,10. Essa estratégia é rápida e eficiente; no entanto, resulta em material genético extra na forma do gene marcador que pode produzir efeitos inesperados. Além disso, há um limite superior prático para o número de transgenes que pode ser introduzido limitado pelo número de marcadores selecionáveis únicos disponíveis. As estratégias transitórias de seleção dominante (TDS) abordaram essa questão facilitando a geração de vírus recombinantes "sem carro"11. Usando essa estratégia, um plasmídeo contendo um gene VACV mutante e um gene marcador selecionável são integrados ao genoma viral, mas sem dna vacv adicional. Esta abordagem resulta na integração transitória de todo o plasmídeo e duplicação do gene VACV como resultado da integração por um único evento de crossover. Este intermediário é estável desde que seja mantido sob pressão de seleção, permitindo o enriquecimento deste construto. Quando a seleção é removida, a duplicação vacv permite um segundo evento de crossover que resulta na remoção do plasmídeo e posterior formação do tipo selvagem (wt) ou do vírus recombinante em uma razão aproximada de 50:50. Enquanto o TDS gera vírus recombinantes sem exigir a introdução estável de DNA estranho, vários clones de vírus devem ser rastreados para a mutação esperada por meio da análise de sequenciamento, um passo potencialmente demorado e caro.
Aqui, apresentamos uma abordagem para a geração de porcavírus recombinantes combinando os melhores aspectos de cada uma dessas abordagens, semelhante a uma abordagem que foi descrita para a replicação incompetente da vaccinia modificada Ancara12,,13,14. Essa estratégia combina a seleção visual e de alcance do host para gerar rapidamente vírus recombinantes por eventos de crossover duplo e, posteriormente, eliminar o gene marcador selecionável por recombinação homóloga. Esta abordagem permite a rápida geração de mutantes mediados pela recombinação homóloga, com a natureza "sem brilho" das abordagens TDS, ao mesmo tempo em que não requer um passo de triagem subsequente para distinguir vírus selvagens e mutantes. Nosso método também utiliza a seleção da faixa do hospedeiro no lugar da seleção de antibióticos, eliminando o risco de alterações pheotípicas quimicamente induzidas na linha celular. Para esta abordagem, optamos por usar o host antiviral protein kinase R (PKR) como agente seletivo para gerar VACV recombinante. PKR é expresso como um monômero inativo na maioria dos tipos de células15. Ao vincular o RNA de dupla revasão (dsRNA) nos domínios n-terminal de ligação de dsRNA, o PKR dimeriza e é autofosforilado16. Esta forma ativa de PKR fosforila a subunidade alfa do fator de iniciação eucariótica 2 (eIF2), inibindo, em última análise, a entrega do iniciador methionyl-tRNA ao ribossomos, impedindo assim a tradução intracelular e inibindo amplamente a replicação de muitas famílias de vírus17,18.
Em resposta à ampla e potente atividade antiviral do PKR, muitos vírus desenvolveram pelo menos uma estratégia para impedir a ativação do PKR. A maioria dos vírus da varíola expressa dois antagonistas PKR, codificados pelos genes E3L e K3L no VACV, que antagonizam o PKR através de dois mecanismos distintos19. O E3 previne a homodimerização do PKR vinculando o RNA de dupla recadenação20,21, enquanto o K3 atua como um inibidor pseudosubstrato, vinculando-se diretamente ao PKR ativado e, assim, inibindo a interação com o seu substrato eIF2α22. É importante ressaltar que esses dois antagonistas pkr não necessariamente inibem o PKR de todas as espécies. Por exemplo, o homolog K3 do vírus da ovelhapox inibiu fortemente o PKR das ovelhas, enquanto o homolog sheeppox E3 não mostrou uma inibição considerável de PKR23,24. Neste estudo, apresentamos um método para usar a pressão seletiva mediada por PKR combinada com a seleção de fluorescência para gerar um REcombinante VACV excluído para E3L e K3L (VC-R4), que não pode se replicar em células competentes pkr derivadas de espécies diversas. Este vírus recombinante fornece um excelente fundo para a geração rápida de vírus recombinantes que expressam genes sob controle do promotor Nativo E3L.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Gerar o vetor de recombinação
- Projete primers para gerar o de seleção. Projete cada amplicon individual com seqüências sobrepostas com amplicons vizinhos e o vetor para facilitar a montagem enzimática isotérmica de moléculas de DNA, também chamada de montagem gibson, usando qualquer uma das várias ferramentas de design de primer on-line.
NOTA: Este protocolo também pode ser concluído usando métodos tradicionais de clonagem baseadaem endonuclease. Nesse caso, os primers de design com os locais de restrição apropriados em vez de com seqüências sobrepostas. - Utilizando os primers projetados na etapa 1.1, o PCR amplifica os seguintes elementos em ordem de 5' a 3'(Figura 1): ~500 nucleotídeos da região genômica VACV 5' do braço E3L (5'), EGFP ou o gene de interesse, ~150 nucleotídeos da região genômica VACV imediatamente 3' de E3L (braço curto de 3'),um promotor de poxvirus precoce/tardio sintético25, o gene de fusão mCherry-E3L, e ~500 nucleotídeos da região genômica VACV 3' de E3L, incluindo o braço curto de 3' (braço longo de 3').
- Em um tubo PCR, adicione os reagentes na seguinte ordem para cada amplicon: 17 μL de água livre de DNase, 1,2 μL de cada primer (concentração inicial = 10 μM, concentração final = 0,5 μM), 5 μL de tampão de reação PCR de 5x, DNA do modelo (10 ng para amplicons amplificados a partir de plasmídeos: EGFP e E/L promotor-mCherry-E3L cassete; 100 ng para amplicons amplificados a partir de DNA genômico viral: 5' e 3' braços), e 0,5 μL de DNA polimerase. Ajuste o volume de água adicionado para um volume de reação final de 50 μL.
NOTA: A concentração do DNA do modelo deve ser empiricamente determinada, mas geralmente começamos com 10 ng/reação. - Coloque o tubo em um termociclador e derreta o DNA a 98 °C por 30 s e use 25 rodadas de um protocolo PCR de três etapas: 98 °C para 5 s, 55 °C para 10 s e 72 °C para 1 min.
NOTA: Determine a temperatura de fusão com base no Tm sugerido pelo fabricante para cada conjunto de primer. Determine o tempo de extensão apropriado com base no comprimento de cada amplicon (1 minuto/kb).
- Em um tubo PCR, adicione os reagentes na seguinte ordem para cada amplicon: 17 μL de água livre de DNase, 1,2 μL de cada primer (concentração inicial = 10 μM, concentração final = 0,5 μM), 5 μL de tampão de reação PCR de 5x, DNA do modelo (10 ng para amplicons amplificados a partir de plasmídeos: EGFP e E/L promotor-mCherry-E3L cassete; 100 ng para amplicons amplificados a partir de DNA genômico viral: 5' e 3' braços), e 0,5 μL de DNA polimerase. Ajuste o volume de água adicionado para um volume de reação final de 50 μL.
- Visualize os produtos de amplificação em um gel de agarose de 1%. Adicione 10 μL de cada produto de DNA e 2 μL de tampão de carga para cada poço, e execute a 8 V/cm por 1 h.
- O gel purifica cada amplicon usando um kit de extração de gel de DNA e o protocolo do fabricante. Eluie os amplicons da coluna adicionando 50 μL de água livre de DNase e imediatamente centrifugando.
- Linearize o vetor de clonagem pUC19 utilizando digestão de endonuclease EcoRI. A um tubo, adicione 1 μg de pUC19, água a um volume de 17 μL, 2 L de tampão de reação e 1 μL (20 unidades) de EcoRI. Incubar a 37 °C por 1h.
- Visualize os produtos de amplificação em um gel de agarose de 1% executado a 8 V/cm por 1 h. Excise a banda do gel e purifique o produto usando o kit de extração de gel de DNA conforme descrito na etapa 1.4.
- Ligatodos os amplicons individuais, purificados em gel e o vetor linearizado usando um kit de mistura mestre.
- Em um tubo PCR, adicione 0,2 pmol de pUC19 linearizado e cada amplicon (braço de 5', EGFP, braço curto de 3'', E/L promoter-mCherry-E3L, 3'arm). Adicione água livre de DNase a um volume final de 10 μL e, em seguida, adicione 10 μL de mistura mestre de montagem de DNA. Incubar amostras a 50 °C por 1 h.
- Transforme e. coli quimicamente competente com 2 μL do produto montado a partir da etapa 1.6 como descrito anteriormente26,27. Placa de 100 μL das células transformadas em placas de agarose LB contendo ampicillina de 100 μg/mL. Incubar as placas durante a noite a 37 °C.
- Escolha colônias bem isoladas e transfira colônias individuais para tubos contendo caldo luria com ampicillina de 100 μg/mL. Incubar os tubos durante a noite a 37 °C enquanto agita a 225 rpm.
- Isole os plasmídeos da cultura durante a noite usando um miniprep plasmídeo. Verifique a concentração e pureza do DNA usando um espectrofotômetro. Uma razão A260/A280 entre 1,8 e 2,0 é aceitável.
- Envie os plasmídeos para sequenciamento de Sanger para determinar se o produto de clonagem desejado está correto. Armazene o DNA a -20 °C.
2. Gerar o vírus recombinante
- Infectar uma monocamada confluente de células adequadas com o vírus a ser recombinada em uma multiplicidade de infecção de 1,0 (MOI = 1,0) em uma placa de 6 poços. Incubar as células infectadas a 37 °C e 5% de CO2 por 1 h. Em seguida, aspirar o meio e substituí-lo por DMEM fresco. Incubar as células infectadas a 37 °C e 5% de CO2.
NOTA: Para a replicação, é apropriado para a replicação de vírus competentes, como um vírus de vacinação que não tenha K3L22, uma linha celular como a linha europeia de células renais de coelho RK13 (ATCC #CCL-37) ou BSC-40. No entanto, para a replicação de vírus deficientes, como o vírus descrito neste artigo, sem ambos os antagonistas PKR E3L e K3L, uma linha celular complementar expressando esses dois genes em células trans ou PKR knock-down ou knock-out são necessárias. - Transfect as células infectadas com 500 ng do vetor gerado e validado na etapa 1.10 utilizando um reagente de transfecção comercialmente disponível seguindo o protocolo do fabricante. Incubar as células a 37 °C e 5% de CO2 por 48 h.
NOTA: Se o uso de um vírus de vaccinia sem E3L e K3L, a pressão seletiva mediada por PKR conduzirá a seleção de vírus recombinados e manterá a expressão da proteína de fusão mCherry-E3L nessas células. Se desejar, também deve ser possível amplificar apenas a inserção para uso para transfecção em vez de todo o plasmídeo. - 48 horas após a infecção, colher a monocamada infectada. Em alguns casos, as células podem ser colhidas por pipetting, mas se ainda estiverem firmemente aderidas, colhe-as com um raspador de células. Congele as células três vezes e, em seguida, sônica os lisatos por 15 s a 50% de amplitude. Guarde este lysate a -80 °C até estar pronto para usar.
- Diluir em série 10 vezes o lysato colhido na etapa 2.3 de 10-1 a 10-6 adicionando 120 μL do lysate a 1080 μL de DMEM (10-1), e depois adicionando 120 μL desta diluição a 1080 μL de DMEM (10-2), e repetindo este processo mais quatro vezes. Adicione 1 mL de cada diluição a um indivíduo, confluente bem de uma linha celular competente PKR, neste caso células RK13.
- Incubar as células infectadas a 37 °C e 5% de CO2 por 1 h. Em seguida, aspirar o meio e substituí-lo por dmem fresco Incubar as células infectadas a 37 °C e 5% CO2.
- 24 a 48 horas após a infecção, identificar vírus recombinantes por microscopia de fluorescência. Placas de vírus recombinantes expressam fluorescência vermelha devido à integração do gene de fusão mCherry-E3L(Figura 2). Se um vírus desprovido de inibidores de PKR foi usado inicialmente, todas as placas conterão vírus recombinante.
- Placa purifica vírus recombinantes três vezes em células RK13. Após a rodada final de purificação da placa, todas as placas devem expressar fluorescência vermelha.
- Infectar uma placa de 6 poços confluente de células RK13 expressando os inibidores DE PKR VACV E3L e K3L (células RK13+E3L+K3L28) com o vírus fluorescing vermelho purificado da placa do passo 2.6. Aponte para aproximadamente 50-100 placas por poço.
NOTA: Essas células fornecem os antagonistas do VACV PKR em trans e aliviam a pressão seletiva mediada pelo PKR para manter o gene de fusão mCherry-E3L, promovendo assim a geração "sem carro" do vírus recombinante. - Identificar vírus colapsados por microscopia de fluorescência usando um microscópio EVOS2, e um cubo de filtro GFP (Excitação: 470/22, Emissão: 525/50) e um cubo de filtro RFP (Excitação: 531/40, Emissão: 593/40).
NOTA: A frequência em que o gene de fusão mCherry-E3L é perdido é de aproximadamente 2,5%(Tabela 2). Se o EGFP não for incluído como um gene marcador, placas de vírus mutantes que perderam o gene de fusão mCherry-E3L serão incolores. - Placa purificar placas verdes (VC-R4) ou incolores (E3L) três vezes em células RK13+E3L+K3L. Certifique-se de que nenhuma placa fluoresce vermelho.
- Confirme a perda de mCherry-E3L e a presença da mutação esperada pelo sequenciamento PCR e Sanger.
NOTA: Se o gene ou mutação de interesse não tiver atividade inibitória de PKR, os vírus recombinantes devem ser cultivados em células RK13+E3L+K3L ou em uma linha celular deficiente de PKR equivalente(Figura 3).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Utilizou-se o procedimento diagramado na Figura 1 para gerar um VACV sem antagonistas PKR E3L e K3L, substituindo E3L por EGFP em um vírus já excluído para K3L (vP872). A Figura 2 apresenta placas fluorescentes vermelhas em células RK13 competentes do PKR indicativas de expressão viral de mCherry-E3L, bem como EGFP expressas em células RK13+E3L+K3L confirmando a perda de E3L e o colapso do marcador de seleção mCherry-E3L. A Figura 3 confirma que este vírus recombinante, VC-R4, sem ambos os antagonistas PKR não pode se replicar em células RK13 competentes pkr, enquanto o vírus pai, vP872 expressando E3L, é competente em replicação. Para confirmar que essa incapacidade de replicação em células RK13 foi apenas devido à perda de E3L, substituímos o EGFP em VC-R4 por E3L, para gerar um vírus revertante usando o mesmo protocolo de seleção. A Figura 3 também valida que este vírus revertante se replica tão eficientemente quanto vP872 em células RK13. Curiosamente, placas incolores consistentes com o colapso do marcador de seleção mCherry-E3L foram identificadas antes da seleção em células RK13+E3+K3 que são geralmente necessárias para selecionar recombinantes sem esa, provavelmente devido à identidade de seqüência estendida entre o de recombinação mCherry-E3L e o gene E3L sendo inserido em VC-R4. Portanto, para determinar a eficiência da recombinação e a taxa de colapso, elegemos para produzir vírus que expressem o antagonista PKR do poxvirus K3L para evitar o problema do colapso precoce23. A Figura 4 indica o aparecimento de placas incolores (pontas de flecha) após a infecção das células RK13+E3L+K3L. A Tabela 1 mostra os resultados de três experimentos independentes, onde, em média, 12,6% das virions descendentes foram submetidas à recombinação com o plasmídeo transinfectado, semelhante às frequências relatadas anteriormente29,30,31. A Tabela 2 detalha a frequência de placas incolores relativas ao total de placas em células RK13+E3L+K3L, demonstrando a taxa de colapso e perda do marcador de seleção mCherry-E3L ocorrido em uma frequência de aproximadamente 1,8%.
Figura 1: Diagrama do p837-GOI-mCherry-E3L, bem como a estratégia de recombinação visual e alcance de host. (A) braço de 5' (preto) e braço de 3' (cinza) flanqueiam o lócus E3L (marrom) em VACV. (B) Em p837-GOI-mCherry-E3L, estes braços flanqueiam um contendo o gene de interesse (GOI), neste caso EGFP, (verde) separado de um gene de fusão mCherry-E3L (vermelho) sob controle do sintético promotor de poxvirus precoce/tardio25 azul) por um braço curto de 3'' (cinza). Estas recombinações homólogas de acionamento de braços externos entre o VACV e o p837-GOI-mCherry-E3L. Pontas de flechas pretas indicam os locais dos primers sobrepostos usados para gerar este plasmídeo pela clonagem gibson. (C)Quando a pressão seletiva pkr é removida, vírus que sofreram recombinação intramolecular entre os braços curto e longo de 3' podem ser selecionados. (D) Resultando em um vírus (VC-R4) contendo apenas o gene de interesse no lócus E3L. Clique aqui para ver uma versão maior desta figura.

Figura 2: Micrografias fluorescentes de (superior) uma placa de vírus recombinante 24 horas após a recombinação com p837-GOI-mCherry-E3L expressando tanto mCherry (esquerda) quanto EGFP (direita) em células RK13. (Embaixo) Micrografo de uma placa de vírus recombinante 48 horas após a pressão seletiva mediada por PKR ter sido removida em células RK13++, expressando EGFP (direita) mas não mCherry (esquerda). A barra de escala indica 650 μm para todos os painéis. Clique aqui para ver uma versão maior desta figura.
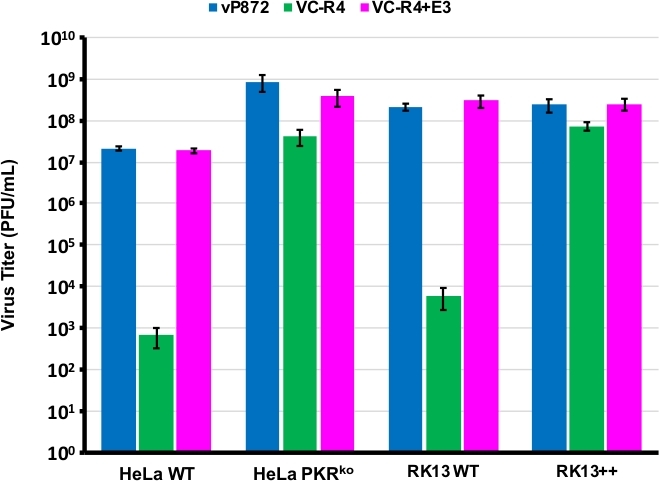
Figura 3: VC-R4 não pode se replicar em células competentes pkr. As linhas celulares indicadas foram infectadas com vP872 (azul), VC-R4 (verde) ou VC-R4+E3L (magenta) em MOI = 0,1. 48 horas após a infecção, as células infectadas foram colhidas e por diluição serial em células RK13+E3L+K3L. Os títulos são relatados em PFU/mL, as barras de erros representam o desvio padrão de três experimentos de réplica. Clique aqui para ver uma versão maior desta figura.

Figura 4: Perda da expressão mCherry-E3L em células RK13+E3L+K3L. Sobreposição de micrografos fluorescentes e de contraste de fase de células K-R4+K3L-mCherry-E3L infectadas RK13+E3L+K3L. Três placas não expressam mais mCherry (círculos) devido ao colapso do de seleção que produz VC-R4+K3L. Clique aqui para ver uma versão maior desta figura.
| Experimento 1 | Experimento 2 | Experimento 3 | |
| Placas vermelhas (RK13) | 30 | 11 | 18 |
| Placas totais (RK13+E3L+K3L) | 225 | 64 | 249 |
| Taxa de recombinação | 13.30% | 17.20% | 7.20% |
Tabela 1: Frequência de recombinação de VACV com o plasmídeo p837-K3L-mCherry-E3L.
| Experimento 1 | Experimento 2 | Experimento 3 | |
| Placas totais (RK13+E3L+K3L) | 115 | 44 | 210 |
| Placas incolores (RK13+E3L+K3L) | 3 | 1 | 1 |
| Taxa de recombinação | 2.60% | 2.30% | 0.50% |
Tabela 2: Freqüência de perda de mCherry-E3L de células VC-R4+K3L-mCherry-E3L em células RK13+E3+K3.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Aqui apresentamos uma variação de uma estratégia transitória de seleção de marcadores 32 para gerar vírus de vaccinia recombinantes sem reter DNA estranho no vírus recombinante final. Nossa estratégia usa pressão seletiva mediada pelo PKR de proteína antiviral hospedeira em vez de outras formas de pressão seletiva, como antibióticos. O uso de genes antivirais hospedeiros elimina a possibilidade de alterações pheotípicas quimicamente induzidas nas células, ou aumento do risco de mutação devido a drogas de seleção. Além disso, ao contrário da seleção de medicamentos, não há fase de defasagem para nossa abordagem, pois o PKR é expresso constitutivamente em todas as células. A seleção visual secundária baseada na expressão mCherry também melhora a especificidade deste método, garantindo que apenas placas expressando o transgene sejam escolhidas durante a primeira fase, e é igualmente eficiente como um marcador seletivo negativo ao selecionar vírus recombinantes maduros que perderam o gene mCherry-E3L.
As etapas mais críticas para essa estratégia de recombinação são a geração do vetor de recombinação apropriado e a purificação de placa apropriada para garantir que o vírus selecionado seja clonal. Neste artigo sugerimos "Conjunto Gibson" para gerar o vetor de recombinação. Essa estratégia é extremamente eficiente e permite a montagem de todos os fragmentos que compõem o vetor de recombinação em um único dia. No entanto, como o braço curto de 3' e o braço longo de 3' compartilham sequências idênticas, esses fragmentos têm o potencial de serem unidos durante a reação de clonagem, e alguns vetores podem não conter o mCherry-E3L. Em nossa experiência isso é raro, mas confirmar a estrutura do vetor após a clonagem é necessário. Também geramos vetores de recombinação para essa estratégia usando métodos tradicionais de endonuclease e ligase. Essa estratégia evita o problema descrito acima, mas pode ser mais trabalhosa. A purificação da placa é geralmente simples e depende principalmente do uso de células permissivas apropriadas para a recombinação inicial, células competentes para a purificação inicial da placa para garantir que apenas vírus recombinantes possam se replicar e, em seguida, células permissivas novamente para facilitar a recombinação intramolecular e a perda do marcador selecionável. A atenção das linhas celulares é, portanto, fundamental para a aplicação bem-sucedida e eficiente dessa estratégia.
Neste estudo, demonstramos o uso deste método para gerar um VACV recombinante excluído tanto para os antagonistas pkr E3L e K3L quanto para expressar EGFP sob controle do promotor E3L. Daqui para frente, esse vírus servirá como um fundo eficiente para futuros vírus recombinantes, pois é incapaz de se replicar em células competentes do PKR. Portanto, haverá forte pressão seletiva mediada por PKR para conduzir o de recombinação mCherry-E3L em virions progeny e, ao mesmo tempo, essencialmente impedindo a replicação de vírus não recombinantes. Além disso, a perda de EGFP pela captação do de recombinação é um marcador de seleção secundário útil para garantir que as placas escolhidas não sejam co-infectadas com um vírus não recombinante. Observamos taxas de recombinação consistentes com as taxas relatadas anteriormente para VACV, mas os marcadores fluorescentes visuais aumentam a eficiência da geração de vírus recombinantes, garantindo que o aumento da probabilidade de que os vírus recombinantes apropriados sejam selecionados. Nossa observação de placas incolores após duas rodadas de seleção em células competentes para PKR, presumivelmente devido ao aumento do comprimento da seqüência idêntica entre e3L e o gene marcador mCherry-E3L, sugere que a taxa de perda de mCherry-E3L pode ser "sintonizada" aumentando ou diminuindo o comprimento do braço curto de 3'. A principal limitação desta técnica é o uso de PKR como pressão seletiva para recombinantes. O uso mais eficiente dessa estratégia de recombinação é gerar esses vírus em um plano de fundo sem antagonistas de PKR. No entanto, o marcador de seleção colorimétrica permite que essa estratégia de recombinação seja usada mesmo sem a seleção mediada por PKR, simplesmente por placas que purificam placas expressas por mCherry. Embora a falta de pressão seletiva mediada por PKR reduza a eficiência da primeira etapa de triagem, a porcentagem de placas expressas mCherry ainda é alta o suficiente para que a seleção baseada em cores seja viável. Assim, este método pode ser usado para inserir quase qualquer gene no genoma do poxvirus.
Como demonstrado pela inserção do EGFP, com esta abordagem, qualquer gene pode ser rapidamente inserido no lócus E3L sob controle do promotor nativo, desde que células nulas de PKR ou linhas celulares de elogio sejam usadas para experimentos a jusante se o transgene não for um antagonista PKR. Esta estratégia, combinada com o vírus VC-R4 que relatamos aqui, adiciona um método novo e potente para gerar rapidamente e de forma confiável vírus de vaccinia recombinante usando pressão seletiva mediada por hospedeiro e identificação visual de recombinantes no início do processo.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores não declaram interesses financeiros concorrentes.
Acknowledgments
Este projeto foi financiado pelos Institutos Nacionais de Saúde (AI114851) à RS.
Materials
| Name | Company | Catalog Number | Comments |
| 2X-Q5 Master Mix | NEB | M0492L | High-fidelity polymerase used in PCR |
| Ampicillin | ThermoFisher Scientific | 11593027 | Bacterial selective agent |
| Disposable Cell Scrapers | ThermoFisher Scientific | 08-100-242 | Cell scraper to harvest infected cells |
| EVOS FL Auto 2 Cell imaging system | ThermoFisher Scientific | AMAFD2000 | Fluorescent microscope |
| EVOS Light Cube, GFP | ThermoFisher | AMEP4651 | GFP Cube |
| EVOS Light Cube, RFP | ThermoFisher | AMEP4652 | RFP Cube |
| GenJet | SignaGen Laboratories | SL100489 | Transfection reagent |
| Luria Bertani (LB) Broth | Gibco | 10855021 | Bacterial growth medium |
| Monarch DNA gel extraction kit | NEB | T1020L | Gel purification kit used to purify amplicons and linearized vectors |
| Monarch Plasmid Miniprep kit | NEB | T1010L | Miniprep kit ussed to purify plasmids |
| NanoDrop One | ThermoFisher Scientific | ND-ONE-W | Spectrophotometer used to measure RNA and DNA concentration |
| NEBuilder Master Mix | NEB | E2621L | Isothermal enzymatic assembly kit used to generate the recombination vector |
| Q500 Sonicator | Qsonica | Q500-110 | Sonicator for virus lysates |
| RK13 cells | ATCC | CCL-37 | Rabbit kidney cells |
| VWR Multiwell Cell Culture plates | VWR | 10062-892 | Cell culture plates |
References
- Brochier, B., et al. Large-scale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature. 354 (6354), 520-522 (1991).
- Pastoret, P. P., Brochier, B. The development and use of a vaccinia-rabies recombinant oral vaccine for the control of wildlife rabies; a link between Jenner and Pasteur. Epidemiology and Infection. 116 (3), 235-240 (1996).
- Chan, W. M., McFadden, G. Oncolytic Poxviruses. Annual review of virology. 1 (1), 119-141 (2014).
- Nguyen, D. H., et al. Vaccinia virus-mediated expression of human erythropoietin in tumors enhances virotherapy and alleviates cancer-related anemia in mice. Molecular Therapy. 21 (11), 2054-2062 (2013).
- Frentzen, A., et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proceedings of the National Academy of Sciences of the United States of America. 106 (31), 12915-12920 (2009).
- Pastoret, P. P., Vanderplasschen, A. Poxviruses as vaccine vectors. Comparative Immunology, Microbiology and Infectious Diseases. 26 (5-6), 343-355 (2003).
- COLLIER, L. H. The development of a stable smallpox vaccine. The Journal of Hygiene. 53 (1), 76-101 (1955).
- Weir, J. P., Bajszár, G., Moss, B. Mapping of the vaccinia virus thymidine kinase gene by marker rescue and by cell-free translation of selected mRNA. Proceedings of the National Academy of Sciences of the United States of America. 79 (4), 1210-1214 (1982).
- Mackett, M., Smith, G. L., Moss, B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proceedings of the National Academy of Sciences of the United States of America. 79 (23), 7415-7419 (1982).
- Nakano, E., Panicali, D., Paoletti, E. Molecular genetics of vaccinia virus: demonstration of marker rescue. Proceedings of the National Academy of Sciences of the United States of America. 79 (5), 1593-1596 (1982).
- Falkner, F. G., Moss, B. Transient dominant selection of recombinant vaccinia viruses. Journal of Virology. 64 (6), 3108-3111 (1990).
- Staib, C., Drexler, I., Ohlmann, M., Wintersperger, S., Erfle, V., Sutter, G. Transient Host Range Selection for Genetic Engineering of Modified Vaccinia Virus Ankara. BioTechniques. 28 (6), 1137-1148 (2000).
- Staib, C., Drexler, I., Sutter, G. Construction and Isolation of Recombinant MVA. Vaccinia Virus and Poxvirology. , 77-99 (2004).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Pfaller, C. K., Li, Z., George, C. X., Samuel, C. E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Current Opinion in Immunology. 23 (5), 573-582 (2011).
- Bevilacqua, P. C., George, C. X., Samuel, C. E., Cech, T. R. Binding of the protein kinase PKR to RNAs with secondary structure defects: Role of the tandem A - G mismatch and noncontigous helixes. Biochemistry. 37 (18), 6303-6316 (1998).
- Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E., Hinnebusch, A. G. Tight Binding of the Phosphorylated Subunit of Initiation Factor 2 (eIF2) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor eIF2B Is Required for Inhibition of Translation Initiation. Molecular and Cellular Biology. 21 (15), 5018-5030 (2001).
- Rothenburg, S., Georgiadis, M. M., Wek, R. C. Evolution of eIF2α kinases: Adapting translational control to diverse stresses. Evolution of the Protein Synthesis Machinery and Its Regulation. , 235-260 (2016).
- Bratke, K. A., McLysaght, A., Rothenburg, S. A survey of host range genes in poxvirus genomes. Infection, Genetics and Evolution. 14, 406-425 (2013).
- Chang, H. W., Watson, J. C., Jacobs, B. L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proceedings of the National Academy of Sciences. 89 (11), 4825-4829 (1992).
- Romano, P. R., et al. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Molecular and Cellular Biology. 18 (12), 7304-7316 (1998).
- Beattie, E., Tartaglia, J., Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology. 183 (1), 419-422 (1991).
- Park, C., Peng, C., Brennan, G., Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Annals of the New York Academy of Sciences. 1438 (1), 18-29 (2019).
- Rothenburg, S., Brennan, G. Species-Specific Host-Virus Interactions: Implications for Viral Host Range and Virulence. Trends in Microbiology. , (2019).
- Chakrabarti, S., Sisler, J. R., Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques. 23 (6), 1094-1097 (1997).
- Chung, C. T., Niemela, S. L., Miller, R. H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution (recombinant DNA). Biochemistry. 86, 2172-2175 (1989).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Methods in Enzymology. 218, 621-627 (1993).
- Rahman, M. M., Liu, J., Chan, W. M., Rothenburg, S., McFadden, G. Myxoma Virus Protein M029 Is a Dual Function Immunomodulator that Inhibits PKR and Also Conscripts RHA/DHX9 to Promote Expanded Host Tropism and Viral Replication. PLOS Pathogens. 9 (7), 1003465 (2013).
- Evans, D. H., Stuart, D., McFadden, G. High levels of genetic recombination among cotransfected plasmid DNAs in poxvirus-infected mammalian cells. Journal of Virology. 62 (2), 367-375 (1988).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. Journal of Virology. 61 (6), 1788-1795 (1987).
- Spyropoulos, D. D., Roberts, B. E., Panicali, D. L., Cohen, L. K. Delineation of the viral products of recombination in vaccinia virus-infected cells. Journal of Virology. 62 (3), 1046-1054 (1988).
- Liu, L., et al. Transient dominant host-range selection using Chinese hamster ovary cells to generate marker-free recombinant viral vectors from vaccinia virus. BioTechniques. 62 (4), 183-187 (2017).
